
Abstract
Background
Most of the previous research work had focused on the epidemiology and prevention of duck enteritis virus (DEV). Whilst with the development of protocols in molecular biology, nowadays more and more information about the genes of DEV was reported. But little information about DEV UL53 gene and glycoprotein K(gK) was known except our reported data.
Results
In our paper, the fluorescent quantitative real-time PCR(FQ-RT-PCR) assay and nucleic acid inhibition test were used to study the transcription characteristic of the DEV UL53 gene. Except detecting the mRNA of DEV UL53 gene, the product gK encoded by UL53 gene was detected through the expression kinetics of UL53 gene by the purified rabbit anti-UL53 protein polyclonal antibodies. Western-blotting and indirect immunofluorescence assays were used to detect gK. From the results of these experiments, the UL53 gene and gK were respectively identified as a late gene and a really late protein. On the other hand, the indirect immunofluorescence assay provided another information that the intracellular localization of DEV gK was mainly distributed in cytoplasm.
Conclusions
By way of conclusions, we conceded that DEV UL53 gene is a really late gene, which is coincident with properties of UL53 homologs from other herpesvirus, such as ILTV(Infectious Laryngotracheitis virus) and HSV-1(Herpes simplex virus type 1). The properties of intracellular localization about gK protein provided a foundation for further functional analysis and further studies will be focused on constructing of the UL53 gene DEV mutant.
Similar content being viewed by others

Background
Duck enteritis virus (DEV) is an alphaherpesvirinae that causes an acute, contagious and highly lethal disease in all ages of birds from the order Anseriformes (ducks, geese, and swans) [1–4]. DEV leads to heavy economic losses to the commercial duck industry due to its high mortality rate and decreased duck egg production [1].
Whilst most of the previous research work had focused on the epidemiology and prevention of this disease [5, 6]. With the development of protocols in molecular biology, nowadays more and more information about the genes of DEV was reported, such as UL5 [7], gC [8–10], UL24 [11–13], UL31 [14, 15], UL35 [16, 17], UL46 [18], UL38 [19], gE [20], UL51 [21], TK gene [22] and so on. While no information about DEV UL53 gene was known except our reported data [23, 24], UL53 gene encoded gK, one of DEV glycoproteins localized in the virion envelope, which played a major role in virus entry by mediating attachment of virions to cell-surface receptors and fusion of the viral envelope with the plasma membrane during penetration according to UL53 homologenes of other alphaherpesvirinae [25, 26]. In order to investigate the roles that UL53 gene played in DEV replication and detect characterization of intracellular localization of DEV gK that was the product of UL53 gene, we carried out the fluorescent quantitative real-time PCR (FQ-RT-PCR) method, nucleic acid inhibition test and expression phase study to analyze the gene category of DEV UL53 and intracellular localization of DEV gK.
To begin dealing with the research project on the properties or functions of DEV UL53 gene and gK, we constructed the pET32b/UL53 plasmid and pMD18-T/β-actin plasmid, used the raised anti-DEV gK serum that specificly recognized the gK protein and revealed its temporal transcription course and intracellular localization in DEV-infected DEF cells. The research will provide useful data for DEV UL53 gene's properties or gK functional analysis, and also will be useful for further understanding the localization properties of alphaherpesvirus UL53 homologs.
Results
Fluorescent quantitative real-time PCR (FQ-RT-PCR) detect the UL53 gene transcript during DEV replication
Detect the specificity of the primers and the integrality or purity of the total RNA of each sample
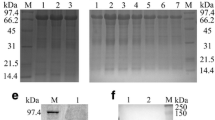
The primers P1,P2 for amplifying 164 bp of UL53 gene and primers P3,P4 for amplifying 178 bp of β-actin gene were detected the specificity by traditional PCR. The PCR products were fractionated on 1.5% agarose gel electrophoresis and stained with golden view. From the result (Figure 1A), the two pairs of primers had good specificity and no primer dimmer. The amplified products were the same with the predicted size.

Specific detection of the primers and integrality analysis of RNA samples. A. Specific detection of the primers for DEV UL53 gene and the endogenous control β-actin gene. M, DNA marker--Marker I; 1, the specific amplification of the primers for DEV UL53 gene; 2, the specific amplification of the primers for the endogenous control β-actin gene. B. The integrality of RNA samples tested by agarose gel electrophoresis. Total RNA was isolated three bands(28S, 18S and 5S).
The total RNA was extracted from mock or DEV-infected cells at given times using Trizol Reagent (Tiangen Biotech). After removing DNA in the RNA, the total RNA integrality analysis of each sample was detected by the agarose gel electrophoresis. From the figure 1B, the bands of 28S, 18S and 5S were clearly seen. At the same time, the purity of RNA was detected by nucleic acid-protein detecting instrument (Bio-Rad). The value of OD260/OD280 fluctuated between 1.8 and 2.0. Both results indicated the total RNA had good integrality and high purity.
Construction the double standard curves of FQ-RT-PCR
The pET32b/UL53 plasmid was used to establish one of the standard curves of FQ-RT-PCR. Firstly, we diluted the initiate concentration of the pET32b/UL53 plasmid in order to contain the scope of cDNA sample products by the whole concentration gradients. Moreover, the appropriate diluted plasmids used as standard preparations were ten-fold dilution from 10-1 to 10-5. According to the results of FQ-RT-PCR(Figure 2A), the standard curve equation was Y = -3.305X-0.641, the UL53 gene amplification efficiency was 100.7% and the correlation coefficient was 0.998. It's also clear that the crest value of melt curves is sole about 83.5°C(Figure 2B). Meanwhile the pMD18-T/β-actin plasmid was used to establish the other standard curve of FQ-RT-PCR. Using the same way to dilute the initiate concentration of the pMD18-T/β-actin plasmid, the result indicated that the β-actin gene amplification efficiency is 98.5%, the correlation coefficient is 0.999 (Figure 2C) and the crest value of melt curves is also sole about 89.5°C(Figure 2D). The standard curve equation of β-actin gene is Y = -3.358X+6.066.

The standard curves and melt curves of DEV UL53 gene and β-actin gene by FQ-RT-PCR. A: Standard curve of DEV UL53 gene. The x-axis represented ten-fold dilutions of pET32b/UL53 plasmids, and the y-axis represented corresponding cycle threshold (Ct) values. The standard curve equation is Y = -3.305X-0.641. The correlation coefficient and the slope value of the regression curve were 0.998 and -3.305, respectively; B: Melt curve of DEV UL53 gene. There was only a singlet; C: Standard curve of β-actin gene. The x-axis represented ten-fold dilutions of pMD18-T/β-actin plasmids, and the y-axis represented corresponding cycle threshold (Ct) values. The standard curve equation is Y = -3.358X+6.066. The correlation coefficient and the slope value of the regression curve were 0.999 and -3.358, respectively; D: Amplification curve of β-actin gene. There was only a singlet, too.
Transcription analysis of DEV UL53 gene in DEF detecting by FQ-RT-PCR
The isolated and processed RNA at given times was reverse transcribed into cDNA by Quantscript RT Kit(TIANGEN BIOTECH CO., LTD.). The quantitive primers(P1, P2) of DEV UL53 gene and primers(P3, P4) of the endogenous control gene β-actin were used to respectively detect the reverse transcribed sample, cDNA by FQ-RT-PCR. The fluorescent quantitation amplification curves of DEV UL53 gene and endogenous control gene β-actin were shown in figure 3A. In accordance with the endogenous control gene β-actin, the method of delta-delta Ct was used to analyze the relative transcripted quantity of DEV UL53 gene, and the data were calculated by iQ™5 Optical System Software Version 2.1. From the calculated result(Figure 3B), it's manifest that DEV UL53 gene begins to transcribe in DEF at 10 h.p.i.

FQ-RT-PCR was used to detect the transcript of UL53 gene. A. The amplification curves of DEV UL53 gene and β-actin gene to detect the samples by FQ-RT-PCR. B. Gene expression analysis of UL53 gene by iQ™5 Optical System Software Version 2.1. The x-axis represented the conditions of different infected time, and the y-axis represented UL53 gene relative expressing quantity.
Detection of the UL53 gene transcript during DEV replication in the presence of a specific inhibitor of herpesvirus DNA polymerase
To characterize the temporal class of UL53 gene expression, DEF were infected with DEV in the presence or absence of ganciclovir. The total RNA was separately isolated from the mock and the test specimen that adding with ganciclovir, the specific inhibitor of herpesvirus DNA polymerase at different times(24 h, 36 h). The isolated RNA was processed with to remove the DNA within the sample RNA, detected the integrality of RNA by 1.5% agarose gel electrophoresis. From the figure 4A, it's obviously seen that the integrality of RNA is pretty good. RT-PCR analysis of total RNA harvested at different times by the primers(P1, P2) of DEV UL53 gene revealed a specific PCR product(164 bp) which was detected at 24 h.p.i and persisted in the subsequent infection stages(36 h) in the absence of ganciclovir, whereas no product was detected in infected cells in the presence of ganciclovir(Figure 4B). In contrast, the endogenous control gene β-actin always had a specific PCR product(178 bp) in the absence or presence of ganciclovir(Figure 4B).

Transcript detection of the UL53 by nucleic acid inhibition test. A. The integrality of RNA samples tested by agarose gel electrophoresis. Total RNA was isolated three bands(28S, 18S and 5S). B. Detection of the UL53 transcript by nucleic acid inhibition test. M: DNA marker--Marker I. DEF were infected with DEV in the absence (Mock) or presence of 300 μg/ml ganciclovir and total RNA of infected cells were harvested at 24 h and 36 h postinfection followed by RT-PCR detection using specific primers to UL53 gene and β-actin gene respectively.
Characterization analysis of DEV UL53 gene and gene product gK
Western-blotting assay to study the expression kinetics of UL53 gene
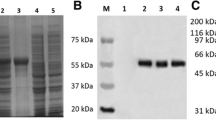
In order to study the expression kinetics of UL53 gene, DEF cells were infected with DEV. Cell lysates were harvested at 10 h, 14 h, 24 h, 36 h and 48 h postinfection (p.i). Equal amounts of cell lysates were resolved by SDS-PAGE, and proteins in the gel were electrophoretically transferred to PVDF membrane and subjected to Western blot analysis with the purified rabbit anti-UL53 protein polyclonal antibodies. As seen in Figure 5A, DEV gK (molecular mass, approximately 37.7 kDa), was firstly detected in DEF cells till 14 h.p.i. The UL53 gene expression increased over time, reached a maximum at approximately 36 h.p.i and decreased in 48 h.p.i; this finding showed that gK is expressed in the viral replication cycle.

Expression kinetics of DEV UL53 gene and intracellular location of the DEV gK in infected DEF by IIF. A. Proteins isolated from mock or DEV-infected cells at different times (10 h.p.i. to 48 h.p.i.) were analyzed by Western blot. The specific band demonstrated the expected position for DEV gK (about 38 kDa). M represented standard protein molecular weight markers. B. Expression kinetics and intracellular location of the DEV gK in infected DEF. Mock-infected(a) and DEV infected DEF(b to f) were fixed as described in Methods. The samples were incubated with the purified rabbit anti-UL53 protein polyclonal antibodies(a, c to f) or preimmune serum(b), and reacted with fluorescein isothiocyanate (FITC)-conjugated secondary antibody, and then counter-stained with DAPI (the cell nuclei was stained with blue). The merged fluorescence microscopy images of DEF are shown from a to f with high magnification (400 ×).
Indirect immunofluorescence assay(IIF) to detect the expression kinetics of UL53 gene and characterization of intracellular localization of DEV gK
The intracellular localization of gK protein was examined by indirect immunofluorescence staining. DEF were cultivated on coverslips and infected with DEV. The cells were processed at 8 h, 12 h, 24 h, and 36 h.p.i., and gK was detected by using the purified rabbit anti-UL53 protein polyclonal antibodies and fluorescein isothiocyanate (FITC)-conjugated secondary antibody. As can be seen from the Figure 5B, the gK mainly distributed in the cytoplasmic of infected DEF at 12 h.p.i. At the same time, the specific fluorescence increased over time of DEV infection(Figure 5B).
Discussions
The expression of Herpesviridae genes is temporally controlled and coordinated in a cascade fashion [27]. In order to affirm the transcription characteristics of the DEV UL53 gene, the fluorescent quantitative real-time PCR(FQ-RT-PCR) assay and nucleic acid inhibition test were used to detect the mRNA transcribed by DEV UL53 gene. The technology of fluorescent quantitative real-time PCR is divided into relative quantitation method and absolute quantitation method according to final distinct data. In our paper, the relative quantitation method(delta-delta Ct) was used to analyze the transcription characteristics. But this method was based on the coincidental amplification efficiency between DEV UL53 gene and the endogenous control gene β-actin that was used as standardization of the other gene's expressing research. Before the beginning of relative quantitation method, the primers of the target gene DEV UL53 and endogenous control gene β-actin were specific and no primer dimmer through the PCR, which was also improved by melting curves of two pairs of primers. In preliminary experiment, it's necessary to respectively establish standard curves of DEV UL53 gene and β-actin gene and the Rotor-gene software would automatically generate the value of slope, amplification efficiency, correlation coefficient. If the slope and the correlation coefficient of the two standard curves are close to the theoretical values of -3.322 and 1.000, respectively, the consequence demonstrated the amplification efficiency of two genes was really approximate and the relative quantitation method of delta-delta Ct could be applied to analyze the transcription characteristics [28, 29]. Otherwise this method could not be used. In our research, the difference of two standard curves slope between the target DEV UL53 gene and β-actin gene was not notable, therefore this method was valid. From the consequence of FQ-RT-PCR, we could clearly see that the DEV UL53 gene is firstly transcribed in 10 h.p.i.. According to the previous research, two identified immediate early products, namely protein kinase pUS3 and dUTPase, were firstly detected at 2 h.p.i. and 4 h.p.i., respectively [30, 31]. In contrary, two another identified late products, namely tegument protein pUL31 and pUL51 were first detected at 6 h.p.i. and 8 h.p.i., respectively [15, 32]. Hence, we concluded that UL53 was a late gene because the mRNA of UL53 could be firstly detected in 10 h.p.i.
On the background of gene category theory, gene temporal regulation condition should be analyzed when the infected cell was dealt with some canonical medicine [33]. Such as γ (late) gene could not be detected in the cell dealt with phosphonoacetate(DNA synthesis inhibition factor); α (immediate-early) gene can be detected in the cell dealt with protein synthesis inhibitor; β (early) gene can be inhibited by cycloheximide (protein synthesis inhibitor) while can't be inhibited by phosphonoacetate [34]. In nucleic acid inhibition test, no specific PCR product was detected in infected cells in the presence of ganciclovir(the specific inhibitor of herpesvirus DNA polymerase). So the DEV UL53 gene encoded a late transcript.
Except detecting the mRNA of DEV UL53 gene, the product gK encoded by UL53 gene was detected by the purified rabbit anti-UL53 protein polyclonal antibodies through the expression kinetics of UL53 gene. Western-blotting and indirect immunofluorescence assays were used to detect gK. From the results of both assays, the gK was respectively detected in 14 and 12 h.p.i. According to the recent reports of Wei Xie, Chanjuan Shen and Jun Xiang, the gK was a really late product [19, 31, 32].
On the other hand, the indirect immunofluorescence assay provided another information about the intracellular localization of DEV gK protein. That was valuable and useful data because little information about the intracellular localization of the DEV gK is known. However, due to the difficulties in the experimental determination of protein's cellular localization, the methods of theoretical prediction on the known sequence are becoming more important [35]. The analysis of the protein intracellular localization predicted by computer indicated that the gK locate in cytoplasm with 1.0%, cytoplasmic membrane with 92.6% and extracellular with 3.2%[36]. But the results of our research showed that cytoplasm fluorescence first appeared in DEV-infected cells at 12 h.p.i. At later times of infection, the specific fluorescence predominantly in cytoplasm region was increased. That was different between the predicted consequence and our experimental result. We presumed the gK maybe locate in the Golgi apparatus and endoplasmic reticulum (ER) membranes.
Conclusions
By way of conclusions, through the experiments of the fluorescent quantitative real-time PCR, nucleic acid inhibition and the expression kinetics of UL53 gene, we conceded that DEV UL53 gene is a really late gene, which is coincident with properties of herpesvirus UL53 homologs, such as ILTV(Infectious Laryngotracheitis virus) UL53 gene and HSV-1(Herpes simplex virus type 1) UL53 gene [37, 38]. From the result of indirect immunofluorescence assay, the gK protein showed obviously cytoplasmic staining in infected DEF. These properties of the gK protein provided a foundation for further functional analysis and further studies will be focused on constructing of the UL53 gene DEV mutant.
Methods
Detection of the UL53 gene transcript during DEV replication by fluorescent quantitative real-time PCR (FQ-RT-PCR)
Detecting the primers specificity of fluorescent quantitative real-time PCR (FQ-RT-PCR)
According to the DEV-UL53 gene(GenBank accession no.: EU071035), the primers of fluorescent quantitative real-time PCR (FQ-RT-PCR) was designed by IQ5(Bio-Rad). The forward primer(P1) was 5'-CGTATTGTATCTGCGGCT-3' and the reverse primer(P2) was 5'-CGAGTGGGCGAAATGAAC-3', which amplified a 164-base pair (bp) fragment of DEV-UL53 gene. As for the endogenous control gene β-actin, the forward primer(P3) was 5'-CCGGGCATCGCTGACA-3' and reverse primer(P4) was 5'-GGATTCATCATACTCCTGCTTGCT-3', which amplified a 178-base pair (bp) fragment [33]. Both primers were synthesized by Ivitrogen.
Using the pET32b/UL53 and pMD18-T/β-actin as templates, the specificity of the primers P1, P2 and P3, P4 were detected respectively. The 25 μl PCR reaction system was as follows: PrimerSTAR(Premix) DNA polymerase (0.625 Units) 12.5 μl, template 1.5 μl, each primer (20 pmol) 0.5 μl and the sterile ultrapure water 10 μl. Meanwhile the PCR reaction conditions were: 95°C for 4 min, 30 cycles of 94°C for 1 min, 59°C for 1 min, 72°C for 2 min, and then a final extension at 72°C for 10 min. The PCR products were fractionated on 1.5% agarose gel electrophoresis and stained with golden view.
Construction the double standard curves of FQ-RT-PCR
The pET32b/UL53 plasmid and pMD18-T/β-actin plasmid were constructed by our laboratory, which were used to establish the standard curves of FQ-RT-PCR. Firstly, according to the initiate concentration of two various plasmids, we diluted the initiate concentration in order to contain the scope of cDNA sample products by the whole concentration gradients. Moreover, the appropriate diluted plasmids used as standard preparations were ten-fold dilution from 10-1 to 10-5. Lastly, the FQ-RT-PCR assay was performed to build the standard curves by an iCycler iQ™ real-time PCR detection system (Bio Rad Lab., Hercules, CA, USA). The program of FQ-RT-PCR was as follows: 1 min at 95°C followed by 40 three-step cycles of 30 sec at 94°C, 30 sec at 60°C and 30 sec at 72°C. The reaction system was a 20 μl reaction mixture containing 9 μl of POWER High-Capacity cDNA Reverse Transcription Kits SYBR Green PCR master mix (Applied Biosystems), 0.5 μl of each primer, 1 μl of plasmid template and 9 μl of sterile ultrapure water [9]. Each reaction had three replicates. Homogeneity of products from each reaction was confirmed by melt curve analysis. Analysis of the real-time PCR data was carried out using the comparative ΔΔCt method [39].
Duck embryo fibroblasts(DEF) culture and total RNA extraction
Duck embryo fibroblasts (DEF) was cultured at 37°C with 5% CO2 in minimal essential medium(MEM) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (FBS). After infecting DEV, DEF either mock-infected or infected were harvested at 1, 2, 3, 4, 6, 8, 10, 24, 36, 48 and 60 h.p.i. Total RNA was isolated from mock or DEV-infected cells at given times according to the instructions of Trizol Reagent (Tiangen Biotech Co., LTD).
Remove DNA in the RNA, and detect the integrality and purity of RNA
DNase I (RNase free) was added 10 U into 20-50 μg total RNA in order to remove the DNA within the sample RNA [40]. The reaction system was in a 10 μl mixture including 1 μl DNase buffer, 1 μl DNase I (RNase free), 0.5 μl RNase inhibitors and 7.5 μl sample RNA. After mixing the mixture, the 10 μl mixture was digested in 37°C about 30 min.
In order to detect integrality of the RNA, 5 μl sample RNA was detected by 1.5% agarose gel electrophoresis. At the same time, the purity of RNA was detected by nucleic acid-protein detecting instrument (Bio-Rad).
Prepare the sample cDNA through reverse transcription and detect the sample cDNA by FQ-RT-PCR
Total RNA extracted from various time was reverse transcribed into cDNA by Quantscript RT Kit (TIANGEN BIOTECH CO., LTD.) and the products were reserved at -20°C. The reaction system of reverse transcription was in 20 μl mixture containing 2 μl 10 × RT mix, 2 μl dNTP, 2 μl Random primers, 1 μl Quant Reverse Transciptase, 11 μl RNase-free H2O and 2 μl RNA template. When mixing the mixture, the mixture was incubated for 60 min at 37°C for reverse transcription.
After the reverse transcription, the cDNA products from mock or DEV-infected cells at various times were detected by fluorescent quantitative real-time PCR assay. The primers(P1, P2) and (P3, P4) were used to amplify 164-base pair (bp) fragment of DEV UL53 gene and 178 bp fragment of the endogenous control gene β-actin, respectively. The reaction system and procedures were the same with construction of the double standard curves of FQ-RT-PCR. The fold change in expression of UL53 gene relative to the endogenous control gene (β-actin) at various time points was calculated as Fold change = Log(2-ΔΔCt), where ΔΔCt = (Ct,Target-Ct,Reference)Time x- (Ct, Target-Ct, Reference)Time 0[9, 33].
Detection of the UL53 gene transcript during DEV replication in the presence of a specific inhibitor of herpesvirus DNA polymerase
Cell culture and sample preparation
Duck embryo fibroblasts (DEF) was cultured at 37°C with 5% CO2 in minimal essential medium(MEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin. When the DEF were grew into cell monolayer in 20 ml culture flask, we respectively added the ganciclovir (a specific inhibitor of herpesvirus DNA polymerase) with 300 μg/ml into the DEF infected with 150 μl DEV(TCID50 = 10-6.334/100 μl) [32, 41]. In contrary, the mock was only infected with 150 μl DEV(TCID50 = 10-6.334/100 μl). We separately harvested three different samples at 24 h.p.i or 36 h.p.i. Each sample has three replications.
RNA extraction and cDNA preparation
After dumpaging supernatant, the remaining cells were used to extracted total RNA by Trizol Reagent (Tiangen Biotech) from various samples, which was carried out according to the manufacturer's instructions. Moreover, the reaction procedures and reaction conditions of removing DNA within the total RNA and implementing reverse transcription were the same with that of fluorescent quantitative real-time PCR (FQ-RT-PCR). And then, the products were storaged at -20°C for use.
Conventional PCR was used to detect the targeted fragment
Using the above products as templates, the primers(P1,P2) and primers(P3,P4) were respectively used to detect the UL53 gene and β-actin gene. The reaction system was carried out in a 25 μl reaction mixture containing 0.5 μl of each primer(20 pmol), 1.5 μl cDNA template, 12.5 μl PrimerSTAR(Premix) DNA polymerase (0.625 Units) and 10 μl water (all reagents were purchased from TaKaRa). The PCR conditions were: 95°C for 4 min, 30 cycles of 94°C for 1 min, 59°C for 1 min, 72°C for 2 min, and then a final extension at 72°C for 10 min. The PCR products were fractionated on 1.5% agarose gel electrophoresis and stained with golden view.
Expression phase analysis of duck enteritis virus UL53 gene
Western-blotting assay to study the expression kinetics of UL53 gene
To study the expression kinetics of UL53 gene, DEF were either mock or infected with DEV as described above and harvested at 10, 14, 24, 36, and 48 h.p.i. Cell lysates were in SDS sample buffer, which were separated by SDS-PAGE, and simultaneously electrophoretic transfered to polyvinylidene difluoride (PVDF) membranes (Bio-Rad Lab., Hercus, CA, USA) at 120 V for 90 min [9, 19]. Non-specific protein binding was blocked through dealing with PVDF membranes with TBST (20 mmol Tris-HCl, 150 mmol NaCl, pH 7.4, and 0.05% Tween-20) containing 3% bovine serum albumin (BSA). And then, the purified rabbit anti-UL53 protein polyclonal antibodies were used as primary antibody at a dilution of 1:50. The PVDF membranes were overnightly incubated at 4°C. After washing membranes three times with TBST, the goat anti-rabbit peroxidase-labeled antibody (KPL Inc., Gaithersburg, Maryland, USA) was used as secondary antibody to incubate the membranes. Lastly, the membranes were developed in diaminobenzidine (DAB) substrate buffer and photos were taken by a digital camera.
Indirect immunofluorescence assay(IIF) to detect the expression kinetics of UL53 gene and characterization of intracellular localization of DEV gK
DEF cells were cultivated on coverslips in six-well plates, which were mock or infected with DEV. The coverslips were harvested at different times (8, 12, 24, 36 h.p.i) and then fixed with 4% paraformaldehyde for 12 h at 4°C. The coverslips were washing three times with PBS, and then blocked the coverslips with PBS containing 3% BSA at 37°C for 1 h [9]. The purified rabbit anti-UL53 protein polyclonal antibodies (1:100 dilution) were used to incubate coverslips at 37°C for 2 h. After washing three times with PBS including 0.1% Tween-20, the coverslips were incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG(1:150 dilution) (Sino-American Biotechnology Co., Shanghai, China) for 1 h at 37°C. At last 4,6-Diamidino-2-phenylindole (DAPI; Sigma) staining was used to visualize the cell nuclei [21]. Immunofluorescence photos were taken and recorded by the Bio-Rad MRC 1024 imaging system.
References
Sandhu , Metwally SamiaA: Duck virus enteritis(Duck Plague). In Diseases of poultry. 12th edition. Edited by: Saif YM, Fadly AM, Glisson JR, McDougald LR, Nolan LK, Swayne DE. Oxford, Blackwell Publishing; 2008:384-393.
Montali RJ, Bush M, Greenwell GA: An epornitic of duck viral enteritis in a zoological park. J Am Vet Med Assoc 1976, 169: 954-958.
Proctor SJ: Pathogenesis of duck plague in the bursa of Fabricius, thymus, and spleen. Am J Vet Res 1976, 37: 427-431.
Davison S, Converse KA, Hamir AN, Eckroade RJ: Duck viral enteritis in Muscovy ducks in Pennsylvania. Avian Dis 1993, 37: 1142-1146. 10.2307/1591927
Spieker JO, Yuill TM, Burgess EC: Virulence of six strains of duck plague virus in eight waterfowl species. Journal of Wildlife Diseases 1996, 32: 453-460.
Sarmah R, Sarmah AK: Development of interference against duck plague virus. Indian Journal of Virology 1996, 12: 113-114.
Pana Huaqi, Cao Ruibing, Liu Lei, Niu Mingfu, Zhou Bin, Chen Puyan, Jiangchun Hu: Molecular cloning and sequence analysis of the duck enteritis virus UL5 gene. Virus Res 2008, 136: 152-156. 10.1016/j.virusres.2008.05.003
Shen Fuxiao, Cheng Anchun, Wang Mingshu, Huang Haibin, Li Chuanfeng, Jiang Jinfeng, Jia Renyong, Zhu Dekang, Chen Xiaoyue: Factors affecting synonymous codon usage bias in the gC gene of DPV CHv strain. African Journal of Microbiology Research 2010, 4: 343-353.
Lian Bei, Xu Chao, Cheng Anchun, Wang Mingshu, Dekang Zhu, Qihui Luo, Renyong Jia, Fengjun Bi, Chen Zhengli, Zhou Yi, Yang Zexia, Chen Xiaoyue: Identification and characterization of duck plague virus glycoprotein C gene and gene product. Virol J 2010, 7: 349. 10.1186/1743-422X-7-349
Zou Qing, Sun Kunfeng, Anchun Cheng, Mingshu Wang, Xu Chao, Zhu Dekang, Jia Renyong, Luo Qihui, Yi Zhou, Zhengli Chen, Xiaoyue Chen: Detection of anatid herpesvirus 1 gC gene by TaqMan™ fluorescent quantitative real-time PCR with specific primers and probe. Virology Journal 2010, 7: 37. 10.1186/1743-422X-7-37
Jia Renyong, Anchun Cheng, Mingshu Wang, Xin Hongyi, Guo Yufei, Zhu Dekang, Qi Xuefeng, Lichan Zhao, Ge Han, Xiaoyue Chen: Analysis of synonymous codon usage in the UL24 gene of duck enteritis virus. Virus Genes 2009, 38: 96-103. 10.1007/s11262-008-0295-0
Jia Renyong, Anchun Cheng, Mingshu Wang, Zhu Dekang, Ge Han, Xin Hongyi, Liu Fei, Luo Qihui, Guo Yufei, Qi Xuefeng, Yin Zhongqiong, Xiaoyue Chen: Cloning, expression, purification and characterization of UL24 partial protein of duck enteritis virus. Intervirology 2009, 52: 326-334. 10.1159/000242354
Jia Renyong, Anchun Cheng, Mingshu Wang, Zhu Dekang, Ge Han, Xin Hongyi, Liu Fei, Luo Qihui, Guo Yufei, Qi Xuefeng, Yin Zhongqiong, Xiaoyue Chen: Development and evaluation of an antigen-capture ELISA for detection of the UL24 antigen of the duck enteritis virus, based on a polyclonal antibody against the UL24 expression protein. J Virol Methods 2009, 161: 38-43. 10.1016/j.jviromet.2009.05.011
Xie Wei, Anchun Cheng, Mingshu Wang, Hua Chang, Zhu Dekang, Luo Qihui: Molecular cloning and characterization of the UL31 gene from Duck enteritis virus. Molecular Biology Reports 2010, 37: 1495-1503. 10.1007/s11033-009-9546-y
Xie Wei, Anchun Cheng, Mingshu Wang, Hua Chang, Zhu Dekang, Luo Qihui, jia Renyong, Xiaoyue chen: Expression and characterization of the UL31 protein from Duck enteritis virus. Virology Journal 2009, 6: 19. 10.1186/1743-422X-6-19
Cai Mingsheng, Anchun Cheng, Mingshu Wang, Lichan Zhao, Zhu Dekang, Luo Qihui, Liu Fei, Xiaoyue Chen: His6-tagged UL35 protein of duck plague virus: expression, purification, and production of polyclonal antibody. Intervirology 2009, 52: 141-151. 10.1159/000221833
Cai Mingsheng, Anchun Cheng, Mingshu Wang, Lichan Zhao, Zhu Dekang, Luo Qihui, Liu Fei, Xiaoyue Chen: Characterization of synonymous codon usage bias in the duck plague virus UL35 gene. Intervirology 2009, 52: 266-278. 10.1159/000231992
Lu Liting, Anchun Cheng, Mingshu Wang, Jinfeng Jiang, Zhu Dekang, Jia Renyong, Luo Qihui, Liu Fei, Zhengli Chen, Xiaoyue Chen, Jinlong Yang: Polyclonal antibody against DPV UL46M protein can be a diagnostic candidate. Virology Journal 2010, 7: 83. 10.1186/1743-422X-7-83
Xiang Jun, Ma Guangpeng, Zhang Shunchuan, Cheng Anchun, Wang Mingshu, Dekang Zhu, Renyong Jia, Qihui Luo, Chen Zhengli, Chen Xiaoyue: Expression and intracellular localization of duck enteritis virus pUL38 protein. Virol J 2010, 7: 162. 10.1186/1743-422X-7-162
Chang Hua, Cheng Anchun, Wang Mingshu, Guo Yufei, Xie Wei, Lou Kunpeng: Complete nucleotide sequence of the duck plague virus gE gene. Arch Virol 2009, 154: 163-165. 10.1007/s00705-008-0284-6
Shen Chanjuan, Guo Yufei, Cheng Anchun, Wang Mingshu, Zhou Yi, Lin Dan, Xin Hongyi, Zhang Na: Characterization of subcellular localization of duck enteritis virus UL51 protein. Virol J 2009, 6: 92. 10.1186/1743-422X-6-92
Wen YongPing, Cheng Anchun, Wang Mingshu, Ge Han, Shen ChanJuan, Xiang Jun, RenYong Jia, Zhu Dekang, Chen Xiaoyue, Lian Bei, Chang Hua, Zhou Yi: A TK recombinant protein-based ELISA for detecting antibodies to Duck Plague Virus. Virology Journal 2010, 7: 77. 10.1186/1743-422X-7-77
Zhang Shunchuan, Ma Guangpeng, Xiang Jun, Cheng Anchun, Wang Mingshu, Zhu Dekang, Jia Renyong, Luo Qihui, Chen Zhengli, Chen Xiaoyue: Expressing gK gene of duck enteritis virus guided by bioinformatics and its applied prospect in diagnosis. Virol J 2010, 7: 168. 10.1186/1743-422X-7-168
Zhang Shunchuan, Xiang Jun, Cheng Anchun, Wang Mingshu, Li Xin, Li Lijuan, Cheng Xiwen, Zhu Dekang, Luo Qihui, Chen Xiaoyue: Production, purification and characterization of polyclonal antibody against the truncated gK of the duck enteritis virus. Virol J 2010, 7: 241. 10.1186/1743-422X-7-241
Spear PG: Entry of alphaherpesviruses into cells. Seminars in Virology 1993, 4: 167-180. 10.1006/smvy.1993.1012
Dietz P, Klupp BG, Fuchs W, Köllner B, Weiland E, Mettenleiter TC: Pseudorabies Virus Glycoprotein K Requires the UL20 Gene Product for Processing. J Virol 2000, 74: 5083-5090. 10.1128/JVI.74.11.5083-5090.2000
Honess RW, Roizman B: Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J Virol 1974, 14: 8-19.
Arocho A, Chen B, Ladanyi M, Pan Q: Validation of the 2-DeltaDeltaCt calculation as an alternate method of data analysis for quantitative PCR of BCR-ABL P210 transcripts. Diagn Mol Pathol 2006, 15: 56-61. 10.1097/00019606-200603000-00009
Li Yubao, Bao Endong, Wang Zhiliang, Zhao Ruqian: Detection of HSP mRNA Transcription in Transport Stressed Pigs by Fluorescence Quantitative RT-PCR. Agricultural Sciences in China 2007, 6: 361-367. 10.1016/S1671-2927(07)60057-1
Xin Hongyi, Cheng Anchun, Mingshu Wang, Renyong Jia, Chanjuan Shen, Hua Chang: Identification and characterization of a duck enteritis virus US3-like gene. Avian Dis 2009, 53: 363-369. 10.1637/8643-020409-Reg.1
Zhao Lichan, Cheng Anchun, Wang Mingshu, Yuan Guiping, Jia Renyong, Zhou Dengchun, Qi Xuefeng, Ge Han, Sun Tao: Identification and characterization of duck enteritis virus dUTPase gene. Avian Dis 2008, 52: 324-331. 10.1637/8169-110607-ResNote.1
Shen Chanjuan, Guo Yufei, Cheng Anchun, Wang Mingshu, Zhou Yi, Lin Dan, Xin Hongyi, Zhang Na: Identification and characterization of the duck enteritis virus UL51 gene. Arch Virol 2009, 154: 1061-1069. 10.1007/s00705-009-0407-8
Shen Aimei, Ma Guangpeng, Cheng Anchun, Wang Mingshu, Luo Dandan, Lu Liting, Zhou Tao, Zhu Dekang, Luo Qihui, Jia Renyong, Chen Zhengli, Zhou Yi, Chen Xiaoyue: Transcription phase, protein characteristics of DEV UL45 and prokaryotic expression, antibody preparation of the UL45 des-transmembrane domain. Virol J 2010, 7: 232. 10.1186/1743-422X-7-232
Gale MJ, Tan SL, Katze MG: Translational control of viral gene expression in eukaryotes. Microbiol Mol Biol Rev 2000, 64: 239-280. 10.1128/MMBR.64.2.239-280.2000
Nakai K: Protein sorting signals and prediction of subcellular localization. Adv Protein Chem 2000, 54: 277-344.
Zhang Shunchuan, Cheng Anchun, Wang Mingshu, Xiang Jun, Jia Renyong, Luo Qihui, Cui Hengmin, Zhou Yi, Wang Yin, Xu Zhiwen, Chen Zhengli, Chen Xiaoyue, Wang Xiaoyu: Bioinformatics Analysis and Characteristics of Glycoprotein K Encoded by the Newly Identified UL53 Gene of Duck Enteritis Virus. IEEE, the 4th International Conference on Bioinformatics and Biomedical Engineering (iCBBE 2010) 2010.
Moyal Michal, Becker Yechiel: Analysis of the Transcription Pattern of HSV-I UL52 and UL53 Genes. Virus Genes 1993, 9: 15-21.
Johnson MA, Prideaux CT, Kongsuwan K, Tyack SG, Sheppard M: ICP27 immediate early gene, glycoprotein K (gK) and DNA helicase homologues of infectious laryngotracheitis virus (gallid herpesvirus 1) SA-2 strain. Arch Viro 1995, 140: 623-634. 10.1007/BF01309954
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25: 402-408. 10.1006/meth.2001.1262
Ziegler BL, Lamping C, Thoma S, Thomas CA: Single-cell cDNA-PCR: removal of contaminating genomic DNA from total RNA using immobilized DNase I. Biotechniques 1992, 13: 726-729.
Huang Chienjin, Wu Chingying: Characterization and expression of the pseudorabies virus early gene UL54. J Virol Methods 2004, 119: 129-136. 10.1016/j.jviromet.2004.03.013
Acknowledgements
The research was supported by Changjiang Scholars and Innovative Research Team in University(PCSIRT0848), China Agricultural Research System (CARS-43-8) and China 973 program(2011CB111606).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SCZ, JX carried out most of the experiments and drafted the manuscript. ACC, MSW, YW, XYY, DKZ, RYJ, QHL, ZLC and XYC helped in experiments and drafted the manuscript.. All authors read and approved the final manuscript.
Shunchuan Zhang, Jun Xiang contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhang, S., Xiang, J., Cheng, A. et al. Characterization of duck enteritis virus UL53 gene and glycoprotein K. Virol J 8, 235 (2011). https://doi.org/10.1186/1743-422X-8-235
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-8-235