
Abstract
Background
Eurytrema pancreaticum is one of the most common flukes, which mainly infects ruminants globally and infects human beings accidentally; causing eurytremiasis that has high veterinary and economic importance. MicroRNAs (miRNAs) are small non-coding RNAs and are now considered as a key mechanism of gene regulation at the post-transcription level.
Methods
We investigated the global miRNA expression profile of E. pancreaticum adults using next-generation sequencing technology combined with real-time quantitative PCR.
Results
By using the genome of the closely-related species Schistosoma japonicum as reference, we obtained 27 miRNA candidates out of 16.45 million raw sequencing reads, with 13 of them found as known miRNAs in S. japonicum and/or S. mansoni, and the remaining 14 miRNAs were considered as novel. Five out of the 13 known miRNAs coming from one family named as sja-miR-2, including family members from miR-2a to miR-2e. Targets of 19 miRNAs were successfully predicated out of the 17401 mRNA and EST non-redundant sequences of S. japonicum. It was found that a significant high number of targets were related to “chch domain-containing protein mitochondrial precursor” (n = 29), “small subunit ribosomal protein s30e” (n = 21), and “insulin-induced gene 1 protein” (n = 9). Besides, “egg protein cp3842” (n = 2), “fumarate hydratase” (n = 2), “ubiquitin-conjugating enzyme” (n = 2), and “sperm-associated antigen 6” (n = 1) were also found as targets of the miRNAs of E. pancreaticum.
Conclusions
The present study represents the first global characterization of E. pancreaticum miRNAs, which provides novel resources for a better understanding of the parasite, which, in turn, has implications for the effective control of the disease it causes.
Similar content being viewed by others


Background
The pancreatic fluke Eurytrema pancreaticum is one of the most common flukes in the pancreas and bile ducts of ruminants[1]. It is closely related to Schistosoma japonicum, which has more than 40 species of mammals serving as potential zoonotic reservoirs, which com-plicates parasite transmission dynamics[2]. As a member of the Trematoda, E. pancreaticum also has a broad range of hosts and mainly infects ruminants, including cattle, buffaloes, pigs, sheep, and goats[3–6]. Some reports indicated that it can accidentally infect humans through dietary habits[7]. E. pancreaticum infection causes eurytremiasis with high veterinary and economic importance, which is marked by gastrointestinal dis-turbances and progressive weight loss, diarrhea or constipation, and death, with economic losses in meat and milk production[3, 8, 9]. Eurytremiasis is distributed globally, in South America, Europe and Asia, including countries such as Brazil, China, Japan and Thailand[8, 10], and it might be under evaluated due to the current investigating methods[11].
MicroRNAs (miRNAs) are small non-coding RNAs regulating gene expression at the post-transcriptional level and resulting in post-transcriptional repression. miRNAs are conserved in metazoans and can be used as biomarkers[12]. miRNAs were reported in diverse organisms from viruses to mammals[13–15], and are now considered as a key mechanism of gene regulation and are essential for the complex life cycle of pathogenic parasites[13]. The miRNAs of some members of the Trematoda, including Schistosoma japonicum, S. mansoni, Orientobilharzia turkestanicum, Fasciola hepatica and F. gigantica have been reported[16–20]. However, there was no miRNAs identified from E. pancreaticum despite its veterinary and economic importance.
As a member of the Trematoda, E. pancreaticum may also have miRNAs involved in its gene regulation in the pancreatic fluke. Here we investigated the expression profile of miRNAs and detected potential novel miRNAs in E. pancreaticum adults. Due to the similarity in morphology, life cycle and modes of transmission among members of the Trematoda[5, 21], miRNA profile research in E. pancreaticum will shed light on the miRNA studies of other species such as Dicrocoelium dendriticum and E. coelomaticum.
Methods
Ethics statement
This study was approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (Approval No. LVRIAEC2011-006). The sheep from which E. pancreaticum adults were collected, was handled in accordance with good animal practices required by the Animal Ethics Procedures and Guidelines of the People’s Republic of China.
Parasites
Adults of E. pancreaticum were collected from the pancreas of a sheep (Northeast Merino) with a naturally acquired infection in December 2011 in Daqing City, Heilongjiang Province, China. Worms were randomly selected, confirmed as the adult stage with mature eggs based on microscopic examination after staining with carmine[22]. After being washed extensively with sterile physiological saline (37°C) in a sterile beaker, the parasites were transferred to Dulbecco’s modification of Eagle’s medium (DMEM) and incubated at 37°C (10% CO2) for 3 h to allow the flukes to regurgitate all the gut contents from their digestive tracts, and then stored at −80°C until use.
Total RNA and small RNA isolation
Total RNA of ten worms was prepared with Trizol Reagent according to the manufacturer’s protocol (Invitrogen Co. Ltd). Small RNA was prepared as previously[23]. Briefly, RNA fragments of 20–35 bases in length were isolated from 10 μg total RNA with a Novex 15% TBE-Urea gel. These fragments were then reverse transcribed and purified using a 6% TBE PAGE gel. All gels and kits were purchased from Invitrogen Co. Ltd.
High-throughput sequencing and computational analysis
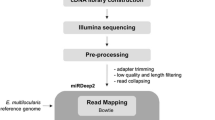
Samples were sequenced using a Solexa (Illumina) sequencer. Adaptors, low quality reads and reads smaller than 18 nucleotides (nt) were firstly removed from the raw dataset. Rfam database (version 10.1) (http://rfam.sanger.ac.uk/) was searched with BLAST software[24] to remove non-coding RNA, including rRNA, tRNA, snRNA, snoRNA. RepeatMasker (http://www.repeatmasker.org) was used to identify repetitive sequences. Because no publically-available genome is currently accessible for Eurytrema spp., the genome of the related schistosome and the closely-related species in genetic distance, S. japonicum (http://lifecenter.sgst.cn/schistosoma) was used as a reference genome, using the SOAP software[25]. The software Mfold (http://www.bioinfo.rpi.edu/applications/mfold) was used for the prediction of miRNA candidates. The identified miRNA candidates were then searched against the Sanger miRBase (version 17.0) to identify known or conserved miRNAs.
The mRNA and EST data of S. japonicum were downloaded from the CHGC database (http://www.chgc.sh.cn/japonicum/Resources.html). Potential targets of known miRNAs were predicated with RNAhybrid software[26]. To reduce false-positive results, two extra parameters were performed on the analyzed result: 1) the △△G was set as lower than −25 kcal/mol; 2) P-value was set as ≦0.01. The Gene Ontology (GO, http://www.geneontology.org/) database was used for functional analysis of predicted targets.
Analysis of novel miRNA transcription
Novel miRNAs were analyzed using a modified stem-loop real-time RT-PCR (ABI PRISM® 7300 Sequence Detection System). All of the primers were synthesized by Shenggong Co, Ltd., China. All reactions were carried out in triplicate. Synthetic lin 4 was used as the endogenous control[27]. The primer pairs were as follows: forward 5′-ACACTCCAGCTGGGTCCCTGAGACCTCAAGTG-3′ and reverse 5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCACACTT-3′. The amplification cycle conditions were as follows: 95°C for 5 min, followed by 30 cycles of 95°C for 15 s, 65°C for 15 s, and 72°C for 32 s. The quantification of each miRNA relative to the cel-lin-4 was calculated using the equation: N = 2-ΔCt, ΔCt = CtmiRNA-Ctlin4[28].
Results
Profile characteristics of short RNAs
High throughput sequencing yielded 16.45 million raw reads from the total RNA of E. pancreaticum. After removing low quality reads, adaptors and poly-A sequences, there were 15.35 million reads left with high quality. Length distribution analysis showed that most of the reads were significantly focused on the length of 20 nt with 23.32%; while those around it, including reads of 19, 21 and 22 nt, had only 4.86%, 7.62%, and 7.23%, respectively.
Those representing exons and introns accounted only for a very small percentage of the clean reads (0.18% of unique siRNA), which indicated high integrity of the RNA in the sample. Repeat analysis revealed hundreds of repeat sequences (0.02%), including two types as LINE/RTE:0 (274 reads) and LINE/RTE:1 (558 reads). Other non-coding RNA, including tRNA, rRNA, snRNA and snoRNA, represented a 4.51% of the total.
Analysis of miRNA profiles
By mapping with the S. japonicum genome, we obtained 27 miRNA candidates with the precursors having standard stem-loop structures, from the total RNA of the parasite. By matching the miRNA candidates with known Trematoda miRNAs, including 56 S. japonicum miRNAs (Sja-miR) and 20 S. mansoni miRNAs (Sma-miR), deposited in the miRBase database, 13 miRNA candidates were previously identified, with 14 novel candidates (Table 1).
Two conserved miRNAs named as bantam and let-7 were found in both S. japonicum and S. mansoni miRNAs. The miRNAs bantam and let-7 are conserved miRNAs found in 27 other organisms with miRNAs deposited in the miRBase database, while the let-7 was found distributed in a larger range of 67 other organisms, including invertebrates and vertebrates.
One of the distinguished characteristics of the known miRNAs was that 5 miRNAs were members of one family named miR-2, including members from miR-2a to miR-2e. All of these miRNAs are located at the 3p arm of their precursors. Besides the miR-2 family, another two miRNAs, miR-71 and miR-71b-5p, were also found from one family, which is located on two different scaffolds named as SJC_S000054 and SJC_S000102. Except for miR-10 that had two mature miRNAs located at the 5p and 3p arms of its precusor, all of the other miRNAs had only one mature miRNA found at 5p or 3p arms of their precursors.
Among the novel miRNAs, 3 miRNAs (Epa-miR-05, Epa-miR-06, and Epa-miR-07) were from one scaffold named as SJC_S000102. Another two miRNAs named Epa-miR-09 and Epa-miR-10 came from another scaffold named as SJC_S000254. Only Epa-miR-14 had both mature miRNAs found at the 5p and 3p of its precursors, and all the others had only one mature miRNA found. The detailed blast information and stem-loop structure of Epa-miR-14 is shown in Figure 1.

The stem-loop structure of Epa-miR-14. The first line including gene location, length of precursor, and energy of stem-loop structure. The mature miRNA in the precursor was shown in red letter (5p) and blue letter (3p). Color from red to black in the stem-loop structure of the miRNA indicated the 5′ to 3′ direction.
Target prediction and functional analysis
A total of 17,401 mRNA and EST non-redundant sequences of S. japonicum were downloaded from the CHGC website (See Methods) and used for target prediction for the 27 miRNAs identified in the E. pancreaticum. Targets of 19 miRNAs were successfully predicted, with numbers ranged from one (Epa-miR-03) to 33 (Epa-miR-01) with 226 in total and an average of 12 (Additional file1: Table S1).
Out of 226 total targets, 130 targets were successfully illustrated with Blast analysis (Additional file2: Table S2). Among which, a significant high number of targets were related to “chch domain-containing protein mitochondrial precursor” (n = 29), which were followed by “small subunit ribosomal protein s30e” (n = 21), “insulin-induced gene 1 protein” (n = 9) and “kh domain-RNA-signal transduction-associated protein 1” (n = 7). Besides, “egg protein cp3842” (n = 2), “fumarate hydratase” (n = 2), “ubiquitin-conjugating enzyme” (n = 2), and “sperm-associated antigen 6” (n = 1) were also found as targets of the miRNAs of E. pancreaticum.
miRNAs quantification
Four representative novel miRNAs named Epa-miR-05, Epa-miR-06, Epa-miR-08, and Epa-miR-14 were verified using qRT-PCR. The 4 miRNAs had higher sequencing numbers than others and/or had both mature miRNAs on both arms of their precursors. The standard stem-loop structure of Epa-miR-14 precursors is shown in Figure 1.
All the 4 miRNAs could be successfully amplified with qRT-PCR. The relative expression level of Epa-miR-06 (9.06 ± 1.14) was 9.06 fold higher than that of the inner reference gene; the other 2 miRNAs, including Epa-miR-07 and Epa-miR-05 showed similar expression levels of 1.43 ± 0.22 and 1.0 ± 0.19, respectively. For Epa-miR-14, we detected the relative expression level of its mature miRNA at the 5p arm, which was 1.32 ± 0.48.
Discussion
The objective of this study was to characterize the miRNA profiles of E. pancreaticum. We obtained 27 miRNA candidates from 16.45 million raw sequencing reads with 13 of them previously known and 14 of them novel. Trematode miRNAs deposited in the miRBase database included 56 S. japonicum miRNAs and 20 S. mansoni miRNAs. However, among the 13 known miRNAs only bantam and let-7 were found from both S. japonicum and S. mansoni. The remaining 11 known miRNAs were from S. japonicum miRNAs only. This observation indicated that miRNAs profiles vary between different species in the same class.
miRNAs are known to regulate gene expression at the post-transcriptional level by binding to the 3′ UTR of messenger RNA (mRNA) resulting in gene repression, cleavage or destabilization[29, 30]. Therefore, miRNAs are essential for the regulation of the complex life cycles of parasites, allowing them to respond to environmental and developmental signals[23, 31]. Thus, the novel miRNAs identified in the present study provided novel resources for better understanding of the biology of E. pancreaticum.
For target prediction and functional analysis, a total of 226 targets with 12 or so in average were obtained for the 19 out of 27 E. pancreaticum miRNAs. The target number for each gene is low ranging from one to 33, although as many as 17,401 mRNA and EST non-redundant sequences were used. Normally, we found hundreds of miRNA targets for some miRNAs in other species, such as Ascaris suum, A. lumbricoides, and Toxoplasma gondii (data not shown). Especially for Ascaris spp., where two to three thousands were found for some miRNAs (unpublished observations). The phenomenon of a higher number of targets for one miRNA can also be found in other animals, such as humans, Caenorhabditis elegans, and Drosophilidae spp. as indicated by popular target predicting websites at present, including TargetScan[32] and Pictar[33]. For the few target phenomenon of E. pancreaticum, one reason might be that mRNA dataset for target prediction of miRNAs of E. pancreaticum was from another trematode, S. japonicum, instead of the parasite itself, for which the transcriptome data is not available at present; another reason might be that this is a specific character of the miRNAs of E. pancreaticum. However, more experimental information is needed to verify these possibilities.
Of the predicted targets, a significant high number of targets were related to the “chch domain-containing protein” (n = 29). The “chch domain” is also called “churchill domain”, belongs to a zinc finger transcriptional activator. It was reported that the protein regulated the transition between gastrulation and neurulation, and regulates cell ingression[34]. Besides, it was interesting to find that both “egg protein” (n = 2) and “sperm-associated antigen” (n = 1) were found as targets of the miRNAs of E. pancreaticum.
Conclusions
In the present study, the miRNA profiles of the pancreatic fluke E. pancreaticum were investigated and 27 miRNAs were identified from the pancreatic fluke. Furthermore, we also investigated the potential targets and their functions of 19 of the 27 miRNAs. The present study represented the first global characterization of E. pancreaticum miRNAs, which provides novel resources for better understanding of the biology of the parasite, which, in turn, has implications for the effective control of the disease it causes.
References
Sakamoto T, Oikawa T: Cubic crystal protein inclusions in the neodermis of the pancreatic fluke, Eurytrema pancreaticum, and Eurytrema coelomaticum. Parasitol Res. 2007, 101: 1393-1399. 10.1007/s00436-007-0658-9.
Lu DB, Wang TP, Rudge JW, Donnelly CA, Fang GR, Webster JP: Evolution in a multi-host parasite: chronobiological circadian rhythm and population genetics of Schistosoma japonicum cercariae indicates contrasting definitive host reservoirs by habitat. Int J Parasitol. 2009, 39: 1581-1588. 10.1016/j.ijpara.2009.06.003.
Basch PF: Completion of the life cycle of Eurytrema pancreaticum (Trematoda: dicrocoeliidae). J Parasitol. 1965, 51: 350-355. 10.2307/3275951.
Graydon RJ, Carmichael IH, Sanchez MD, Weidosari E, Widjayanti S: Mortalities and wasting in Indonesian sheep associated with the trematode Eurytrema pancreaticum. Vet Rec. 1992, 131: 443-
Rojo-Vazquez FA, Meana A, Valcarcel F, Martinez-Valladares M: Update on trematode infections in sheep. Vet Parasitol. 2012, 189: 15-38. 10.1016/j.vetpar.2012.03.029.
Zhou YB, Liang S, Jiang QW: Factors impacting on progress towards elimination of transmission of Schistosomiasis japonica in China. Parasit Vectors. 2012, 5: 275-10.1186/1756-3305-5-275.
Ishii Y, Koga M, Fujino T, Higo H, Ishibashi J, Oka K, Saito S: Human infection with the pancreas fluke, Eurytrema pancreaticum. AmJTrop Med Hyg. 1983, 32: 1019-1022.
Ilha MR, Loretti AP, Reis AC: Wasting and mortality in beef cattle parasitized by Eurytrema coelomaticum in the State of Parana, southern Brazil. Vet Parasitol. 2005, 133: 49-60. 10.1016/j.vetpar.2005.02.013.
Jiraungkoorskul W, Sahaphong S, Tansatit T, Kangwanrangsan N, Pipatshukiat S: Eurytrema pancreaticum: the in vitro effect of praziquantel and triclabendazole on the adult fluke. Exp Parasitol. 2005, 111: 172-177. 10.1016/j.exppara.2005.07.004.
Bassani CA, Sangioni LA, Saut JP, Yamamura MH, Headley SA: Epidemiology of eurytrematosis (Eurytrema spp. Trematoda: Dicrocoeliidae) in slaughtered beef cattle from the central-west region of the State of Parana, Brazil. Vet Parasitol. 2006, 141: 356-361. 10.1016/j.vetpar.2006.06.003.
Sukhdeo MV: Where are the parasites in food webs?. Parasit Vectors. 2012, 5: 239-10.1186/1756-3305-5-239.
Wang J, Liu X, Jia B, Lu H, Peng S, Piao X, Hou N, Cai P, Yin J, Jiang N, Chen Q: A comparative study of small RNAs in Toxoplasma gondii of distinct genotypes. Parasit Vectors. 2012, 5: 186-10.1186/1756-3305-5-186.
Hussain M, Taft RJ, Asgari S: An insect virus-encoded microRNA regulates viral replication. J Virol. 2008, 82: 9164-9170. 10.1128/JVI.01109-08.
Zhang B, Wang Q, Pan X: MicroRNAs and their regulatory roles in animals and plants. J Cell Physiol. 2007, 210: 279-289. 10.1002/jcp.20869.
Blahna MT, Hata A: Smad-mediated regulation of microRNA biosynthesis. FEBS Lett. 2012, 586: 1906-1912. 10.1016/j.febslet.2012.01.041.
Wang Z, Xue X, Sun J, Luo R, Xu X, Jiang Y, Zhang Q, Pan W: An “in-depth” description of the small non-coding RNA population of Schistosoma japonicum schistosomulum. PLoS Negl Trop Dis. 2010, 4: e596-10.1371/journal.pntd.0000596.
Xue X, Sun J, Zhang Q, Wang Z, Huang Y, Pan W: Identification and characterization of novel microRNAs from Schistosoma japonicum. PLoS One. 2008, 3: e4034-10.1371/journal.pone.0004034.
Simões MC, Lee J, Djikeng A, Cerqueira GC, Zerlotini A, da Silva-Pereira RA, Dalby AR, LoVerde P, El-Sayed NM, Oliveira G: Identification of Schistosoma mansoni microRNAs. BMC Genomics. 2011, 12: 47-10.1186/1471-2164-12-47.
Wang CR, Xu MJ, Fu JH, Nisbet AJ, Chang QC, Zhou DH, Huang SY, Zou FC, Zhu XQ: Characterization of microRNAs from Orientobilharzia turkestanicum, a neglected blood fluke of human and animal health significance. PLoS One. 2012, 7: e47001-10.1371/journal.pone.0047001.
Xu MJ, Ai L, Fu JH, Nisbet AJ, Liu QY, Chen MX, Zhou DH, Zhu XQ: Comparative characterization of microRNAs from the liver flukes Fasciola gigantica and F. hepatica. PLoS One. 2012, 7: e53387-10.1371/journal.pone.0053387.
Zheng Y, Luo X, Jing Z, Hu Z, Cai X: Comparison of 18S ribosomal RNA gene sequences of Eurytrema coelmaticum and Eurytrema pancreaticum. Parasitol Res. 2007, 100: 645-646.
Wang CR, Qiu JH, Zhu XQ, Han XH, Ni HB, Zhao JP, Zhou QM, Zhang HW, Lun ZR: Survey of helminths in adult sheep in Heilongjiang Province, People’s Republic of China. Vet Parasitol. 2006, 140: 378-82. 10.1016/j.vetpar.2006.04.008.
Xu MJ, Liu Q, Nisbet AJ, Cai XQ, Yan C, Lin RQ, Yuan ZG, Song HQ, He XH, Zhu XQ: Identification and characterization of microRNAs in Clonorchis sinensis of human health significance. BMC Genomics. 2010, 11: 521-10.1186/1471-2164-11-521.
Mount DW: Using the basic local alignment search tool (BLAST). CSH Protoc. 2007, 10.1101/pdb.top17. pdb.top17
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J: SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009, 25: 1966-1967. 10.1093/bioinformatics/btp336.
Kruger J, Rehmsmeier M: RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34: W451-W454. 10.1093/nar/gkl243.
Gilad S, Meiri E, Yogev Y, Benjamin S, Lebanony D, Yerushalmi N, Benjamin H, Kushnir M, Cholakh H, Melamed N, Bentwich Z, Hod M, Goren Y, Chajut A: Serum microRNAs are promising novel biomarkers. PLoS One. 2008, 3: e3148-10.1371/journal.pone.0003148.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001, 25: 402-408. 10.1006/meth.2001.1262.
Lewis BP, Burge CB, Bartel DP: Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005, 120: 15-20. 10.1016/j.cell.2004.12.035.
Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM: Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005, 433: 769-773. 10.1038/nature03315.
Lin WC, Li SC, Lin WC, Shin JW, Hu SN, Yu XM, Huang TY, Chen SC, Chen HC, Chen SJ, Huang PJ, Gan RR, Chiu CH, Tang P: Identification of microRNA in the protist Trichomonas vaginalis. Genomics. 2009, 93: 487-493. 10.1016/j.ygeno.2009.01.004.
Friedman RC, Farh KK, Burge CB, Bartel DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19: 92-105.
Betel D, Wilson M, Gabow A, Marks DS, Sander C: The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008, 36: D149-D153.
Sheng G, Dos RM, Stern CD: Churchill, a zinc finger transcriptional activator, regulates the transition between gastrulation and neurulation. Cell. 2003, 115: 603-613. 10.1016/S0092-8674(03)00927-9.
Acknowledgements
This work was supported by The International Science & Technology Cooperation Program of China (Grant No. 2013DFA31840), The Science Fund for Creative Research Groups of Gansu Province (Grant No. 1210RJIA006), the National S & T Major Program (Grant No. 2012ZX10004220), the China Postdoctoral Science Foundation (Grant Nos. 201104363 and 20090460064), the Yunnan Provincial Program for Introducing High-level Scientists (Grant No. 2009CI125) and the Fund for Imported Talents in Heilongjiang Bayi Agricultural University (2012YB-04).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XQZ and MJX conceived and designed the study, and critically revised the manuscript. MJX, CRW and JHF performed the experiments, analyzed the data and drafted the manuscript. SYH, DHZ, QCC and XZ helped in study design, study implementation and manuscript revision. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Xu, MJ., Wang, CR., Huang, SY. et al. Identification and characterization of microRNAs in the pancreatic fluke Eurytrema pancreaticum. Parasites Vectors 6, 25 (2013). https://doi.org/10.1186/1756-3305-6-25
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-6-25