
Abstract
Background
Lignin is an integral component of the plant cell wall matrix but impedes the conversion of biomass into biofuels. The plasticity of lignin biosynthesis should permit the inclusion of new compatible phenolic monomers such as flavonoids into cell wall lignins that are consequently less recalcitrant to biomass processing. In the present study, epigallocatechin gallate (EGCG) was evaluated as a potential lignin bioengineering target for rendering biomass more amenable to processing for biofuel production.
Results
In vitro peroxidase-catalyzed polymerization experiments revealed that both gallate and pyrogallyl (B-ring) moieties in EGCG underwent radical cross-coupling with monolignols mainly by β–O–4-type cross-coupling, producing benzodioxane units following rearomatization reactions. Biomimetic lignification of maize cell walls with a 3:1 molar ratio of monolignols and EGCG permitted extensive alkaline delignification of cell walls (72 to 92%) that far exceeded that for lignified controls (44 to 62%). Alkali-insoluble residues from EGCG-lignified walls yielded up to 34% more glucose and total sugars following enzymatic saccharification than lignified controls.
Conclusions
It was found that EGCG readily copolymerized with monolignols to become integrally cross-coupled into cell wall lignins, where it greatly enhanced alkaline delignification and subsequent enzymatic saccharification. Improved delignification may be attributed to internal trapping of quinone-methide intermediates to prevent benzyl ether cross-linking of lignin to structural polysaccharides during lignification, and to the cleavage of ester intra-unit linkages within EGCG during pretreatment. Overall, our results suggest that apoplastic deposition of EGCG for incorporation into lignin would be a promising plant genetic engineering target for improving the delignification and saccharification of biomass crops.
Similar content being viewed by others


Background
Lignin serves a vital role as an inter- and intra-molecular glue strengthening plant cell walls, but it hinders numerous agro-industrial processes such as the chemical pulping of woody crops, forage digestion by livestock, and the enzymatic saccharification and fermentation of lignocellulosic biomass into liquid biofuels. As a result, considerable effort has been directed towards reducing or altering the biosynthesis of lignin in plants to permit more efficient utilization of plant cell walls [1–7]. In angiosperms, lignin is normally formed by the oxidative copolymerization of monolignols, principally coniferyl alcohol (CA) and sinapyl alcohol (SA), Figure 1. Perturbing single or multiple genes in the monolignol pathway can lead to massive structural changes in the polymer due to dramatic shifts in the deposition of normal monolignols [8–11], and/or incorporation of pathway intermediates and other phenolic compounds [12–16]. The inherent malleability of plant lignification is further illustrated by the natural incorporation of various γ-acylated monolignols [13, 17, 18] and ferulate arabinoxylan esters [19, 20] into lignin and the recent discovery of a seed-coat lignin surprisingly formed solely from caffeyl alcohol [21]. These findings support the notion that plants could be genetically engineered to make use of precursors from alternate phenolic pathways to form lignins that are more amenable to processing [20, 22–30].

Structures of conventional monolignols, coniferyl alcohol (CA) and sinapyl alcohol (SA), and flavonols and gallate derivatives used in this study, epigallocatechin gallate (EGCG), epigallocatechin (EGC), and ethyl gallate (EG).
In order to identify the most promising genetic engineering targets for modifying lignin, we have used a biomimetic cell wall model system to test a variety of plant-derived phenolics as alternative precursors for lignification [31–34]. These studies have demonstrated that copolymerization of hydroxycinnamate conjugates such as coniferyl ferulate [34] and rosmarinic acid [31] with normal monolignols dramatically improves the alkali extractability of lignin and the subsequent enzymatic hydrolysis of fiber. Accompanying in vitro lignification studies demonstrated that these conjugates readily participate in peroxidase-catalyzed copolymerization reactions with normal monolignols. The resulting lignin contains readily cleaved ester linkages in the backbone of the polymer which permit lignin depolymerization under mild alkaline conditions [31]. Subsequent cell wall studies revealed that several flavonoid and gallate derivatives hold promise as monolignol substitutes for modulating the adverse effects of lignin to enhance the inherent fermentability of cell walls [32, 33]. Among these, epigallocatechin gallate (EGCG, Figure 1) was particularly attractive because it readily formed wall-bound polymers with normal monolignols and enhanced the fermentability of non-pretreated cell walls by 25% [32]. Similarly to the aforementioned hydroxycinnamate conjugates, incorporation of EGCG could introduce easily cleaved ester linkages into the lignin backbone via oxidative coupling of its epigallocatechin and gallate moieties with monolignols. However, the involvement of these EGCG moieties in coupling reactions with monolignols is not known. It is also not known whether EGCG incorporation into lignin could enhance the delignification of cell walls by chemical pretreatment and/or their saccharification by hydrolytic enzymes.
Therefore in the present study, we examined the copolymerization of EGCG and CA into dehydrogenation polymers (synthetic lignins, DHPs), utilizing an in vitro horseradish peroxidase (HRP)-catalyzed polymerization system that models lignin polymerization in vivo[35–38]. Two-dimensional nuclear magnetic resonance (NMR) experiments with the DHPs revealed one major cross-coupling mode between CA and both epigallocatechin and gallate moieties of EGCG. We then subjected cell wall dehydrogenation polymers (CWDHPs) formed by artificially lignifying maize cell walls with CA, SA, and EGCG [32] to alkaline pretreatment and enzymatic hydrolysis. Wet chemical and NMR analyses of CWDHPs, alkali-insoluble residues, and enzyme hydrolysates revealed that EGCG incorporation into lignin dramatically enhanced the delignification and enzymatic saccharification of cell walls.
Results and discussion
In vitro lignin polymerization with EGCG
In these experiments, we examined HRP/H2O2-mediated coupling reactions of EGCG and simplified models of its gallate and gallocatechin moieties with CA, a conventional plant monolignol (Figure 1). To avoid excess formation of insoluble polymers that are difficult to analyze by NMR, most copolymerization reactions were quickly quenched after 10 min of reaction time. Soluble fractions consisting mainly of low molecular weight polymerization products were then extracted with ethyl acetate or acetone in yields ranging from 42-55% for subsequent NMR analysis (see Materials and Methods). Based on thin layer chromatography, the soluble fractions contained only coupling products and no unreacted monomers.
Detailed chemical structures of the polymerization products were elucidated by 2D NMR methods. The HSQC spectra resolved signatures of the various inter-unit linkage types in the oxidation products and clearly revealed the participation of EGCG, epigallocatechin (EGC), and ethyl gallate (EG) in lignin polymerization with CA (Figures 2A-D). In agreement with literature data [39, 40], the polymerization products prepared only with CA contained mainly phenylcoumaran units II with moderate levels of β-aryl ether units I and resinol units III (Figure 2A). Signals from the complete side-chains of these units were seen in the 2D HSQC-TOCSY spectrum (Figure 2E). Such typical lignin units, representing these standard linkage types, were also visible in the spectra of the oxidation products prepared with EGCG, but the most striking difference was the appearance of benzodioxane units IV (Figure 2B), which were totally absent in the control (Figure 2A). The α-, β-, and γ-correlations from trans-benzodioxane-ring units could be assigned by comparison with literature data of analogous benzodioxanes [21, 31] and were further authenticated by TOCSY (Figure 2F). Less pronounced correlations from presumed cis-benzodioxane-ring units were also visible. Benzodioxane structures were also evident in oxidations carried out with EG and EGC (Figure 2, C and D), demonstrating that both epigallocatechin and gallate moieties comprising EGCG could participate in cross-coupling reactions with CA. It is likely that the gallate moiety participated in lignin polymerization primarily via the benzodioxane pathway, because EG polymerization products contained only benzodioxane signals besides the typical lignin signals from CA (Figure 2C). On the other hand, the spectrum of EGC polymerization products (Figure 2D) was complex with numerous unidentified signals (colored in grey), suggesting that the epigallocatechin moiety in EGCG might be involved in several cross-coupling modes besides the pathway to benzodioxanes. In both EGCG and EGC spectra, the two signals from unreacted free resorcinol A rings (EC6 and EC8) were especially prominent, suggesting that reactions of pyrogallol B and gallate rings far exceed reactions of the resorcinol A-ring. Previous studies examining chemical and enzymatic oxidations of flavonols in the absence of monolignols reported lower reactivity of A-ring in comparison to B-ring for EGCG and analogous compounds [41–44].

(A-D) Short-range13C–1H correlation (HSQC) NMR spectra of in vitro peroxidase-catalyzed polymerization products from coniferyl alcohol only (A), and from coniferyl alcohol with epigallocatechin gallate (B), epigallocatechin (C), and ethyl gallate (D). (E and F) Short-range 13C–1H total correlation (HSQC-TOCSY) spectra of the polymerization products from coniferyl alcohol only (E), and from coniferyl alcohol with epigallocatechin gallate (F).
We also examined synthetic lignins (DHPs) prepared by the conventional in vitro lignin polymerization method (end-wise polymerization method), in which the monomers and hydrogen peroxide solutions were slowly added (~20 h) to the peroxidase solution to facilitate polymer chain elongation [45, 46]. These experiments produced DHPs from CA and EGCG (10–20 mol%, in the monomer feed) in good yields (70-80%) but, unlike traditional DHPs prepared only with CA, DHPs prepared with EGCG were mostly insoluble in common lignin solvents used for solution-state NMR. Nevertheless, the HSQC spectrum of the DHP acquired in a suspension-state in dimethylsulfoxide-d6/pyridine-d5 (4:1, v/v) contained signals diagnostic of EGCG aromatic rings and benzodioxane units (Additional file 1: Figure S1); weak signals are likely due to the poor solubility of EGCG-containing polymers. Overall these studies suggest that EGCG readily undergoes oxidative coupling with normal monolignols to produce polymeric lignin.
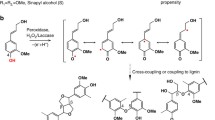
Various flavonol and gallate derivatives including EGCG are known to undergo oxidative homo-coupling reactions in the presence of enzymatic and chemical oxidants [41–44]. When such oxidations are carried out in the presence of monolignols, preferential cross-coupling of galloyl phenoxy radicals from EGCG with monolignol β-radicals and subsequent internal trapping of the quinone methide intermediates (QM) generates benzodioxanes; a logical pathway of such EGCG-incorporation is shown in Figure 3. Cross-coupling between monolignols and galloyl compounds have been implicated but not demonstrated in previous in vivo and in vitro studies [47, 48]. Thus, we are the first to confirm that gallate and pyrogallyl compounds readily participate in lignin polymerization reactions to generate benzodioxane units. Analogous benzodioxanes have also been authenticated as products of lignification with other atypical lignin precursors with o-diphenolic structures such as caffeyl alcohol [16, 21], 5-hydroxyconiferyl alcohol [14, 15, 24, 49], and rosmarinic acid [31], either in vivo or in vitro, or both. Overall, the data presented here support our earlier contention that EGCG readily undergoes oxidative coupling reactions with monolignols to produce polymers with readily cleavable ester linkages in the polymer backbone [32, 33]. Extensive formation of benzodioxane structures should also block the formation of lignin-carbohydrate cross-links formed via lignin quinone methide intermediates because quinone methide intermediates are readily trapped internally by the ortho-OH (Figure 3) and are therefore not available for trapping by external nucleophiles such as water or hydroxyls on polysaccharides [19, 50].

Generation of benzodioxane units during lignification via β–O–4′-cross-coupling reactions between a monolignol (shown here with coniferyl alcohol only) and pyrogallol or gallate units in epigallocatechin gallate. Internal trapping of the intermediate quinone methide by the o-phenol allows rapid rearomatization without requiring external nucleophilic attack (by water, polysaccharide hydroxyls, or other nucleophiles).
Alkaline delignification of cell walls lignified with EGCG
To examine the impact of EGCG on the formation of lignin in cell walls and its removal by alkaline pretreatments, we prepared cell wall-dehydrogenation polymer (CWDHP) complexes by adding dilute H2O2 and lignin precursors to non-lignified primary walls of maize containing natively bound peroxidase. When added, EGCG comprised about 45% by weight of the precursor mixture. The resulting CWDHP control prepared with CA and SA and the CWDHP-EGCG complex prepared with CA, SA, and EGCG contained similar amounts of lignin (averaging of 19.3%) as determined by the acetyl bromide method (Table 1). Comparable lignin concentrations were previously obtained by the Klason lignin procedure and by mass balance calculations [32], indicating that EGCG readily formed wall-bound polymers with normal monolignols. Both CWDHPs contained about 30% glucose (derived mainly from cellulose) and high proportions of the non-cellulosic sugars (xylose, arabinose, and galactose) that are characteristic of primary cell walls in grasses [51].
To assess EGCG effects on cell wall delignification and carbohydrate recovery, CWDHPs were subjected to pretreatment with aqueous NaOH (15% w/w loading on CWDHPs) at 70, 100, and 130°C and alkali-insoluble residues were analyzed by wet-chemical methods and by gel-state 2D NMR. Alkaline pretreatments substantially decreased lignin and non-cellulosic sugar concentrations and increased glucose concentrations in residues recovered from CWDHP-EGCG, whereas comparatively modest compositional shifts were observed following alkaline pretreatment of the CWDHP control (Table 1).
Recovery of alkali-insoluble residues from CWDHP-EGCG was 15 to 20 percentage points lower than from CWDHP-control (Table 2). Based on the recovery of residues and their composition, the incorporation of EGCG increased the extractability of lignin at each pretreatment temperature by an average of 32 percentage points compared to the control (Table 2). Thus ECGC incorporation into lignin permitted more extensive delignification of cell walls under milder conditions that what would be possible for cell walls lignified with normal monolignols. For example, alkaline pretreatment at 130°C removed 62% of the lignin from CWDHP controls, while a less severe alkaline pretreatment at 70°C removed 72% of the lignin from CWDHP-EGCG. Increasing pretreatment temperature to 100 or 130°C boosted delignification of CWDHP-EGCG to about 90%, far in excess of that realized for CWDHP controls.
Gel-state NMR of CWDHPs and their alkali-insoluble residues confirmed that copolymerization of monolignols with EGCG dramatically enhanced the alkaline extractability of lignin (Figure 4). Alkaline pretreatment slightly reduced all visible lignin signals in the CWDHP-control spectra (Figure 4 A and B), but substantially reduced lignin signals in CWDHP-EGCG spectra (Figure 4 C and D). Sliced 1D F2 (1H) spectra showing relative signal intensities of syringyl lignin (S2/6) and glucans (β-D-Glcp1) more clearly illustrates the enhanced extraction of lignin relative to alkali-insoluble cellulosic glucose in CWDHP-EGCG vs CWDHP controls (Figure 4 E-J). In addition, EGCG-derived aromatic signals (EC6, EC8, and GA2″/6″), which were apparent (but perhaps underestimated as noted below) before alkaline pretreatment (Figure 4C), are no longer visible after pretreatment (Figure 4D). This implies that EGCG-rich lignin fractions might be preferentially removed during alkali pretreatment of cell walls. As we previously observed [33], EGCG units were under-represented in gel NMR spectra of CWDHPs; poor NMR responses might be attributed to poor dissolution of EGCG-containing lignin (as we observed for DHPs) or dispersion of signals due to couplings at the 6,8-positions of the epigallocatechin moiety that was targeted in our 13C–1H correlation NMR experiments [32].

Gel-state 2D13C–1H correlation (HSQC) NMR spectra of maize cell walls lignified with coniferyl alcohol and sinapyl alcohol only (CWDHP-control), and in combination with epigallocatechin gallate (CWDHP-EGCG). (A-D) Partial HSQC spectra of CWDHP-control (A and B) and CWDHP-EGCG (C and D) before and after the alkali pretreatment at 100°C. (E-J) Expanded HSQC spectra and sliced 1D F2 (1H) spectra (at 103.3 ppm in F1, 13C) showing syringyl lignin aromatic (S2/6) and glucan anomeric (β-D-Glcp1) signals, of CWDHP-control (E-G) and CWDHP-EGCG (H-J) before and after the alkali pretreatment at 70°C, 100°C, and 130°C.
Overall, these results provide compelling evidence that EGCG incorporation into lignin substantially improves the delignification of cell walls during alkaline pretreatment, even under relatively mild conditions. The most plausible explanation is that EGCG enhances lignin depolymerization via cleavage of the ester linkage between its epigallocatechin and gallate moieties. Improved alkaline solubility of lignin could also be attributed to less frequent cross-linking of lignin to carbohydrate due to EGCG’s inherent ability to readily trap quinone methide intermediates to form benzodioxane structures (Figure 3). Ionization of abundant hydroxyl groups on EGCG might also contribute to lignin dissolution in alkali. Thus if successfully bioengineered into plants, a modified lignin containing EGCG could be very desirable for reducing input costs for delignifying lignocellulose during the chemical pulping of paper or the chemical pretreatment of biomass for biofuel production.
Incorporation of EGCG into lignin also enhanced the extraction of carbohydrates from the cell walls with alkali; the recovery of total sugars in alkali-insoluble residues of CWDHP-EGCG was approximately 9, 21, and 23 points lower than that of CWDHP-control after alkaline pretreatment at 70, 100, and 130°C (Table 2). Both types of CWDHPs had similar glucose recovery at 70°C, but CWDHP-EGCG had lower glucose recovery than CWDHP-control at higher pretreatment temperatures. By contrast, CWDHP-EGCG always had lower recoveries of xylose, arabinose, and galactose in alkaline insoluble residues than CWDHP controls, but differences were again more pronounced at higher pretreatment temperatures. Thus high pretreatment severity (temperature) enhanced the extraction of carbohydrates, particularly from CWDHP-EGCG where more efficient removal of lignin likely aided carbohydrate exposure to and dissolution in alkali. It is also reasonable that the incorporation of EGCG reduced the level of chemical linkages between lignin and hemicelluloses and made the hemicelluloses easier to remove. It must, however, be emphasized that the alkaline extractability of carbohydrate from primary-walled CWDHPs containing high proportions of hemicelluloses and pectin will likely be more extensive than from normal plant biomass containing secondary cell walls enriched in alkali-insoluble cellulose.
Cell wall saccharification
Finally, the enzymatic saccharification of CWDHPs, before and after alkaline pretreatment, was evaluated with cellulases supplemented with β-glucosidase and hemicellulases. The relative abundance of sugars released during saccharification was always in the order of glucose > arabinose > galactose > xylose (data not shown). Typical enzymatic hydrolysis profiles were obtained for non-pretreated and alkali-pretreated CWDHPs; saccharification was rapid during the first 3 to 6 h of hydrolysis, and continued incubation released comparatively small amounts of additional sugar (Figure 5). As illustrated in Figure 5, glucose and total sugar yields were influenced by a pretreatment X hydrolysis time interaction, with greater and more rapid sugar production from alkali-insoluble residues than from non-pretreated CWDHPs. Prior to pretreatment, average glucose and total sugar yields in the 6 to 24 h time period of maximal saccharification were similar for both types of CWDHPs (Table 3). Following pretreatment, however, glucose and total sugar yields from CWDHP-EGGC residues exceeded those from CWDHP-control residues by 10 to 21 percentage points, with the greatest differences occurring following 100 and 130°C pretreatments. Based on linear regression analysis of data in Tables 1 and 3, acetyl bromide lignin concentration on average accounted for 97% of the variation in glucose and total sugar yields from CWDHP-EGCG and their alkaline insoluble residues (P < 0.05); thus EGCG clearly improved saccharification by enhancing lignin removal by alkali. In contrast, sugar yields were poorly related to the lignin content of CWDHP-controls and their alkali-insoluble residues (P > 0.10). Removal of lignin (and possibly hemicelluloses) by alkali probably enhanced the exposure of structural polysaccharides to hydrolytic enzymes and reduced non-productive binding of these enzymes to lignin [52–56].

Time-course of total sugar yield (% of total sugar in the residues) and glucose yield (% glucose in the residues) during enzymatic hydrolysis of maize cell walls artificially lignified with coniferyl alcohol and sinapyl alcohol only (CWDHP-control, A and C), and in combination with epigallocatechin gallate (CWDHP-EGCG, B and D).
When data are expressed as a proportion of sugars originally contained in CWDHPs, maximal glucose yields from alkali-insoluble residues were obtained following a mild 70°C pretreatment (Table 3). Under these conditions, glucose yields from CWDHP-EGCG residues exceeded those from CWDHP-control residues by 9.5 percentage points; both CWDHPs had a similar recovery of glucose in alkali-insoluble residues (Table 2), so yield differences were due to more extensive saccharification of CWDHP-EGCG residues (Table 3). This pretreatment also tended to maximize total sugar yields from residues, but differences between CWDHP treatments were not significant because gains in residue saccharification were offset by a lower recovery of carbohydrate in residues. More severe alkaline pretreatments at 100 and 130°C decreased glucose and total sugars yields from both CWDHPs, because gains in residue saccharification were more than offset by losses of alkali-soluble carbohydrate. If, however, alkali-soluble carbohydrate could be recovered and utilized with enzymatically released sugars, then yields of glucose and total sugars increased with pretreatment temperature and yields from CWDHP-EGCG exceeded the CWDHP-control by 9 to 16 percentage points (Table 3). Thus optimal pretreatment conditions were dependent on whether alkali-soluble carbohydrate could be utilized along with enzymatically released sugars. Optimal pretreatment conditions for a biomass crop would also likely differ from CWDHPs, but our results provide compelling evidence that EGCG incorporation into lignin substantially enhanced the ease of cell saccharification following pretreatment.
Conclusions
In the first study, we examined the compatibility of EGCG in in vitro HRP-catalyzed polymerizations with CA to model how EGCG undergoes oxidative copolymerization with conventional monolignols. These studies revealed that EGCG readily copolymerized with CA to become integrally cross-coupled into the lignin polymer. Additional copolymerization studies with ethyl gallate and epigallocatechin, used to model the gallate and flavonol moieties in EGCG, suggested the former coupled with CA to mainly form benzodioxane structures, while the latter possibly participated in several coupling modes in addition to the pathway for benzodioxanes. In the second study, we artificially lignified primary maize cell walls to investigate the impact of EGCG on the lignification of cell walls and their subsequent delignification by alkali and saccharification by hydrolytic enzymes. When added with CA and SA, EGCG readily formed wall-bound lignin that was much more extensively removed by alkaline pretreatment (73 to 90%) than lignin formed only with normal monolignols (44 to 62%). Improved delignification may be attributed to cleavage of ester intra-unit linkages within EGCG, and therefore in the backbone of the modified lignin polymer, and to efficient internal trapping of quinone methide intermediates by EGCG to out-compete benzyl ether cross-linking of lignin to structural polysaccharides. Incorporation of EGCG into lignin did not influence the degradability of cell walls prior to pretreatment. Following pretreatment, alkali-insoluble residues from EGCG-lignified walls yielded up to 30% more glucose and up to 40% more total sugars than lignified controls during enzymatic saccharification. Overall, our results suggest that genetic engineering of plant metabolic pathways to permit EGCG incorporation into lignin could significantly improve the effectiveness of alkaline pretreatments and the subsequent saccharification of biomass crops.
Methods
General
EGCG and EGC were obtained from Biopurify (Chengdu, China) and EG was from MP Biomedicals (Solon, OH, USA). CA and SA were synthesized according to literature methods [57]. HRP (Type II, 188 purpurogallin units/mg) was from Sigma-Aldrich (St. Louis, MO, USA). Novozymes (Franklinton, NC, USA) generously provided cellulase (NS50013), β-glucosidase (NS50010), a multi-carbohydrase complex (NS50012), and xylanase (NS50030) for cell wall hydrolyses. Other chemicals were purchased from Sigma-Aldrich or Fisher Scientific (Pittsburgh, PA, USA) and were used as received.
NMR Methods
NMR spectra were acquired on Bruker Biospin (Billerica, MA, USA) AVANCE 500 (500 MHz) or AVANCE 700 (700 MHz) spectrometers fitted with cryogenically-cooled gradient probes having inverse geometry, i.e., with the proton coils closest to the sample. Spectra were processed with Bruker’s Topspin 3.1 (Mac) software, using the central solvent peaks as internal references [δH/δC: acetone, 2.04/29.8; dimethylsulfoxide, 2.49/39.5 ppm]. Adiabatic 2D-HSQC (‘hsqcetgpsisp2.2’) and 2D-HSQC-TOCSY (‘hsqcetgpml’) experiments for DHP samples in the solution-state [58, 59], and maize cell wall samples in a gel-state [60, 61], were carried out as described previously. Processing used typical matched Gaussian apodization in F2 (LB = −0.3, GB = 0.001), and squared cosine-bell and one level of linear prediction (32 coefficients) in F1. The TOCSY mixing time was 60 ms.
HRP-catalyzed polymerization
Monolignol solutions were prepared by dissolving CA (108 mg, 0.6 mmol) or CA (72 mg, 0.4 mmol) plus EGCG (46 mg, 0.1 mmol), EGC (61 mg, 0.2 mmol), or EG (40 mg, 0.2 mmol) in acetone-water (50 ml, 1:4, v/v). After adding HRP (2 mg), monolignols in each solution were polymerized by the dropwise addition of aqueous 0.1 M hydrogen peroxide (7.2 ml, 0.72 mmol) for 1 min followed by stirring for 9 min at room temperature. Reaction mixtures from CA or CA plus EG reactions were acidified with 0.1 M HCl (10 ml) and extracted with ethyl acetate (200 mL) to recover low molecular weight synthetic lignins (DHPs). Extracts were washed with brine (3 × 50 mL), dried over sodium sulfate, and evaporated under reduced pressure to give brownish solid residues in 42% yield from the CA oxidation and 55% yield from the CA plus EG oxidation. Oxidation products from CA plus EGCG or CA plus EGC reactions were poorly soluble in ethyl acetate, so these products were quenched with ethanol (200 ml) and evaporated to dryness. Recovered solids were stirred in acetone (100 ml), filtered, and the filtrate was evaporated to recover low molecular weight DHPs as brownish solids in 55% yield from the CA plus EGCG oxidation and 50% yield from the CA plus EGC oxidation. The DHPs were dissolved in acetone-d6 for NMR analysis (500 MHz). A high molecular weight DHP was also prepared by the “end-wise” polymerization method as described previously [59]. Polymerization conditions was as follows: CA (0.8 mmol) and EGCG (0.2 mmol) in 240 ml of acetone/sodium phosphate buffer (0.1 M, pH 6.5) (1:9, v/v) and a separate solution of hydrogen peroxide (1.44 mmol) in 240 ml of water were added by peristaltic pump over a 20 h period at 25°C to 60 ml of buffer containing HRP (5 mg), and further stirred for 4 h. The reaction mixture was then acidified (pH ~2) with 0.1 M HCl and the precipitates were collected by centrifugation, washed with 0.01 M HCl (100 ml x 3) and ultrapure water (100 ml), and lyophilized to afford DHPs (77.6% yield, w/w). The DHP was poorly soluble in general organic solvents and NMR (700 MHz) was acquired in a suspension-state (~30 mg DHP in 600 μl DMSO-d6:pyridine-d5, 4:1, v/v).
Cell wall lignification
As described previously [32], CWDHPs were prepared by adding separate monolignol and hydrogen peroxide (1.1 eq) solutions over a 9 h period to peroxidase-containing maize cell walls (125 g fresh weight, ~3.2 g dry weight) stirred in water. A normal lignin control (CWDHP-control) was prepared by adding a two-component equimolar mixture of CA (1.3 mmol) and SA (1.3 mmol) to cell walls. Similarly, an EGCG-containing lignin (CWDHP-EGCG) was prepared by adding a three-component mixture of CA (0.75 mmol), SA (0.75 mmol) and EGCG (0.5 mmol) to cell walls. After formation, CWDHPs were washed with 9:1 (v/v) acetone: water to remove non-bound dehydrogenation products, dehydrated with acetone, and then air dried. Both treatments were replicated twice in independent experiments.
Alkaline pretreatment
Duplicate CWDHP samples (200 mg) in 50 ml Oak Ridge thermal resistant tubes were soaked for 2 h at 25°C in 0.075 M sodium hydroxide (15% w/w loading on cell wall, 50:1 v/w liquid to cell wall ratio). Tubes were then heated at 70, 100, or 130°C for 1 h, cooled, and centrifuged (1500 × g; 15 min) to collect alkaline insoluble residues. Residues were thoroughly washed by repeated suspension in water followed by centrifugation (1500 × g; 15 min) and then freeze-dried for analysis.
Cell wall analyses
For carbohydrate analyses, CWDHPs and alkaline insoluble residues (10 mg) were treated with 0.5 ml of 72% H2SO4 at room temperature (2°C) for 2 h, and then the mixture was diluted to 3% acid concentration followed by autoclaving (121°C, 15 psi) for 1 h. After cooling to room temperature, the hydrolysate was analyzed for neutral sugars by High-performance Ion Chromatography (HPIC) [62]. In brief, the sugars were qunatified on a Dionex (Sunnyvale, CA) ICS-3000 system equipped with an integrated amperometry detector using Dionex PA1 analytic column and PA1 guard column under the conditions of column temperature 20°C, eluent flow rate 0.7 mL/min with gradient (0 → 25 min, 100% water; 25 → 35 min, 40% water and 60% 0.1 M NaOH; 35 → 40 min, 100% water), and post-column eluent (0.5 M NaOH) flow rate 0.3 mL/min for maintaining detector cell pH > 12.5. Lignin content was determined by the acetyl bromide method [63]. Briefly, cell wall samples (~10 mg) were treated with 25% acetyl bromide/glacial acetic acid solution (v/v, 1.5 mL) at 50°C for 2 h in a sonicator. After heating, the samples were ice cooled for few minutes following centrifugation at 12,000 rpm for 5 min. About 0.5 mL of the clarified supernatant was transferred to a 10-mL glass tube with stopper that contained 2 mL of 2 M NaOH, 2.4 mL of glacial acetic acid and 350 μL of 0.5 M hydroxylamine. Finally, each sample was expanded to total 10 mL with glacial acetic acid followed by vortex for few minutes. The lignin content was determined with absorption at 280 nm using 17.7 L·cm-1·g-1 as the extinction coefficient. For gel-state NMR analyses, dried CWDHPs (100 mg) were ball milled in ZrO2 vessels (50 ml) containing ZrO2 ball bearings (10 mm × 10) using a Retsch PM100 ball mill vibrating at 600 rpm (four cycles of 5 min milling followed by 5 min of cooling). The recovered ball-milled CWDHPs were then transferred into NMR tubes, swollen in DMSO-d6:pyridine-d5 (4:1, v/v), and subjected to 2D HSQC NMR (700 MHz) experiments [60, 61].
Enzymatic hydrolysis
Prior to enzymatic hydrolysis, duplicate CWDHPs and alkali-insoluble residues were shaken in 10 ml Falcon tubes at 300-rpm for 24 h at 50°C with 50 mM sodium acetate buffer (pH 4.8, 0.2%, w/v cell wall to liquid ratio). The hydrolysis was then carried out in duplicate over a 24 h period by adding a mixture of cellulase (15 filter paper unit/g glucan), β-glucosidase (30 cellobiose unit/g glucan), multi-carbohydrase complex (15 fungal β-glucanase unit/g CWDHP), and xylanase (15 Farvet xylan unit/g CWDHP). Tetracycline (0.16%, w/v) was added to prevent microbial contamination. To create enzymatic hydrolysis time profile, sample of the hydrolysate at 1, 3, 6, 12 and 24 h was taken, respectively, and supernatant was collected after centrifugation (1500 × g; 5 min) and analyzed for neutral sugars by HPIC.
Statistical analysis
Data from compositional analyses, alkali pretreatment, and enzymatic hydrolyses were subjected to an analysis of variance by the PROC mixed procedure (SAS Institute Inc., NC). Differences among treatment means were tested by the pdiff procedure at P = 0.05.
Authors’ information
SE is a Postdoc with interest in the areas of chemical delignification, enzymatic saccharification, simultaneous saccharification and fermentation, and catalytic conversion of biomass for the production of biofuels. YT was a Postdoctoral Fellow supported by the Japan Society for the Promotion of Science (JSPS), and is currently an assistant scientist. He has interests in lignification reactions, including those producing novel lignins in specialized tissues and in transgenic plants, fluorescence-tagged molecules, radical coupling reactions, and bioenergy in general. JG is a Research Agronomist whose interests include the development and use of biomimetic cell wall models to investigate the formation of lignin and the effect of lignin composition, structure, and cross-linking on cell wall utilization. Other studies by JG are aimed at improving the yield and forage quality of alfalfa and enhancing the utilization of protein by ruminants through the use of protein-binding polyphenols in forages. XP is an Associate Professor of Bioenergy and Biomaterials. XP’s areas of interest include pretreatment and fractionation of lignocellulose, chemical and enzymatic saccharification of lignocellulose, biofuels (e.g. ethanol and hydrocarbon) from lignocellulose, and cellulose, hemicellulose and lignin based materials. JR is a Professor of Biochemistry and the Plants Area Leader of the DOE Great Lakes Bioenergy Research Center. His interests are in: general plant cell wall (CW) chemistry/biochemistry; lignin biosynthesis (including pathway delineation); lignin chemistry and reactions; synthesis of biosynthetic products, precursors, intermediates, molecular markers, cell wall model compounds, etc.; solution-state NMR (particularly of CW components, especially lignins); cell wall cross-linking mechanisms; methods for wall structural analysis (chemical/degradative, NMR, GC-MS, etc.).
Abbreviations
- CWDHP:
-
Cell wall-dehydrogenation polymer
- CA:
-
Coniferyl alcohol
- DHP:
-
Dehydrogenation polymer
- EGC:
-
Epigallocatechin
- EGCG:
-
Epigallocatechin gallate
- EG:
-
Ethyl gallate
- HSQC:
-
Heteronuclear single quantum coherence spectroscopy
- HPIC:
-
High-performance Ion Chromatography
- HRP:
-
Horseradish peroxidase
- NMR:
-
Nuclear magnetic resonance
- QM:
-
Quinone methide intermediates
- SA:
-
Sinapyl alcohol
- TOCSY:
-
Total coherence spectroscopy.
References
Hu W-J, Lung J, Harding SA, Popko JL, Ralph J, Stokke DD, Tsai C-J, Chiang VL: Repression of lignin biosynthesis in transgenic trees promotes cellulose accumulation and growth. Nat Biotechnol 1999, 17: 808-812. 10.1038/11758
Pilate G, Guiney E, Holt K, Petit-Conil M, Lapierre C, Leple JC, Pollet B, Mila I, Webster EA, Marstorp HG, et al.: Field and pulping performances of transgenic trees with altered lignification. Nat Biotechnol 2002, 20: 607-612. 10.1038/nbt0602-607
Chen F, Dixon RA: Lignin modification improves fermentable sugar yields for biofuel production. Nat Biotechnol 2007, 25: 759-761. 10.1038/nbt1316
Jackson LA, Shadle GL, Zhou R, Nakashima J, Chen F, Dixon RA: Improving saccharification efficiency of Alfalfa stems through modification of the terminal stages of monolignol biosynthesis. Bioenergy Res 2008, 1: 180-192. 10.1007/s12155-008-9020-z
Li X, Weng JK, Chapple C: Improvement of biomass through lignin modification. Plant J 2008, 54: 569-581. 10.1111/j.1365-313X.2008.03457.x
Li X, Ximenes E, Kim Y, Slininger M, Meilan R, Ladisch M, Chapple C: Lignin monomer composition affects Arabidopsis cell-wall degradability after liquid hot water pretreatment. Biotechnol Biofuels 2010, 3: 27. 10.1186/1754-6834-3-27
Fu C, Mielenz JR, Xiao X, Ge Y, Hamilton CY, Rodriguez M, Chen F, Foston M, Ragauskas A, Bouton J, et al.: Genetic manipulation of lignin reduces recalcitrance and improves ethanol production from switchgrass. Proc Natl Acad Sci 2011, 108: 3803-3808. 10.1073/pnas.1100310108
Ralph J, Akiyama T, Kim H, Lu F, Schatz PF, Marita JM, Ralph SA, Reddy MSS, Chen F, Dixon RA: Effects of coumarate-3-hydroxylase downregulation on lignin structure. J Biol Chem 2006, 281: 8843-8853. 10.1074/jbc.M511598200
Wagner A, Ralph J, Akiyama T, Flint H, Phillips L, Torr KM, Nanayakkara B, Te Kiri L: Exploring lignification in conifers by silencing hydroxycinnamoyl-CoA:shikimate hydroxycinnamoyltransferase in Pinus radiata . Proc Natl Acad Sci 2007, 104: 11856-11861. 10.1073/pnas.0701428104
Stewart JJ, Akiyama T, Chapple CCS, Ralph J, Mansfield SD: The effects on lignin structure of overexpression of ferulate 5-hydroxylase in hybrid poplar. Plant Physiol 2009, 150: 621-635. 10.1104/pp.109.137059
Weng J-K, Akiyama T, Ralph J, Golden BL, Chapple C: Independent recruitment of an O-methyltransferase for syringyl lignin biosynthesis in Selaginella moellendorffii. Plant Cell 2011, 23: 2708-2724. 10.1105/tpc.110.081547
Kim H, Ralph J, Lu F, Ralph SA, Boudet A-M, MacKay JJ, Sederoff RR, Ito T, Kawai S, Ohashi H, Higuchi T: NMR Analysis of Lignins in CAD-deficient Plants. Part 1. Incorporation of hydroxycinnamaldehydes and hydroxybenzaldehydes into lignins. Org Biomol Chem 2003, 1: 268-281. 10.1039/b209686b
Ralph J, Kim H, Lu F, Grabber JH, Leplé J-C, Berrio-Sierra J, Mir Derikvand M, Jouanin L, Boerjan W, Lapierre C: Identification of the structure and origin of a thioacidolysis marker compound for ferulic acid incorporation into angiosperm lignins (and an indicator for cinnamoyl-CoA reductase deficiency). Plant J 2008, 53: 368-379.
Vanholme R, Ralph J, Akiyama T, Lu F, Rencoret Pazo J, Christensen J, Rohde A, Morreel K, DeRycke R, Kim H, et al.: Engineering traditional monolignols out of lignins by concomitant up-regulation F5H1 and down-regulation of COMT in Arabidopsis. Plant J 2010, 64: 885-897. 10.1111/j.1365-313X.2010.04353.x
Weng J-K, Mo H, Chapple C: Over-expression of F5H in COMT-deficient Arabidopsis leads to enrichment of an unusual lignin and disruption of pollen wall formation. Plant J 2010, 64: 898-911. 10.1111/j.1365-313X.2010.04391.x
Wagner A, Tobimatsu Y, Phillips L, Flint H, Torr KM, Donaldson L, Te Kiri L, Ralph J: CCoAOMT suppression modifies lignin composition in Pinus radiata. Plant J 2011, 67: 119-129. 10.1111/j.1365-313X.2011.04580.x
del Rio JC, Rencoret J, Marques G, Gutierrez A, Ibarra D, Santos JI, Jimenez-Barbero J, Zhang LM, Martinez AT: Highly Acylated (acetylated and/or p-coumaroylated) native lignins from diverse herbaceous plants. J Agr Food Chem 2008, 56: 9525-9534. 10.1021/jf800806h
Martinez AT, Rencoret J, Marques G, Gutierrez A, Ibarra D, Jimenez-Barbero J, del Rio JC: Monolignol acylation and lignin structure in some nonwoody plants: A 2D NMR study. Phytochem 2008, 69: 2831-2843. 10.1016/j.phytochem.2008.09.005
Ralph J, Bunzel M, Marita JM, Hatfield RD, Lu F, Kim H, Schatz PF, Grabber JH, Steinhart H: Peroxidase-dependent cross-linking reactions of p -hydroxycinnamates in plant cell walls. Phytochem Revs 2004, 3: 79-96.
Ralph J: Hydroxycinnamates in Lignification. Phytochem Revs 2010, 9: 65-83. 10.1007/s11101-009-9141-9
Chen F, Tobimatsu Y, Havkin-Frenkel D, Dixon RA, Ralph J: A polymer of caffeyl alcohol in plant seeds. Proc Natl Acad Sci 2012, 109: 1772-1777. 10.1073/pnas.1120992109
Sederoff RR, MacKay JJ, Ralph J, Hatfield RD: Unexpected variation in lignin. Curr Opin Plant Biol 1999, 2: 145-152. 10.1016/S1369-5266(99)80029-6
Ralph J, MacKay JJ, Hatfield RD, O’Malley DM, Whetten RW, Sederoff RR: Abnormal lignin in a loblolly pine mutant. Science 1997, 277: 235-239. 10.1126/science.277.5323.235
Ralph J, Lapierre C, Marita J, Kim H, Lu F, Hatfield RD, Ralph SA, Chapple C, Franke R, Hemm MR, et al.: Elucidation of new structures in lignins of CAD- and COMT-deficient plants by NMR. Phytochem 2001, 57: 993-1003. 10.1016/S0031-9422(01)00109-1
Ralph J, Lundquist K, Brunow G, Lu F, Kim H, Schatz PF, Marita JM, Hatfield RD, Ralph SA, Christensen JH, Boerjan W: Lignins: natural polymers from oxidative coupling of 4-hydroxyphenylpropanoids. Phytochem Revs 2004, 3: 29-60.
Ralph J: What makes a good monolignol substitute? In The Science and Lore of the Plant Cell Wall Biosynthesis, Structure and Function. Edited by: Hayashi T. Boca Raton, FL: Universal Publishers (BrownWalker Press); 2006:285-293.
Weng JK, Li X, Bonawitz ND, Chapple C: Emerging strategies of lignin engineering and degradation for cellulosic biofuel production. Curr Opin Biotechnol 2008, 19: 166-172. 10.1016/j.copbio.2008.02.014
Mansfield SD: Solutions for dissolution-engineering cell walls for deconstruction. Curr Opin Biotechnol 2009, 20: 286-294. 10.1016/j.copbio.2009.05.001
Simmons BA, Loqué D, Ralph J: Advances in modifying lignin for enhanced biofuel production. Curr Opin Plant Biol 2010, 13: 313-320.
Vanholme R, Van Acker R, Boerjan W: Potential of Arabidopsis systems biology to advance the biofuel field. Trends Biotechnol 2010, 28: 543-547. 10.1016/j.tibtech.2010.07.008
Tobimatsu Y, Elumalai S, Grabber JH, Davidson CL, Pan X, Ralph J: Hydroxycinnamate conjugates as potential monolignol replacements: in vitro lignification and cell wall studies with rosmarinic acid. ChemSusChem 2012, 2012: 676-686.
Grabber JH, Ress DK, Ralph J: Identifying new lignin bioengineering targets. Impact of epicatechin, quercetin glycoside, and gallate derivatives on the lignification and fermentation of maize cell walls. J Agr Food Chem 2012, 60: 5152-5160. 10.1021/jf203986a
Grabber JH, Schatz PF, Kim H, Lu F, Ralph J: Identifying new lignin bioengineering targets: 1. Monolignol substitute impacts on lignin formation and cell wall fermentability. BMC Plant Biol 2010, 10: 1-13. 10.1186/1471-2229-10-1
Grabber JH, Hatfield RD, Lu F, Ralph J: Coniferyl ferulate incorporation into lignin enhances the alkaline delignification and enzymatic degradation of maize cell walls. Biomacromolecules 2008, 9: 2510-2516. 10.1021/bm800528f
Fournand D, Cathala B, Lapierre C: Initial steps of the peroxidase-catalyzed polymerization of coniferyl alcohol and/or sinapyl aldehyde: capillary zone electrophoresis study of pH effect. Phytochem 2003, 62: 139-146. 10.1016/S0031-9422(02)00573-3
Barakat A, Winter H, Rondeau-Mouro C, Saake B, Chabbert B, Cathala B: Studies of xylan interactions and cross-linking to synthetic lignins formed by bulk and end-wise polymerization: a model study of lignin carbohydrate complex formation. Planta 2007, 226: 267-281. 10.1007/s00425-007-0479-1
Tobimatsu Y, Takano T, Kamitakahara H, Nakatsubo F: Studies on the dehydrogenative polymerization of monolignol β-glycosides. Part 6: monitoring of horseradish peroxidase-catalyzed polymerization of monolignol glycosides by GPC-PDA. Holzforschung 2010, 64: 173-181.
Tobimatsu Y, Takano T, Kamitakahara H, Nakatsubo F: Reactivity of syringyl quinone methide intermediates in dehydrogenative polymerization. Part 2: pH effect in horseradish peroxidase-catalyzed polymerization of sinapyl alcohol. Holzforschung 2010, 64: 183-192.
Terashima N, Akiyama T, Ralph SA, Evtuguin D, Pascoal Neto C, Parkås J, Paulsson M, Westermark U, Ralph J: 2D-NMR (HSQC) difference spectra between specifically 13C-enriched and unenriched protolignin of Ginkgo biloba obtained in the solution-state of whole cell wall material. Holzforschung 2009, 63: 379-384.
Terashima N, Atalla RH, Ralph SA, Landucci LL, Lapierre C, Monties B: New preparations of lignin polymer models under conditions that approximate cell well lignification. I: Synthesis of novel lignin polymer models and their structural characterization by 13C NMR. Holzforschung 1995, 49: 521-527. 10.1515/hfsg.1995.49.6.521
Bors W, Michel C, Stettmaier K: Electron paramagnetic resonance studies of radical species of proanthocyanidins and gallate esters. Arch Biochem Biophys 2000, 374: 347-355. 10.1006/abbi.1999.1606
Mukai K, Mitani S, Ohara K, Nagaoka SI: Structure-activity relationship of the tocopherol-regeneration reaction by catechins. Free Radical Biol and Med 2005, 38: 1243-1256. 10.1016/j.freeradbiomed.2005.01.011
Itoh N, Katsube Y, Yamamoto K, Nakajima N, Yoshida K: Laccase-catalyzed conversion of green tea catechins in the presence of gallic acid to epitheaflagallin and epitheaflagallin 3-O-gallate. Tetrahedron 2007, 63: 9488-9492. 10.1016/j.tet.2007.06.093
Kusano R, Tanaka T, Mtsuo Y, Kouno I: Structures of epicatechin gallate trimer and tetramer produced by enzymatic oxidation. Chem Pharm Bull 2007, 55: 1768-1772. 10.1248/cpb.55.1768
Tobimatsu Y, Takano T, Kamitakahara H, Nakatsubo F: Studies on the dehydrogenative polymerizations of monolignol beta-glycosides. Part 2: Horseradish peroxidase catalyzed dehydrogenative polymerization of isoconiferin. Holzforschung 2006, 60: 513-518.
Tobimatsu Y, Takano T, Kamitakahara H, Nakatsubo F: Studies on the dehydrogenative polymerizations of monolignol β-glycosides. Part 3: Horseradish peroxidase-catalyzed polymerizations of triandrin and isosyringin. J Wood Chem Technol 2008, 28: 69-83. 10.1080/02773810802124787
Jouin D, Tollier MT, Monties B: Lignification of Oak Wood.1. Lignin Determinations in Sapwood and Heartwood. Cell Chem Technol 1988, 22: 231-243.
Helm RF, Ranatunga TD, Chandra M: Lignin-hydrolyzable tannin interactions in wood. J Agr Food Chem 1997, 45: 3100-3106. 10.1021/jf970083b
Lu F, Marita JM, Lapierre C, Jouanin L, Morreel K, Boerjan W, Ralph J: Sequencing around 5-hydroxyconiferyl alcohol-derived units in caffeic acid O-methyltransferase-deficient poplar lignins. Plant Physiol 2010, 153: 569-579. 10.1104/pp.110.154278
Grabber JH, Hatfield RD, Ralph J: Apoplastic pH and Monolignol Addition Rate Effects on Lignin Formation and Cell Wall Degradability in Maize. J Agr Food Chem 2003, 51: 4984-4989. 10.1021/jf030027c
Grabber JH, Ralph J, Hatfield RD: Ferulate cross-links limit the enzymatic degradation of synthetically lignified primary walls of maize. J Agr Food Chem 1998, 46: 2609-2614. 10.1021/jf9800099
Cullis IF, Mansfield SD: Optimized Delignification of Wood-Derived Lignocellulosics for Improved Enzymatic Hydrolysis. Biotechnol Bioeng 2010, 106: 884-893. 10.1002/bit.22768
Wyman CE, Dale BE, Elander RT, Holtzapple M, Ladisch MR, Lee YY: Coordinated development of leading biomass pretreatment technologies. Bioresource Technol 2005, 96: 1959-1966. 10.1016/j.biortech.2005.01.010
Nakagame S, Chandra RP, Kadla JF, Saddler JN: The isolation, characterization and effect of lignin isolated from steam pretreated Douglas-fir on the enzymatic hydrolysis of cellulose. Bioresource Technol 2011, 102: 4507-4517. 10.1016/j.biortech.2010.12.082
Rahikainen J, Mikander S, Marjamaa K, Tamminen T, Lappas A, Viikari L, Kruus K: Inhibition of Enzymatic Hydrolysis by Residual Lignins From Softwood-Study of Enzyme Binding and Inactivation on Lignin-Rich Surface. Biotechnol Bioeng 2011, 108: 2823-2834. 10.1002/bit.23242
Chandra RP, Bura R, Mabee WE, Berlin A, Pan X, Saddler JN: Substrate pretreatment: The key to effective enzymatic hydrolysis of lignocellulosics? Biofuels 2007, 108: 67-93. 10.1007/10_2007_064
Kim H, Ralph J: Simplified preparation of coniferyl and sinapyl alcohols. J Agr Food Chem 2005, 53: 3693-3695. 10.1021/jf047787n
Holmgren A, Brunow G, Henriksson G, Zhang L, Ralph J: Non-enzymatic reduction of quinone methides during oxidative coupling of monolignols: implications for the origin of benzyl structures in lignins. Org Biomol Chem 2006, 4: 3456-3461. 10.1039/b606369a
Tobimatsu Y, Davidson CL, Grabber JH, Ralph J: Fluorescence-tagged monolignols: Synthesis and application to studying in vitro lignification. Biomacromolecules 2011, 12: 1752-1761. 10.1021/bm200136x
Kim H, Ralph J: Solution-state 2D NMR of ball-milled plant cell wall gels in DMSO-d 6 /pyridine-d 5 . Org Biomol Chem 2010, 8: 576-591. 10.1039/b916070a
Mansfield SD, Kim H, Lu F, Ralph J: Whole plant cell wall characterization using solution-state 2D-NMR. Nat Protocols 2012. in press
Shuai L, Yang Q, Zhu JY, Lu FC, Weimer PJ, Ralph J, Pan XJ: Comparative study of SPORL and dilute-acid pretreatments of spruce for cellulosic ethanol production. Bioresource Technol 2010, 101: 3106-3114. 10.1016/j.biortech.2009.12.044
Hatfield RD, Grabber JH, Ralph J, Brei K: Using the acetyl bromide assay to determine lignin concentrations in herbaceous plants: some cautionary notes. J Agr Food Chem 1999, 47: 628-632. 10.1021/jf9808776
Acknowledgments
The authors thank Christy Davidson and Hoon Kim for providing assistance with preparing and characterizing lignified cell walls. This work was supported primarily by a grant to XP and JR from Stanford University’s Global Climate and Energy Project (GCEP). The authors also acknowledge partial funding from the US DOE Great Lakes Bioenergy Research Center (DOE Office of Science BER DE-FC02-07ER64494). YT gratefully acknowledges Postdoctoral Fellowship support from the Japan Society for the Promotion of Science (JSPS).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SE, YT, JG, XP, and JR designed research; SE, YT, and JG performed research; YT, JG, SE, XP, and JR analyzed data and wrote the paper. SE and YT contributed equally to this work and therefore should be considered as co-first-authors. All authors read and approved the final manuscript.
Sasikumar Elumalai, Yuki Tobimatsu contributed equally to this work.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Elumalai, S., Tobimatsu, Y., Grabber, J.H. et al. Epigallocatechin gallate incorporation into lignin enhances the alkaline delignification and enzymatic saccharification of cell walls. Biotechnol Biofuels 5, 59 (2012). https://doi.org/10.1186/1754-6834-5-59
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1754-6834-5-59