
Abstract
Background
Baylisascaris schroederi is one of the most significant threats to the giant panda’s survival, responsible for half of the deaths reported from 2001 to 2005. MicroRNA (miRNA) has been identified as one of the key factors for gene regulations at the post-transcriptional level, and also considered as a potential control and treatment target against infectious diseases.
Methods
The present study investigated the miRNA profile of B. schroederi via high throughput sequencing and real-time quantitative PCR.
Results
A total of 18.07 million raw reads were obtained and 18.01 million were identified with high quality. By analysis of standard stem-loop structures, 108 miRNA candidates were predicted, including 60 known miRNAs and 48 novel ones. Target prediction revealed that the “chitinase” was the most abundant target with 483 sequences, and 263 targets were related to ovarian and egg development. The ribosomal protein related sequences occupied 449 sequences.
Conclusions
Previous studies have shown that some parasites secrete chitinases for exsheathment and/or for penetrating the peritrophic matrix of the host. It therefore seems that B. schroederi may be effectively regulated by miRNAs for development, invasion, and reproduction. Given that chitinases have been identified as important biological control agents for pests, identification of microRNAs in B. schroederi of the giant panda would provide useful information for the development of biological control strategies and/or vaccines against B. schroederi infection in the giant panda.
Similar content being viewed by others

Background
The giant panda (Ailuropoda melanoleuca) is one of flag and rare species for wildlife conservation, with an estimate of about 1600 restricted to Qinling, Minshan, Qionglai, Daxiangling, Xiaoxiangling and Liangshan mountains in China [1–3]. The giant panda is famous for its cute appearance, important taxonomic status and valuable gene pool for genetic studies [4–7]. Unfortunately, it is also one of the most endangered species due to limited habitats, surprisingly extensive poaching, and parasitic and other infections [8–10].
The roundworms in the family Ascarididae are parasitic nematodes with great medical, veterinary and economic significance [11, 12]. Within this family, Baylisascaris spp. could infect animals and humans, resulting in visceral larval migrants (VLM), ocular larva migrants (OLM) and even neural larva migrants (NLM) that is normally fatal to some wild animals [13]. Among them, B. schroederi is the most common parasite in wild and captive giant pandas, and the VLM caused by B. schroederi infection was identified as the most significant threat to the survival of the giant panda, responsible for half of the deaths from 2001 to 2005 [8, 14].
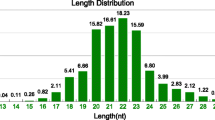
MicroRNAs (miRNAs) are small non-coding RNAs with a length of 18–25 nt which have been identified in various plants, animals and virus. They play key regulatory functions for gene expression at the post-transcriptional level [15–18] and are considered as a potential treatment target against parasitic and other infectious diseases [19, 20]. Therefore, identification and prediction of miRNAs in pathogenic agents have important implications for controlling their infection. In the present study, the miRNA expression profile of B. schroederi was investigated by high throughput sequencing technology and real-time quantitative PCR.
Methods
Ethics statement
The present study was performed strictly according to the Guidelines and Recommendations for the Care and Use of Laboratory Animals of the Ministry of Health, China, and the study protocol was reviewed and approved by the Research Ethics Committee of Northwest A&F University.
Parasites
Adult female nematodes were collected from the faeces of the giant pandas after anthelmintic treatment in Shaanxi Rare Wildlife Rescue Breeding Research Center rescued from Qinling Mountains in Shaanxi province, China. Worms were incubated in physiological saline for 3 h at 37°C and then washed extensively to get rid of contamination from the host. Female adults were identified by morphology and further ascertained as B. schroederi by sequencing the first internal transcribed spacer (ITS-1) of nuclear ribosomal DNA [21]. The parasites were then stored in liquid nitrogen for further study.
Isolation of total RNA and small RNA
Total RNA and small RNA of the worms were prepared as described previously [22]. Briefly, one whole worm was grounded into fine powder under liquid nitrogen. The total RNA was prepared using TRIzol Reagent according to the manufacturer’s protocol (Invitrogen, USA). Ten micrograms of total RNA were used to separate small RNA of 20–40 nt length via a Novex 15% TBE-Urea gel. The purified fragments were ligated with 5′ and 3′ adaptors (Illumina, USA), re-purified on a Novex 10% TBE-Urea gel, and finally reversely transcribed with an RT-PCR kit. All the gels and kits were purchased from Invitrogen Co. Ltd.
High-throughput sequencing and data analysis
The total RNA was deeply sequenced with Illumina Hiseq 2000 sequencer at HuaDa Genomic Co. Ltd, Shenzhen, China. The data was analyzed as described previously [23, 24]. After base-calling, adaptors, reads smaller than 18 nt and those with low qualities were discarded, the remaining sequences were firstly searched against the Rfam databases (http://rfam.sanger.ac.uk/) to identify non-coding RNA, including rRNA, tRNA, snRNA, and snoRNA, and then searched against RepeatMasker (http://www.repeatmasker.org) to identify kinds and numbers of repetitive sequences. The genome sequence of Ascaris suum was used as a reference [25] and mapped with filtered reads via SOAP [26]. The miRNA precursors were identified with Mfold (http://www.bioinfo.rpi.edu/applications/mfold) and checked manually. Only the miRNAs with standard stem-loop structure and energy lower than −18 kcal/mol were saved to form the miRNA expression profile. The predicted mature miRNAs were mapped with A. suum miRNAs deposited in Sanger miRBase (http://www.mirbase.org/) to identify the known miRNAs and novel miRNAs.
The mRNA and EST sequences were downloaded from NCBI and used for target analysis of B. schroederi specific miRNAs via RNAhybrid [27]. The predicated targets were then performed for functional analysis via Gene Ontology (http://www.geneontology.org/) and DAVID database (http://david.abcc.ncifcrf.gov/).
Analysis of miRNA expression level
The modified stem-loop real-time RT-PCR (ABI PRISM® 7300 Sequence Detection System) was used to determine the expressional level of representative miRNAs as described previously [28]. All reactions were carried out in triplicate with a 20 μl reaction mixture. The β-actin gene was used as the endogenous control with primers as follows: forward primer (5′-CTCGAAACAAGAATACGATG-3′) and reverse primer (5′-ACATGTGCCGTTGTATGATG-3′) [29]. Primers were synthesized by Shenggong Co, Ltd., Shanghai, China. The amplification cycles were as follows: 94°C for 32 s, 52°C for 30 s, 72°C for 30 s for 30 cycles. The expression level of miRNA was calculated as: N = 2-ΔCt, ΔCt = CtmiRNA-Ctacin[30].
Results
Profile characteristics of short RNAs
High-throughput sequencing generated 18.07 million raw reads. After base-calling, a total of 18.01 million reads showed high quality. After removing adaptors, poly-A and reads smaller than 18 nt, there were 16.26 million reads left for further analysis. The small non-coding RNAs, including rRNA, tRNA, snRNA and snoRNA, occupied 14.91% of total reads and 5.17% of unique reads, representing high redundancy of these non-coding RNAs. The percentages of rRNA were 13.48% and 4.41% in total and unique reads, respectively. Two types of repeat sequences were identified, namely rRNA: 0 (214 reads) and rRNA: 1 (3758 reads).
Analysis of miRNA profile
Mapping with genomic sequences of A. suum, we obtained 108 miRNA candidates with standard stem-loop structures. For 23 of these miRNAs, multiple locations were detected in the reference genome. Within them, 14 locations were found for the PAA-novel-44 (AGAAGCTGCGTTTTAAATGG), with 10 and 4 in the sense and antisense strands, respectively. The mature miRNA of PAA-novel-44 (Bsc-miR-44) was located at the 5′ arm of its precursors. The other significant miRNA was PAA-novel-45 (Bsc-miR-45), for which 8 locations were found with mature miRNAs at both the 5′ and 3′ arms of the precursor (Figure 1).

Standard stem-loop structure of Bsc-miR-45. First line: name of precursor and energy of stem-loop structure; Others: precursor sequences, mapped reads and stem-loop structure. Color of stem-loop structure: from red to black shows the direction of 5′ to 3′.
Compared with known A. suum miRNA deposited in the Sanger miRBase, 60 miRNAs were previously identified and 48 were novel candidates (see Additional file 1). The mature miRNAs at both 5′ and 3′ arms were identified for 58.3% (35/60) of known miRNAs and 8.3% (4/48) of the novel miRNAs.
Target prediction and functional analysis
The 48 novel miRNAs of B. schroederi were further analyzed for target and function predictions. A total of 58438 mRNA/EST of A. suum (56616 sequences) and A. lumbricoides (1822 sequences), two nematodes being proposed as the same species with different genotypes [31], were downloaded from GenBank.
A total of 6975 targets (137 in average) were predicted for 48 B. schroederi novel miRNAs. Among these targets, 4 117 were well annotated and yielded a 1020 non-repeated dataset (see Additional file 2). The highest number of targets (853) was found for the Bsc-miR-51, but only one target was matched with miRNAs of Bsc-miR-21, Bsc-miR-34 and Bsc-miR-47. For well annotated targets, the most abundant target was the “chitinase” in A. suum, with 483 sequences. The target of “immunosuppressive ovarian message protein” was found within 199 sequences. Two other ovarian related targets were also predicted, namely “vitellogenin precursor-like protein” (33 sequences) and “ovarian abundant message protein” (31 sequences). Therefore, a total of 263 targets were related to ovarian and egg development.
In addition, the ribosomal protein related sequences occupied 449 sequences, including 152 sequences for 40S ribosomal proteins and 228 sequences for 60S ribosomal proteins, such as 40S ribosomal protein s2, 60S ribosomal protein l10, and 60S ribosomal protein l44. For parasite distributions of the annotated sequences, most of them were obtained from nematodes, including Loa loa, Wuchereria bancrofti, Brugia malayi, Ascaris suum, A. lumbricoides and Haemonchus contortus.
miRNAs quantification
Three known miRNAs named miR-100a-5p, miR-44b-3p, and miR-87a-3p, as well as three novel miRNAs of B. schroederi named Bsc-miR-45, Bsc-miR-39 and Bsc-miR-40, were randomly selected for real-time quantitative PCR analysis. All six miRNAs were successfully amplified (Figure 2). The relative expression levels of Bsc-miR-39 (28.43 ± 2.97), miR-87a-3p (12.89 ± 0.72), and miR-100a-5p (8.27 ± 0.43) were significantly higher than that of other miRNAs, while the relative expression levels of miR-44b-3p (1 ± 0.06) and Bsc-miR-45 (1.11 ± 0.23) were similar to that of the endogenous control. Very low expression level (0.03 ± 0) was detected for Bsc-miR-40, which was not shown in Figure 2.

Real-time quantitative PCR analysis for the three known miRNAs as well as three novel miRNAs of B. schroederi . Some miRNAs had significant higher relative expression level than others, such as Bsc-miR-39, miR-87a-3p, and miR-100a-5p. The Bsc-miR-40 had a very low expression level and is not shown in the Figure.
Discussion
The present study characterized the expression profile of miRNAs in B. schroederi from the giant panda in China. We predicted 108 miRNAs for B. schroederi, and this number was similar to that of miRNAs in A. suum predicted by Wang et al. (2011) [32]. However, the number of identified miRNAs herein was much smaller than that for A. suum in our previous study [23]. Given that the functions of miRNAs are mainly in regulating gene expression and ensuring genome maintenance, the expression level of miRNAs is highly sensitive to environmental and developmental signals [33–37]. Therefore, the miRNAs expression level of different parasite species, even the same species or stage, might be reflected by these conditions and infection status.
The regulating functions of miRNAs were played via complementing with target mRNAs to repress expression at protein translation level or affecting target mRNA maintenance. Target prediction for miRNAs of B. schroederi showed that the most abundant sequences were chitinases. The chitinases are enzymes widely distributed in bacteria, virus, fungi, plants and animals [38–40]. Some of the chitinases served non-redundant functions and were essential for survival, molting or development, digestion, molting, defense and pathogenicity of insects [41, 42]. Some parasites, such as Brugia malayi and Plasmodium, can secrete chitinases for exsheathment of microfilaria and/or for penetrating the peritrophic matrix of host [43, 44]. Recently, chitinases have been used to biologically control insect pests on transgenic plants either alone or in combination with other insecticidal proteins, and some chitinases also prove to be possibly useful as biocontrol agents and/or as vaccines [42]. Therefore, predicting microRNA targets would provide useful information for development of biological control strategies and/or vaccines against B. schroederi infection.
The other types of significant targets were proteins related to ovarian or egg development, including “immunosuppressive ovarian message protein” (199 sequences), “vitellogenin precursor-like protein” (33 sequences) and “ovarian abundant message protein” (31 sequences). For adult female B. schroederi, high numbers of ovarian related targets indicated the importance of miRNAs in regulating germ cell development of this nematode.
Ribosomes play a basic housekeeping role in global translation, and some rare developmental phenotypes could be detected in a number of ribosomal-protein-defective mutants [45]. Recent study also showed that the ribosome was emerging as a central hub in sensing the nature of the nascent protein chain, recruiting protein folding and translocation components, and integrating mRNA and nascent chain quality control [46]. In the present study, we predicted redundant high numbers of ribosome related targets (including 40S and 60S ribosomal proteins) complementing with B. schroederi novel miRNAs, indicating active activities and effective regulations of protein expression for this parasite.
Conclusions
We firstly investigated the miRNA profile of B. schroederi from the giant panda in China. A total of 108 miRNA candidates were identified, with 48 representing novel miRNAs. Target analysis showed that high redundant numbers of sequences were related to chitinases, ovarian and ribosomes. These results provided novel resources for in-depth understanding of the biology of B. schroederi, and have profound implications for the development of miRNA-based drugs/vaccines against B. schroederi infection in the giant panda.
References
Wei F, Hu Y, Zhu L, Bruford M, Zhan X, Zhang L: Black and white and read all over: the past, present and future of giant panda genetics. Mol Ecol. 2012, 21: 5660-5674. 10.1111/mec.12096.
Reed F, Gregson R, Girling S, Pizzi R, Clutton RE: Anaesthesia of a captive, male giant panda (Ailuropoda melanoleuca). Vet Anaesth Analg. 2013, 40: 103-104. 10.1111/j.1467-2995.2012.00777.x.
Yang B, Busch J, Zhang L, Ran J, Gu X, Zhang W, Du B, Mittermeier RA: Eco-compensation for giant panda habitat. Science. 2013, 339: 521-10.1126/science.339.6119.521.
Wan QH, Zeng CJ, Ni XW, Pan HJ, Fang SG: Giant panda genomic data provide insight into the birth-and-death process of mammalian major histocompatibility complex class II genes. PLoS One. 2009, 4: e4147-10.1371/journal.pone.0004147.
Swaisgood RR, Wei F, Wildt DE, Kouba AJ, Zhang Z: Giant panda conservation science: how far we have come. Biol Lett. 2010, 6: 143-145. 10.1098/rsbl.2009.0786.
Jin K, Xue C, Wu X, Qian J, Zhu Y, Yang Z, Yonezawa T, Crabbe MJ, Cao Y, Hasegawa M, Zhong Y, Zheng Y: Why does the giant panda eat bamboo? A comparative analysis of appetite-reward-related genes among mammals. PLoS One. 2011, 6: e22602-10.1371/journal.pone.0022602.
Abella J, Alba DM, Robles JM, Valenciano A, Rotgers C, Carmona R, Montoya P, Morales J: Kretzoiarctos gen. nov., the oldest member of the giant panda clade. PLoS One. 2012, 7: e48985-10.1371/journal.pone.0048985.
Zhang JS, Daszak P, Huang HL, Yang GY, Kilpatrick AM, Zhang S: Parasite threat to panda conservation. Ecohealth. 2008, 5: 6-9. 10.1007/s10393-007-0139-8.
Xie Y, Zhang Z, Wang C, Lan J, Li Y, Chen Z, Fu Y, Nie H, Yan N, Gu X, Wang S, Peng X, Yang G: Complete mitochondrial genomes of Baylisascaris schroederi, Baylisascaris ailuri and Baylisascaris transfuga from giant panda, red panda and polar bear. Gene. 2011, 482: 59-67. 10.1016/j.gene.2011.05.004.
Du YJ, Hou YL, Hou WR: Molecular characterization of a gene POLR2H encoded an essential subunit for RNA polymerase II from the Giant Panda (Ailuropoda Melanoleuca). Mol Biol Rep. 2013, 40: 1495-1498. 10.1007/s11033-012-2192-9.
Dold C, Holland CV: Ascaris and ascariasis. Microbes Infect. 2011, 13: 632-637. 10.1016/j.micinf.2010.09.012.
Testini G, Papini R, Lia RP, Parisi A, Dantas-Torres F, Traversa D, Otranto D: New insights into the morphology, molecular characterization and identification of Baylisascaris transfuga (Ascaridida, Ascarididae). Vet Parasitol. 2011, 175: 97-102. 10.1016/j.vetpar.2010.09.017.
Sato H, Matsuo K, Osanai A, Kamiya H, Akao N, Owaki S, Furuoka H: Larva migrans by Baylisascaris transfuga: fatal neurological diseases in Mongolian jirds, but not in mice. J Parasitol. 2004, 90: 774-781. 10.1645/GE-3330.
He G, Wang T, Yang G, Fei Y, Zhang Z, Wang C, Yang Z, Lan J, Luo L, Liu L: Sequence analysis of Bs-Ag2 gene from Baylisascaris schroederi of giant panda and evaluation of the efficacy of a recombinant Bs-Ag2 antigen in mice. Vaccine. 2009, 27: 3007-3011. 10.1016/j.vaccine.2009.02.077.
Carrington JC, Ambros V: Role of microRNAs in plant and animal development. Science. 2003, 301: 336-338. 10.1126/science.1085242.
Du T, Zamore PD: Beginning to understand microRNA function. Cell Res. 2007, 17: 661-663. 10.1038/cr.2007.67.
Zampetaki A, Mayr M: MicroRNAs in vascular and metabolic disease. Circ Res. 2012, 110: 508-522. 10.1161/CIRCRESAHA.111.247445.
Fernandez-Hernando C, Ramirez CM, Goedeke L, Suarez Y: MicroRNAs in metabolic disease. Arterioscler Thromb Vasc Biol. 2013, 33: 178-185. 10.1161/ATVBAHA.112.300144.
Segura MF, Greenwald HS, Hanniford D, Osman I, Hernando E: MicroRNA and cutaneous melanoma: from discovery to prognosis and therapy. Carcinogenesis. 2012, 33: 1823-1832. 10.1093/carcin/bgs205.
Liu X, Liu L, Xu Q, Wu P, Zuo X, Ji A: MicroRNA as a novel drug target for cancer therapy. Expert Opin Biol Ther. 2012, 12: 573-580. 10.1517/14712598.2012.671293.
Lin Q, Li HM, Gao M, Wang XY, Ren WX, Cong MM, Tan XC, Chen CX, Yu SK, Zhao GH: Characterization of Baylisascaris schroederi from Qinling subspecies of giant panda in China by the first internal transcribed spacer (ITS-1) of nuclear ribosomal DNA. Parasitol Res. 2012, 110: 1297-1303. 10.1007/s00436-011-2618-7.
Xu MJ, Fu JH, Nisbet A, Huang SY, Zhou DH, Lin RQ, Song HQ, Zhu XQ: Comparative profiling of microRNAs in male and female adults of Ascaris suum. Parasitol Res. 2013, 112: 1189-1195. 10.1007/s00436-012-3250-x.
Xu MJ, Ai L, Fu JH, Nisbet AJ, Liu QY, Chen MX, Zhou DH, Zhu XQ: Comparative characterization of microRNAs from the liver flukes Fasciola gigantica and F. hepatica. PLoS One. 2012, 7: e53387-10.1371/journal.pone.0053387.
Xu MJ, Wang CR, Huang SY, Fu JH, Zhou DH, Chang QC, Zheng X, Zhu XQ: Identification and characterization of microRNAs in the pancreatic fluke Eurytrema pancreaticum. Parasit Vectors. 2013, 6: 25-10.1186/1756-3305-6-25.
Jex AR, Liu S, Li B, Young ND, Hall RS, Li Y, Yang L, Zeng N, Xu X, Xiong Z, Chen F, Wu X, Zhang G, Fang X, Kang Y, Anderson GA, Harris TW, Campbell BE, Vlaminck J, Wang T, Cantacessi C, Schwarz EM, Ranganathan S, Geldhof P, Nejsum P, Sternberg PW, Yang H, Wang J, Wang J, Gasser RB: Ascaris suum draft genome. Nature. 2011, 479: 529-533. 10.1038/nature10553.
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J: SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009, 25: 1966-1967. 10.1093/bioinformatics/btp336.
Kruger J, Rehmsmeier M: RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34: W451-W454. 10.1093/nar/gkl243.
Xu MJ, Liu Q, Nisbet AJ, Cai XQ, Yan C, Lin RQ, Yuan ZG, Song HQ, He XH, Zhu XQ: Identification and characterization of microRNAs in Clonorchis sinensis of human health significance. BMC Genomics. 2010, 11: 521-10.1186/1471-2164-11-521.
Huang CQ, Gasser RB, Cantacessi C, Nisbet AJ, Zhong W, Sternberg PW, Loukas A, Mulvenna J, Lin RQ, Chen N, Zhu XQ: Genomic-bioinformatic analysis of transcripts enriched in the third-stage larva of the parasitic nematode Ascaris suum. PLoS Negl Trop Dis. 2008, 2: e246-10.1371/journal.pntd.0000246.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C (T)) Method. Methods. 2001, 25: 402-408. 10.1006/meth.2001.1262.
Leles D, Gardner SL, Reinhard K, Iniguez A, Araujo A: Are Ascaris lumbricoides and Ascaris suum a single species?. Parasit Vectors. 2012, 5: 42-10.1186/1756-3305-5-42.
Wang J, Czech B, Crunk A, Wallace A, Mitreva M, Hannon GJ, Davis RE: Deep small RNA sequencing from the nematode Ascaris reveals conservation, functional diversification, and novel developmental profiles. Genome Res. 2011, 21: 1462-1477. 10.1101/gr.121426.111.
Zeiner GM, Norman KL, Thomson JM, Hammond SM, Boothroyd JC: Toxoplasma gondii infection specifically increases the levels of key host microRNAs. PLoS One. 2010, 5: e8742-10.1371/journal.pone.0008742.
Kulkarni S, Savan R, Qi Y, Gao X, Yuki Y, Bass SE, Martin MP, Hunt P, Deeks SG, Telenti A, Pereyra F, Goldstein D, Wolinsky S, Walker B, Young HA, Carrington M: Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature. 2011, 472: 495-498. 10.1038/nature09914.
Png KJ, Halberg N, Yoshida M, Tavazoie SF: A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature. 2012, 481: 190-194.
Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, Kaluza D, Treguer K, Carmona G, Bonauer A, Horrevoets AJ, Didier N, Girmatsion Z, Biliczki P, Ehrlich JR, Katus HA, Muller OJ, Potente M, Zeiher AM, Hermeking H, Dimmeler S: MicroRNA-34a regulates cardiac ageing and function. Nature. 2013, 495: 107-110. 10.1038/nature11919.
TenOever BR: RNA viruses and the host microRNA machinery. Nat Rev Microbiol. 2013, 11: 169-180. 10.1038/nrmicro2971.
Denarie J, Cullimore J: Lipo-oligosaccharide nodulation factors: a minireview new class of signaling molecules mediating recognition and morphogenesis. Cell. 1993, 74: 951-954. 10.1016/0092-8674(93)90717-5.
Frederiksen RF, Paspaliari DK, Larsen T, Storgaard BG, Larsen MH, Ingmer H, Palcic MM, Leisner JJ: Bacterial chitinases and chitin-binding proteins as virulence factors. Microbiology. 2013, 159: 833-847. 10.1099/mic.0.051839-0.
Gauvreau GM: Allergen bronchoprovocation and chitinases in allergic asthma. Clin Exp Allergy. 2013, 43: 149-151. 10.1111/cea.12070.
Loh JT, Stacey G: Feedback regulation of the Bradyrhizobium japonicum nodulation genes. Mol Microbiol. 2001, 41: 1357-1364. 10.1046/j.1365-2958.2001.02603.x.
Arakane Y, Muthukrishnan S: Insect chitinase and chitinase-like proteins. Cell Mol Life Sci. 2010, 67: 201-216. 10.1007/s00018-009-0161-9.
Arnold K, Venegas A, Houseweart C, Fuhrman JA: Discrete transcripts encode multiple chitinase isoforms in Brugian microfilariae. Mol Biochem Parasitol. 1996, 80: 149-158. 10.1016/0166-6851(96)02671-0.
Shahabuddin M, Kaslow DC: Plasmodium: parasite chitinase and its role in malaria transmission. Exp Parasitol. 1994, 79: 85-88. 10.1006/expr.1994.1066.
Horiguchi G, Van Lijsebettens M, Candela H, Micol JL, Tsukaya H: Ribosomes and translation in plant developmental control. Plant Sci. 2012, 191–192: 24-34.
Pechmann S, Willmund F, Frydman J: The ribosome as a hub for protein quality control. Mol Cell. 2013, 49: 411-421. 10.1016/j.molcel.2013.01.020.
Acknowledgements
This work was supported in part by the International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840), the Funds for Basic Research Key Program in Northwest A&F University (Grant No. ZD2012010), and the Science Fund for Creative Research Groups of Gansu Province (Grant No. 1210RJIA006).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XQZ and GHZ conceived and designed the study, and critically revised the manuscript. GHZ and MJX performed the experiments, analyzed the data and drafted the manuscript. All authors read and approved the final manuscript.
Guang-Hui Zhao, Min-Jun Xu contributed equally to this work.
Electronic supplementary material
13071_2013_997_MOESM1_ESM.xlsx
Additional file 1: Known and novel miRNAs of Baylisascaris schroederi . This Excel table describes in detail the sequences of the identified known (n = 60) and novel (n = 48) miRNAs from B. schroederi. (XLSX 14 KB)
13071_2013_997_MOESM2_ESM.xlsx
Additional file 2: Well annotated targets of the 48 novel/specific miRNAs of Baylisascaris schroederi . This Excel table details the predicted targets and their origins of the 48 novel/specific miRNAs identified from B. schroederi. (XLSX 115 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhao, GH., Xu, MJ. & Zhu, XQ. Identification and characterization of microRNAs in Baylisascaris schroederi of the giant panda. Parasites Vectors 6, 216 (2013). https://doi.org/10.1186/1756-3305-6-216
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-6-216