
Abstract
Nearly 50% of HIV-infected individuals suffer from some form of HIV-associated neurocognitive disorders (HAND). HIV-1 Tat (a key HIV t ransa ctivator of t ranscription) protein is one of the first HIV proteins to be expressed after infection occurs and is absolutely required for the initiation of the HIV genome transcription. In addition to its canonical functions, various studies have shown the deleterious role of HIV-1 Tat in the development and progression of HAND. Within the CNS, only specific cell types can support productive viral replication (astrocytes and microglia), however Tat protein can be released form infected cells to affects HIV non-permissive cells such as neurons. Therefore, in this review, we will summarize the functions of HIV-1 Tat proteins in neural cells and its ability to promote HAND.
Similar content being viewed by others

Review
Introduction
The HIV-1 Tat (transactivator of transcription) gene codes for a 14-kDa protein and as its name suggests, it is a key activator of HIV-1 transcription. It is one of the first proteins to be expressed after infection occurs. Unlike typical transcription factors that are DNA binding proteins, Tat is a RNA binding protein that recognizes a specific sequence, TAR (T ransa ctivator R esponse Element), from the HIV-1 RNA molecule [1]. Tat is the protein responsible for the recruitment of the host positive transcription elongation factor b (p-TEFb) to the RNA hairpin formed at the 5’-end of nascent viral RNAs (TAR) [2, 3]. P-TEFb is a complex composed of cdk9 and cyclin T1 (CycT1) subunits that play a key role in regulating RNA polymerase II dependent transcription. Tat mediated recruitment of p-TEFb drives the phosphorylation of the C-terminal domain (CTD) repeats of RNAP II by cdk9. In its inactive form, p-TEFb binds to the inhibitory 7SK snRNP complex, which can be dissociated by Tat in order to activate cdk9. In neurons, Tat has been linked to progressive neuronal deregulation leading to the development of HIV-Associated Neurocognitive Disorders (HAND) and accelerating brain aging [4]. Although the deregulatory effect of Tat protein in the Central Nervous System (CNS) has been studied extensively, the molecular mechanisms involved remain to be elucidated. In this review we aim to summarize not only the conventional functions of HIV-1 Tat, but its contributing role in the overall complex picture of HAND as well.
Conventional functions of HIV-1 Tat protein
Tat stimulates HIV-1 gene expression during transcription initiation and elongation. It contains a very strong transcriptional activation domain composed of a Cys-rich region and a hydrophobic core motif, along with an arginine-rich RNA-binding motif (ARM) that specifies the binding of Tat to a base triple in the bulge region of the TAR RNA structure [5]. Binding of purified Tat to TAR-RNA does not require the cis-acting sequence within the loop of the TAR structure. The interaction of Tat with a transcriptional co-activator is required for high affinity, loop-specific binding to TAR RNA [6]. The Tat transactivation domain can function independently of the ARM when tethered to the DNA-or RNA- binding domain of a heterologous protein [5]. Although HIV-1 transactivation by Tat in most cell types requires intact TAR sequences, previous reports demonstrated that Tat activates HIV-1 long terminal repeat (LTR)-directed gene expression in several central nervous system-derived astrocytic/glial cell lines in the absence of TAR [7]. Furthermore, genetic experiments have suggested that Tat transactivation of the human immunodeficiency virus type 1 (HIV-1) LTR requires functional upstream enhancer sequences: the kappa B and GC-rich regions. Experiments done with HeLa cell nuclear extracts when using matrices containing chemically synthesized or bacterially expressed HIV-1 Tat, revealed the presence of the Sp1 transcription factor as one of the Tat-binding cellular proteins. Other transcription factors (Oct and NF-κB) also bound to Tat matrices but with less affinity [8].
Tat enhances the processivity of RNA polymerase II complexes that would otherwise terminate transcription prematurely and generate short transcripts, and is thus required to stimulate efficient elongation of viral transcripts [6]. Recently, a new elongation factors were identified, as part of two main protein complexes: TATcom1, which includes the p-TEFb (positive transcription elongation factor b), MLL-fusion partners involved in leukemia (AF9, AFF4, AFF1, ENL, and ELL), and the PAF1 complex [5, 9]. This 42-kDa protein was later known as cdk9 which binds to 87-kDa protein, cyclin C-related protein, designated Cyclin T [5]. Neither cdk9 nor cyclin T, cyclin K and Tat activity appear to be cell cycle regulated (this is also true for TFIIH and cdk8 -other factors involved in the phosphorylation elongation phenomenon-) [10]. The addition of Tat to nuclear extracts induces the binding of cdk9-containing TAK/p-TEFb complexes to TAR RNA. Moreover, the nuclear Tat-TAK/p-TEFb complexes did not associate with loop mutant TAR RNA, indicating that the interaction of Tat with TAK/p-TEFb might alter its RNA-binding specificity [5]. While Tat/p-TEFB complexes bind to TAR, cdk9 modifies RNA polymerase II for the efficient copying of the viral genome [11]. The interaction between Tat and cyclin T1 requires zinc and a critical cysteine residue at position 261, and the lack of this residue (C261) greatly reduces the binding of Tat/p-TEFb complexes to TAR-RNA [5]. Acetylation of Tat at residues Lys28 and Lys50 is crucial for Tat function [12]. Mechanistically, acetylation at Lys28 by P/CAF enhanced Tat binding to the Tat-associated kinase, cdk9/p-TEFb, while acetylation by p300 at Lys50 of Tat promoted the dissociation of Tat from TAR RNA that occurs during early transcription elongation [13].
II- effect of HIV-1 proteins on the central nervous system
One of the most studied secondary manifestations of chronic HIV-1 infection is HAND (HIV-Associated Neurocognitive Disease). HAND encompasses a specific group of neuropathological conditions that emerge from the continued exposure of the CNS tissue to the HIV-1, HIV-1 viral proteins (Tat, Vpr, gp120, and Nef), immune inflammation and the combination of antiretroviral therapy (cART) [14]. HAND is generally divided into three main groups depending on the severity of the neurocognitive impairment as well as the impact on everyday lives of the infected individual: HIV-Associated Dementia (HAD) is the most severe form followed by Mild Neurocognitive disorder (MND) and the Asymptomatic Neurocognitive Impairment (ANI). Since the introduction of cART, HAD cases have decreased significantly, however more chronically infected individuals are diagnosed with the milder MND and ANI [15]. This can be attributed to the fact that in the cART era, while viral detection is at its minimum, in low penetration immune privilege system such as the nervous system, the transcription of viral proteins continues [16]. This leads to a constant cytotoxic stress, inflammatory response and tissue integrity damage, all of which are major contributors to HAND development and progression. Recently, it has been shown that viral replication might occur and could evade the innate immune recognition through the recruitment of (CPSF6) and cyclophilins (Nup358 and CypA) factors [17]. Due to its endless mutation, this new discovery might somehow explain the continuous shedding of viral proteins in the brain, even at minimum and undetectable level, leading to neuronal damage.
a- HIV-1 Tat and the BBB endothelial cells
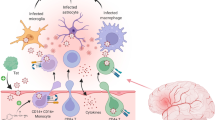
HIV-1 infiltrates the brain soon after the initial infection (Figure 1). The initial “crossing” site of the virus is the Blood Brain Barrier (BBB). The BBB is composed of highly specialized monolayer of Brain Micro-Vascular Endothelial Cells (BMEC) lying on a relatively thick basal lamina. Astrocytes processes extend to the basal lamina and are in direct contact with it. They form a membrane structure that is supported by tight junctions between the cells. The integrity of the BBB is important for the support of brain homeostasis, since it has a selective permeability. It is a physical barrier to pathogenic agents such as bacteria and viruses and to large hydrophilic molecules, but is readily permeable to other small or hydrophobic molecules such as O2, hormones and CO2[18]. Several theories have been proposed that explain the mechanism of HIV-1 entry into the CNS. The “Trojan Horse” theory, a widely accepted, suggests that infected immune cells from the blood stream can accumulate and migrate through the BBB into the brain. Because of the inability of endothelial cells to be productively infected by HIV-1 [19], numerous deleterious vascular effects are thought to be mediated by secondary mediators, including the HIV-1-specific protein Tat. Moreover, cART treatment seems to have little or no effect on the secretion rate of Tat from infected cells in the CNS [20]. Since Tat can be released by infected monocytes and macrophages accumulated at the BBB [21], it can induce those changes either via receptor-mediated pathway or through a direct uptake of the protein in an active endocytosis manner [21, 22]. Tat is responsible for changes in expression pattern of proteins important for the integrity of the endothelial tight junctions: claudins, occludins and junction adhesion molecules (JAMs). Those changes result in an increase permeability of the BBB [23–25]. Recent reports suggest that Tat protein is able to loosen up the tight junctions of the brain endothelial cells through occluding, by inhibiting it’s expression and cleaving it by matrix metalloproteinase 9 (MMP-9) [26]. In addition, Tat can trigger nuclear localization of ZO-1 via Rho signaling pathway which appears to be c-AMP response element-binding protein (CREB)-dependent response [20]. Interestingly, Tat is also able to penetrate a bi-lipid layer in a non-receptor transport mediated mechanism. This important characteristic of the protein is attributed to its transduction domain and it seems to be critical for the trans-endothelial transport of the protein [27–29]. More specifically, Cooper and his colleagues were able to show that CAYGRKKRRQRRR region of Tat is able to induce internalization of high molecular weights molecules [28]. All these studies have been predominantly performed in vitro using primary human brain microvascular endothelial cells (HBMECs) and astrocytes using Tat B peptide. It is known however that HIV-1C infected individuals are less likely to develop neurocognitive decline (see HIV-1 subtypes below). It seems that one reason for that is the reduced ability of Tat C to drive similar changes described above in the BBB as Tat B [24]. The ability of Tat to directly cross the BBB was also confirmed in vivo by Nath and colleagues [23]. They used radioactively labeled Tat (1–72) peptide injected intravenously. Interestingly, the areas of the mouse brain with highest permeably to Tat was the hippocampus, occipital cortex and hypothalamus, areas that are shown to be affected in Tat transgenic animals models as well in postmortem autopsies in HAND patients [30, 31]. This ability of Tat to enter HIV-1 non-permissive cells could open new avenues for research not only in the context of HAND development but drug delivery as well. Tat protein is also considered to be an immune response activator. For example, in Tat treated endothelial cells, cAMP dependent protein kinase pathway is involved in protein kinase C dependent induction of IL-6 [32] which is associated with higher endothelial permeability.

Tat enters the brain through the BBB. Schematic representation of Tat-modulation of the blood brain barrier. Some of the cellular factors involved are also shown.
b- HIV-1 Tat and Microglia
Once in the CNS, beyond the BBB, productive replication of HIV-1 can be supported by two cell types: microglia and the astrocytes. Microglia is a subtype of CNS immune cells that unlike the neuronal cells and astrocytes, which have neuro-ectoderm embryonic lineage, share the same origin as macrophages and other hematopoietic cells [33, 34]. Previously, the involvement of this type of cells in brain diseases was largely seen as secondary to their progression. Currently, more evidence suggests the leading role that microglia cells play in brain pathologies including infections, facial nerve axotomy, Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), HAND and stroke [35–37]. Microglia cells carry a specific role in the progression of HAND and Tat is shown to be cytotoxic and pro-inflammatory in the context of this pathological condition [38]. One of the physiological markers in advanced stages of HAND is microglial activation and multinuclear giant cells nodule formation. This can lead to changes in their immune effector functions, phagocytosis and pro-inflammatory signaling pathways such as TNF-alpha and beta-chemokine production [39, 40]. Recently, novel leucine-rich repeat kinase 2 (LRRK2) was identified as a potential pharmaceutical target for microglia activation inhibitor [41]. Protein-tyrosine phosphatase (PTP), CD45 is another promising molecule, since it is an upstream target of the pro-inflammatory intracellular signaling mediators [42]. Additionally, IL-6 induction in microglia cells is NAPDH dependent and reversible by the use of specific inhibitors [43]. This correlates with recent data showing increase in the release of glutamate, a possible explanation of the neuronal hyper excitability mediated toxicity [44]. Cautious optimism in alleviating HAND symptoms brings the fact that Ibudilast, known non-selective cyclic AMP phosphodiesterase inhibitor, that has recently showed promise as a treatment for neuropathic pain via its ability to attenuate glial cell activation, also seems to attenuate Tat induction of the nuclear factor-kappa B (NF-κB) and TNF-alpha signaling activation [40, 45]. Interestingly subtype C Tat protein was able to modulate the levels of tumor necrosis factor-receptor-associated factor 3 TRAF3 in a miR-32 dependent manner and can change the downstream expression of IRF3 and IRF7 [46]. The last finding might be an important insight, since both molecules are in the base of immune activation in response to various stimuli. Further, recently non-muscular myosin light chain kinase (nmMYLK) was described to be critical for microglial migration in Tat-treated cells and in Tat-transgenic mice, a phenomenon that is important during the innate immune response [47].
c- HIV-1 Tat and astrocytes
Unlike microglia cells, Astrocytes rise from the same neuro-ectoderm embryonic lineage as neurons [48]. They are in direct contact with neuronal cells and play critical supportive role in maintaining their homeostasis. Additionally, astrocytes have mechanical and signaling function in the formation of the Blood Brain Barrier (BBB) [49]. Although, astrocytes support productive HIV-1 infection in the CNS, they remain inaccessible to almost all known anti-retroviral treatments available [50]. Astrocytes are major contributor to the increased MCP-1 levels in the CNS in the context of HAND, Multiple Sclerosis (MS) and other neurodegenerative conditions as well [51–53]. Studies have demonstrated the ability of Tat protein to induce several genes in astrocystes such as MCP-1 through up-regulation of the (PDGF)-B [54, 55]. Tat also activates the EGR-1 promoter via specific serum response sequences within the promoter. This could be interpreted as probably one of the upstream molecular events that initiate Tat-induced astrocyte dysfunction and subsequent Tat neurotoxicity [56, 57]. As a target of MCP-1, Akt and Erk1/2 signaling proteins are indirectly affected by Tat. Additionally, reports suggest that Tat was able to induce COX-2 and PGE2 synthesis in astroglial cells through an NFAT/AP-1-dependent mechanism [58, 59]. Another interesting finding demonstrates the importance of Tat cysteine-rich domain in regulating wnt/β-catenin signaling pathway. As a result wnt/β-catenin cascade is silenced [60, 61]. This leads to abolishment of one of the natural HIV-1 transcriptional suppression mechanisms. Moreover, a clear difference was observed between subtypes B and C Tat proteins regarding their abilities to modify the wnt/β-catenin pathway.
Cell death cascade also appears to be activated as data suggest that the p53 family of proteins plays a suppressing role in HIV-1 Tat acetylation by P/CAF, a required interaction for activation of HIV-1 LTR promoter [65, 66]. This appears to be mostly a defensive response to the HIV-1 infection, since it does not necessarily leads to astrocytes apoptosis.
Further, Tat has been shown to cause cell cycle disturbance through its interaction with p53 protein [67]. Extracellular Tat is rapidly internalized by neurons and astrocytes and this may inhibit p53 ubiquitination leading to p53 accumulation. Also, p53 accumulates in microglia and astrocyte nuclei in a subset of AIDS patients without dementia, while increased neuronal p53 was only observed in HAD cases ([65, 66] and references within). Tat perform these functions through its physical interaction with p53 protein [68]. Finally, while Tat may physically associate with p53, it remains unable to activate p53 directly. Although, direct or indirect mechanisms, which lead to p53 activation in neurons in the context of HIV-1 remain unclear, it is highly likely that, like many other viruses, HIV-1 may stimulate p53 activation, which thereby alters the phenotype of uninfected microglia, and leads to neuronal loss. Note that p53 cannot induce apoptosis in response to DNA damage without p73 [66]. This illustrates that p73 is vital for p53-induced apoptosis and furthermore, that p73 is an important component of the tumor suppressor activity of p53. Finally, using astroglioma cell line, we recently demonstrated that HIV-Tat physically associates and induces the endogenous levels of p73, however, it inhibits its apoptotic activity and vice versae[65, 66]. Tat induction leads to variety of different inclusions characteristic of lysosomes, autophagic vacuoles, and lamellar bodies, which were typically present within distal cytoplasmic processes that correlate with disrupted Long Term Potentiation (LTP) and memory formation in Tat-transgenic Mice [69].
d- HIV-1 Tat and oligodendrocytes
Oligodendrocytes are the myelin producing cells in the CNS. A single cell can maintain the myelin sheath of multiple neuronal cells, which is a key morphological characteristic for the proper function of the neuronal cell. Even though, this subtype of resident glial cells has not been shown to support any active HIV-1 infection, their crucial role suggests that any disruption in their function can potentially contribute to HAND progression. Although the biological significance in vivo is yet to be determined, it has been demonstrated in vitro that oligodendrocytes are susceptible to the HIV-1 Tat protein in a Caspase-3 dependent manner [70]. Further, progressive multifocal leukoencephalopathy (PML) is evident in HIV-1 infected individuals; however reports attributed that to be the result of a co-infection with a JC virus [71, 72]. Recently, evidence emerges that the cART treatment might be further contributing to this condition as well [73, 74]. Unfortunately, much more detailed studies are required to fully understand the effects of chronic HIV-1 infection and the various viral factors on oligodendrocytes in the CNS.
e- HIV-1 Tat and neuronal cells
Unlike microglial and astrocytes, neuronal cells does not support productive HIV-1 infection, however they experience a severe stress in the form of viral proteins, pro-inflammatory cytokines, disrupted BBB and cART [14, 16, 75, 76]. Using Tat transgenic mice, studies have shown the deleterious effect that Tat has on the hippocampal, subcortical and cerebellum areas [4, 30]. Those pathophysiological changes are not only the results of astrocytes and microglia deregulation, but Tat protein can directly affect the function and viability of neurons as well. Two main theories emerge from the literature about the mechanism of direct Tat neurotoxicity: 1) Tat is able to induce changes in the neuronal cell homeostasis via extracellular signaling mechanism including receptors, changes in membrane permeability and composition; 2) Internalization of Tat protein leads to direct interaction with cellular factors involved with Ca2+ regulation, transcription and translation.
Tat has been extensively studied in in vitro systems and has been shown to increase Ca++ flux and to impair synaptic plasticity leading to neuronal deregulation. NMDA receptor for example catches often the researcher’s attention, because of its key importance in Ca++ regulation and membrane polarization [77]. Recent studies suggest that neuronal cell susceptibility relies on the expression levels of NMDAR. Not only that, but different subunits appear also to exacerbate the effect of Tat. For example rat hippocampal neurons appear less susceptible to Tat even though they highly express NMDA receptor. This might be related to low levels of NR2A subunit [78]. Moreover, in differentiated human primary neurons, Tat is able to promote the phosphorylation of Tyr1184, 1325, and Tyr1425 within the NR2A subunit [79].
Another example supporting the extracellular receptor mediated signaling theory is the growing evidence that indicates that HIV-1 Tat protein may affect the function of the dopamine transmission system. In turn, molecular components of dopamine neurotransmission may participate in a complex network of Tat-induced cell responses that result in neurodegenerative conditions. It appears that Tat induced-neurotoxicity has a reverse correlation with the D1 dopamine receptor expression levels and function. Higher levels of D1 will be related to lower apoptosis and blocked function of D1 to increased apoptosis rate in Tat-treated cells [80, 81]. Interestingly, Tat inhibits dopamine (DA) transporter (DAT) function through a PKC and trafficking-dependent mechanism and that Tat impacts the dopaminergic tone by regulating both DAT and vesicular monoamine transporter (VMAT2) proteins [82]. Those changes, at least in DAT function are related to activation of ryanodine receptor (RyRs) via a calcium- and calpain-mediated mechanism, and is independent of DAT protein synthesis, reinforcing the feasibility of RyR and GSK-3β inhibition as clinical therapeutic approaches for HAND [83]. However, the question to which specific RyRs are responsible for this signaling cascade is open for discussion. These findings further provide insight into understanding the mechanisms of HIV-1 viral protein-induced dysfunction of DA neurotransmission in HIV-1 infected patients.
In addition to cell surface receptor mediated signaling, HIV-1 Tat has the unique property of entering the cell in a calveolar and lipid rafts dependent manner. This property of the Tat protein is widely used for mediating the delivery of large protein cargos into cells when present in the extracellular milieu [84]. However, it seems like that this is an understudied area in the subfield of Tat induced-neurotoxicity. In BBB endothelial cells for example, Tat treatment leads to elevated GTP-RhoA levels and its downstream effectors, such as myosin phosphatase target subunit 1 and myosin light chain. In addition, Tat upregulates expression and promoter activity of P-gp gene as well as its efflux function. Inhibition of the Rho signaling cascade effectively blocked P-gp overexpression at the level of promoter activity. Disruption of lipid rafts by depletion of membrane cholesterol by methyl-beta-cyclodextrin, but not caveolin-1 silencing, also abolished Tat-mediated RhoA activation and P-gp upregulation [85]. This shows the critical function of intact lipid rafts and the Rho signaling in HIV-1 Tat-mediated upregulation of P-gp at least in endothelial cells even though it plausible that similar model might be applied to neuronal cells as well. Additional reports suggest that Tat uses numerous receptor- mediated pathways including CD26, CXC chemokine receptor type 4(CXCR4), heparin sulphate proteoglycans and LDL (low-density lipoprotein) receptor-related proteins [86–89]. This also suggests an active endocytosis mediated entry of HIV-1 Tat in neurons. Endocytosis is a fundamental function that plays critical role for the maintenance of neuronal function [90].
Recently, endolysosome pathway has been implicated in a variety of neurological disorders including AD (Alzheimer's disease), Parkinson's disease and HAND [91, 92]. HIV-1 Tat can accumulate in endolysosomes, which leads to endolysosomes size increased, membrane integrity disruption, pH elevation, and autophagy inhibition. Once inside the cells, the protein can be released in the cytoplasm and latter translocated in the nucleus [93]. Moreover, Tat peptide contains binding motifs for two specific protease enzymes: Furin and Calpain (Table 1). Interestingly, both enzymes are Ca++ dependent and while Furin cleavage leads to inactivation of Tat function as LTR activator, Clapain cleavage leads to increased neurotoxic activity. It is an open question whether truncation of Tat leads to increased neurotoxic activity and if so, which parts of the peptide are responsible.
Tat is associated with higher levels of nuclear and mitochondrial genomic DNA damage in the brain. High level of nuclear and mtDNA 8-oxoG damages were identified in the cortex autopsy tissue of HAND patients. Increased accumulation of mtDNA mutations and/or depletion was also detected to occur in brain tissue in a subset of HAND individuals [94]. However, these results do not discriminate between different cell types in the brain and further validation is required. Additionally, HIV-1 Tat can cause a rapid dissipation of the mitochondrial transmembrane potential, as well as cytochrome c release in isolated mitochondria. Pharmacological studies reveal that Tat induces mitochondrial membrane permeabilization (MMP), which is Bax/Bak, Bcl-2 and Bcl-xL independent and can be rescued by the anion-channel inhibitor 4,4’-diisothiocyanostilbene-2,2’-disulfonic acid (DIDS), but not by the ruthenium red, or ryanodine receptor blocker. Moreover, Tat is able to inhibit the cytochrome c oxidase (COX) activity in disrupted mitochondria making it the first viral protein to be a plausible COX inhibitor [95]. All this is an indicator that Tat can induce mitochondrial dysfunction in neurons independently from the Ca++ flux and receptor mediated pathways previously described. Taking into account the central role that mitochondria have in neuronal function, it is easy to see why neuronal cell exhibit high susceptibility to HIV-1 Tat protein. Another important aspect is the synaptic and axonal injury observed in HAND individuals and in Tat transgenic mice as well. In the context of HAND, synaptic loss can be a defense mechanism that allows the neuronal cells to deal with the over excitatory conditions in the CNS [96]. On the other hand it can be a consequence of the physical infiltration of phagocytic leucocytes, which directly target for destruction synapse endings [97]. Although some studies suggest that Tat induced synaptic loss is Ca++ dependent via NMDAR and low-density lipoprotein receptor-related protein (LRP), the molecular mechanism involved is not quite clear [98]. Release of dopamine, glutamate, GABA and acetylcholine neurotransmitters are all shown to be affected by Tat protein and it can lead to diminished LTP, induction of LTD or even an impaired signal transduction between neuronal cells [99–101].
Recent studies reveal that some of the pathways already described to be involved in neuronal deregulation are affected by expression levels of micro RNA molecules (miRNAs). miRNAs introduce a novel concept of regulatory control over gene expression, and there is increasing evidence that they may play a profound role in neuronal cell identity as well as multiple aspects of disease pathogenesis [102]. In support of this observation, a link between miRNAs and neurodegenerative diseases (e.g. Alzheimer, Huntington, and Parkinson) is becoming increasingly evident [103, 104]. Interestingly, Tat treatment of primary human neurons leads to alteration of the expression profile of miRNAs which in term affects critical pathways required for the neuronal cell function [4]. For example, miRs-1, -7, and -34a were examined and validated to deregulate the levels of SERP1 (stress-associated endoplasmic reticulum protein 1 involved in Endoplasmic Reticulum stability), Drp-1 (Dynamin-related protein nvolved in proper mitochondria distribution among the neuronal cell), as well as CREB (a key Ca++ dependent transcription factor in LTP/LTD). The importance of CREB role was also described to be a part of the Pyk2/Erk/CREB pathway, where TRPC channels have been shown to prevent Tat toxicity by inducing the Platelet-derived growth factor-BB (PDGF) [105].
f- HIV-1 Tat outside the CNS additional contributing factors, concerns and alternatives
The deleterious effect of Tat in CNS is not without a precedent and has been shown in different organs. In this regard, it has been shown, that the effect of Tat on osteoclast/ osteoblast crosstalk and homeostasis. HIV-1 infected individuals often suffer from secondary bone remodeling conditions osteopenia/osteoporosis. Key estrogenic factors such as RANKL and M-CSF are deregulated in Tat dependent manner, which leads to hyperactive osteoclast [106].
Another organ system affected is the Urinary system. In the context of HIV-Associated Nephropathy, Tat protein increased albumin permeability and rapidly induced the redistribution and loss of nephrin in isolated glomeruli [107]. Furthermore, studies investigating other HIV-Associated conditions such as HIV-Associated Thrombocytopenia, Pulmonary vesicular remodeling as well as Cancer development suggest that HIV-1Tat protein plays significant role in the pathological progression of those diseases [108–111].
HIV-1 Tat is only one of the many contributing factors that lead to HIV neurocognitive impairment progression. Gp120, Vpr and Nef proteins are all shown to affect normal neuronal function, neurotropic molecules release and immune activation. Gp120 for example is shown to up-regulate Matrix metalloproteinases (MMPs) 2 and 9 which highly correlates with Blood Brain Barrier disruption [112]. VPR is able to induce neuronal cells death and Nef is linked to spatial and recognition memory lost by targeting specifically CA3 Hippocampal neurons in in vivo mouse models [113, 114]. One of the pushbacks whenever investigating the effect of viral proteins in vitro is the use of viral protein concentrations that does not necessarily correspond to physiological conditions in the CNS. This is very critical and one should be extremely careful how to interpret results from in vitro neuronal cells experiments, since there is no active production of viral proteins in this cell type. Studying abnormal function in Astrocytes and Microglia cells, which in most cases include overexpression approach, is much more physiologically relevant since these cell types have been shown to support active HIV-1 infection and viral proteins release. This is why in vitro studies involving viral proteins and neuronal cells should be always validated using appropriate transgenic mice and co-culture assays, which represent more physiologically similar conditions.
Another aspect in studying factors contributing to neurocognitive impairment in HIV-1 positive individuals is the link between the evident immune activation and co-existing predisposing conditions such as alcohol and drugs of abuse.
g- impact of Tat on different subtypes
Analyses of different strains of HIV-1 show that isolates can be subdivided into groups, subtypes, and circulating recombinant forms (CRFs), based on phylogenetic sequence differences. So far, HIV-1 can be divided into three distinct and highly divergent groups: M (major), O (outlier), and N (non-M/O). Several genetic variants can be recognized within group M, including 9 subtypes/clades and 15 major CRFs [115, 116]. Subtype C predominates globally and, in the year 2000, caused 47.2% of all new HIV-1 infections. The second most common clade was A, which caused 30% of all new infections, (including CRF01-AE and CRF02-AG) [115]. Clade D is generally limited to Eastern and Central Africa, E appears as an A/E mosaic detected in South and East Asia [117–120]. F has been reported in Central Africa, South America and Eastern Europe. G and A/G recombinant viruses have been observed in Western and Eastern Africa as well as in Central Europe [120]. H and K have only been detected in central Africa [121–123]. J has been reported in Central America and Central Africa [124].
Since B is the predominant subtype in the western world, antiretroviral drugs used to treat HIV were developed using in vitro studies of subtype-B isolates, and most data on HIV-1 drug resistance mechanisms are from subtype-B viruses. Nevertheless, subtype B is responsible for only 12% of global infections [116]. This leads to the question whether or not HIV-1 subtypes do have any consequences on therapy outcome and the development of drug resistance, especially in regards to the implementation of antiretroviral drugs in areas with high non-B subtypes. Further, one may wonder whether subtype variations affect disease progression. For example, several groups demonstrated that, due to some unknown reasons, patients infected with HIV-1 clade C manifested less neurocognitive disorders than those infected with clade B.
The problem of subtype diversities is becoming more significant in the western world [125], especially with increasing migration and globalization where at least 25% of new infections in Europe are presently non-B African and Asian variants. The relationship between HIV-1 subtype diversities, disease transmissibility and progression is poorly understood especially regarding development of neurocognitive disorders (HAND) that display in the brain as the “white matter disease” due to neuronal degeneration and loss. Even though introduction of HAART diminishes the incidence rates for HIV dementia in the USA where HIV-1 clade B is the predominant genotype however, with continued survival, the prevalence of this disorder has actually increased. All these signify the importance for the development of new therapeutic measures, which will also address the HIV-1 subtype diversities.
Since inter-subtype studies may be complicated by host, sequence variations, societal and virological factors that are difficult to control, several groups suggest that AIDS progression differs as a function of infecting subtype [126, 127]. Their results focused mainly on polymerase (Pol), reverse transcriptase (RT), envelope (gp120, gp41) and the promoter (LTR) sequences variations. Variations in these regions may therefore affect drug susceptibility and development of drug resistance [128, 129]. For example Ethiopian clade C isolates differ (with respect to RT) from clade B by 6.8-10%. Also intra-clade differences of 3.5–5.8% have been reported for strains from Africa, India and South America [127]. It should be stressed that any given percentage variation in nucleotide sequence translates into lower amino acid sequence variation is notable because many genetic mutations are silent. Additionally, it has been shown that the LTR sequences, which contain transcriptional promoters of HIV-1 vary substantially from clade to clade [130]. Each clade has its own LTR copy number as well as an exact nucleotide sequence of enhancer and promoter structures, despite the uniformity in other LTR features, i.e. Sp1 sites, TATA box and TAT-responsive element [131, 132]. However, diversity was observed in numbers of transcriptional promoters. These include the NF-κB binding sites (3 to 4 in C, 2 in B and just 1 in E), the AP-1 transcriptional factor-binding site (1 site in subtypes C, E and G, 2 in A and F, and none in B or D) and the C/EBP-β binding-site (exists only in clade B but not in A or C). Further, subtype discrepancies arise also between the negative regulatory element seen in clades C and E versus that detected in clade B [133]. Given these genetic distinctions between HIV-1 promoters, it is not surprising to find that clades respond differentially to various transcriptional factors. For example, the NF-κB transcription factor stimulates HIV-1 clade C to a far greater extent than clade B or E [134, 135]. Likewise, tumor necrosis factor (TNF-α) activates the LTRs of clade C more impressively than those of clades A, B, D, F and G, with the lowest stimulation seen in clade E [136]. Thus, one might suggest that these intra-sequence variations might implicate the recruitment of different transcription factors through the recruitment of diverse clusters of small RNAs that regulate expression of these factors. If so, these RNAs also play a role in HIV-1 latency and disease progression leading to HAND.
Sequence variations were also observed within the viral genes (e.g. Tat). The transactivator regulatory protein (Tat) plays a major role in viral gene expression and replication. In addition, it has been described to be involved in the process of disruption of neuronal function [137]. Several studies examined the functions of Tat prepared from different clades especially since significant amino acid variations have been observed among the clade-specific Tat proteins. It has been shown that acetylation of subtype-specific Tat proteins may correlate with their transactivation efficiency [136]. Further, Tat proteins derived from HIV-1 clades C and E were strong transactivators of the HIV- promoter compared to other Tat proteins from clades A and B [138]. Tat was also used as a candidate for vaccine. Studies showed that macaques immunized with clade B Tat developed strong antibody responses when compared to Tat prepared from clade C [139]. Moreover, in mice, cross-clade immune responses between HIV-1 subtype B and C Tat proteins mapped to this T-helper epitope was identified [136]. These results led to the conclusion that a cross-clade immune response between subtypes B and C is important for a more rational design of an HIV vaccine. This conclusion was later confirmed when Tat B-clade was similarly recognized by sera from individuals infected by different virus clades A, B, C and D supporting the concept of a cross-clade vaccine [140]. Finally, using primary human neurons, subtype C Tat was shown to be less toxic than subtype B Tat [141]. In addition, clade B Tat protein was shown to increase the level of neuropathogenic agents, such as IDO and KYN in human primary astrocytes when compared to clade C Tat [142]. Taken together, these studies provide further evidence that the prevalence of HAND may be correlated with the difference in clades of HIV-1 especially since Tat has been shown to be involved in AIDS neuropathogenesis.
Concluding remarks
Thirty years of HIV-1 research have led to great advances in the control and treatment of the infection, but there still is a long way ahead in the quest of controlling the virus. The new generation of cART allows HIV-1 infected patients to live longer, almost normal life expectancy lives. Unfortunately secondary complications from the chronic infections and even the drugs themselves are degrading the quality of live for these individuals. Understanding in greater details how the virus and its proteins affect the cells on a basic molecular level will greatly increase the opportunity to design proper defensive strategies that will allow us to alleviate those pathologies. This review summarizes the advancements in only one aspect of the problem: HIV-1 Tat protein. It is surely one of the many factors in the big picture of HIV-1 infection, but as research suggests it is a very important one. With its unique ability to travel between cells and affect fundamental pathways that are important for the proper function of the cell, Tat reveals the very complex and unconventional network of tasks that this protein caries and the potential for future research. The findings from such a research can be applied not only in the field of HIV-1, but in other biological areas as well. One example today is the use of Tat peptides to drive different size molecules internalization. This property can be used in designing new strategies for drug deliveries even beyond the blood brain barrier. The role of small noncoding RNA species in the progression of the disease is another promising topic since there is an increasing body of research describing their fundamental importance in the neurodevelopment. In conclusion we have listed several tables with cellular factors important for Tat functions both canonical and secondary containing endogenous proteins that interact synergistically (Table 2) and cellular factors described to be functionally involved (Table 3) with Tat in general.
References
Feng S, Holland EC: HIV-1 tat trans-activation requires the loop sequence within tar. Nature 1988, 334: 165-167. 10.1038/334165a0
Jones KA, Peterlin BM: Control of RNA initiation and elongation at the HIV-1 promoter. Annual Rev Biochem 1994, 63: 717-743. 10.1146/annurev.bi.63.070194.003441
He N, Zhou Q: New insights into the control of HIV-1 transcription: when Tat meets the 7SK snRNP and super elongation complex (SEC). J Neuroim Pharm 2011., 6:
Chang JR, Mukerjee R, Bagashev A, Del Valle L, Chabrashvili T, Hawkins BJ, He JJ, Sawaya BE: HIV-1 Tat protein promotes neuronal dysfunction through disruption of MicroRNAs. J Biol Chem 2011, 286: 41125-41134. 10.1074/jbc.M111.268466
Wei P, Garber ME, Fang SM, Fischer WH, Jones KA: A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998, 92: 451-462. 10.1016/S0092-8674(00)80939-3
Ott M, Dorr A, Hetzer-Egger C, Kaehlcke K, Schnolzer M, Henklein P, Cole P, Zhou M-M, Verdin E: Tat acetylation: a regulatory switch between early and late phases in HIV transcription elongation. Novartis Found Symp 2004, 259: 182-193.
Taylor JP, Pomerantz RJ, Oakes JW, Khalili K, Amini S: A CNS-enriched factor that binds to NF-kappa B and is required for interaction with HIV-1 tat. Oncogene 1995, 10: 395-400.
Jeang KT, Chun R, Lin NH, Gatignol A, Glabe CG, Fan H: In vitro and in vivo binding of human immunodeficiency virus type 1 Tat protein and Sp1 transcription factor. J Virol 1993, 67: 6224-6233.
Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M: HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell 2010, 38: 439-451. 10.1016/j.molcel.2010.04.012
Gummuluru S, Emerman M: Cell cycle- and Vpr-mediated regulation of human immunodeficiency virus type 1 expression in primary and transformed T-cell lines. J Virol 1999, 73: 5422-5430.
Fujinaga K, Cujec TP, Peng JM, Garriga J, Price DH, Grana X, Peterlin BM: The ability of positive transcription elongation factor b to transactivate human immunodeficiency virus transcription depends on a functional kinase domain, cyclin T1, and Tat. J Virol 1998, 72: 7154-7159.
Marzio G, Tyagi M, Gutierrez MI, Giacca M: HIV-1 Tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc Natl Acad Sci U S A 1998, 95: 13519-13524. 10.1073/pnas.95.23.13519
Kiernan RE, Vanhulle C, Schiltz L, Adam E, Xiao H, Maudoux F, Calomme C, Burny A, Nakatani Y, Jeang KT, Benkirane M, Van Lint C: HIV-1 Tat transcriptional activity is regulated by acetylation. EMBO J 1999, 18: 6106-6118. 10.1093/emboj/18.21.6106
Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, LeBlanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, et al.: HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol 2011, 17: 3-16. 10.1007/s13365-010-0006-1
Woods SP, Moore DJ, Weber E, Grant I: Cognitive neuropsychology of HIV-associated neurocognitive disorders. Neuropsych Rev 2009, 19: 152-168. 10.1007/s11065-009-9102-5
Heaton RK, Clifford DB, Franklin DR, Woods SP, Ake C, Vaida F, Ellis RJ, Letendre SL, Marcotte TD, Atkinson JH, et al.: HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy CHARTER Study. Neurology 2010, 75: 2087-2096. 10.1212/WNL.0b013e318200d727
Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, Towers GJ: HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 2013, 503: 402-405. 10.1038/nature12769
Eugenin EA, Clements JE, Zink MC, Berman JW: Human immunodeficiency virus infection of human astrocytes disrupts blood–brain barrier integrity by a gap junction-dependent mechanism. J Neurosci 2011, 31: 9456-9465. 10.1523/JNEUROSCI.1460-11.2011
Bissel SJ, Wiley CA: Human immunodeficiency virus infection of the brain: pitfalls in evaluating infected/affected cell populations. Brain Pathol 2004, 14: 97-108.
Mediouni S, Darque A, Baillat G, Ravaux I, Dhiver C, Tissot-Dupont H, Mokhtari M, Moreau H, Tamalet C, Brunet C, et al.: Antiretroviral therapy does not block the secretion of the human immunodeficiency virus tat protein. Infect Disord Drug Targets 2012, 12: 81-86. 10.2174/187152612798994939
Rayne F, Debaisieux S, Yezid H, Lin Y-L, Mettling C, Konate K, Chazal N, Arold ST, Pugniere M, Sanchez F, et al.: Phosphatidylinositol-(4,5)-bisphosphate enables efficient secretion of HIV-1 Tat by infected T-cells. EMBO J 2010, 29: 1348-1362. 10.1038/emboj.2010.32
De Marco A, Dans PD, Knezevich A, Maiuri P, Pantano S, Marcello A: Subcellular localization of the interaction between the human immunodeficiency virus transactivator Tat and the nucleosome assembly protein 1. Amino Acids 2010, 38: 1583-1593. 10.1007/s00726-009-0378-9
Banks WA, Robinson SM, Nath A: Permeability of the blood–brain barrier to HIV-1 Tat. Exp Neurol 2005, 193: 218-227. 10.1016/j.expneurol.2004.11.019
Gandhi N, Saiyed ZM, Napuri J, Samikkannu T, Reddy PV, Agudelo M, Khatavkar P, Saxena SK, Nair MPN: Interactive role of human immunodeficiency virus type 1 (HIV-1) clade-specific Tat protein and cocaine in blood–brain barrier dysfunction: Implications for HIV-1-associated neurocognitive disorder. J Neurovirol 2010, 16: 294-305. 10.3109/13550284.2010.499891
Banerjee A, Zhang X, Manda KR, Banks WA, Ercal N: HIV proteins (gp120 and Tat) and methamphetamine in oxidative stress-induced damage in the brain: potential role of the thiol antioxidant N-acetylcysteine amide. Free Radic Biol Med 2010, 48: 1388-1398. 10.1016/j.freeradbiomed.2010.02.023
Xu R, Feng X, Xie X, Zhang J, Wu D, Xu L: HIV-1 Tat protein increases the permeability of brain endothelial cells by both inhibiting occludin expression and cleaving occludin via matrix metalloproteinase-9. Brain Res 2012, 1436: 13-19.
Arese M, Ferrandi C, Primo L, Camussi G, Bussolino F: HIV-1 Tat protein stimulates in vivo vascular permeability and lymphomononuclear cell recruitment. J Immunol 2001, 166: 1380-1388.
Cooper ISK, Teichberg VI, Schnaider-Beeri M, Fridkin M, Shechter Y: Peptide derived from HIV-1 TAT protein, destabilizes a monolayer of endothelial cells in an in vitro model of the blood–brain barrier, and allows permeation of high molecular weight proteins. J Biol Chem 2012, 44676-44683.
Jeang KT, Xiao H, Rich EA: Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J Biol Chem 1999, 274: 28837-28840. 10.1074/jbc.274.41.28837
Kim BO, Liu Y, Ruan YW, Xu ZC, Schantz L, He JJ: Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol 2003, 162: 1693-1707. 10.1016/S0002-9440(10)64304-0
Thompson PM, Dutton RA, Hayashi KM, Toga AW, Lopez OL, Aizenstein HJ, Becker JT: Thinning of the cerebral cortex visualized in HIV/AIDS reflects CD4(+) T lymphocyte decline. Proc Natl Acad Sci U S A 2005, 102: 15647-15652. 10.1073/pnas.0502548102
Zidovetzki R, Wang JL, Chen PJ, Jeyaseelan R, Hofman F: Human immunodeficiency virus Tat protein induces interleukin 6 mRNA expression in human brain endothelial cells via protein kinase C– and cAMP-dependent protein kinase pathways. Aids Res Hum Retroviruses 1998, 14: 825-833. 10.1089/aid.1998.14.825
Perry VH, Nicoll JAR, Holmes C: Microglia in neurodegenerative disease. Nat Rev Neurol 2010, 6: 193-201. 10.1038/nrneurol.2010.17
Prinz M, Priller J, Sisodia SS, Ransohoff RM: Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci 2011, 14: 1227-1235.
Kettenmann H, Hanisch U-K, Noda M, Verkhratsky A: Physiology of Microglia. Physiol Rev 2011, 91: 461-553. 10.1152/physrev.00011.2010
Davoust N, Vuaillat C, Androdias G, Nataf S: From bone marrow to microglia: barriers and avenues. Trends Immunol 2008, 29: 227-234. 10.1016/j.it.2008.01.010
Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dahnau I, Finsen B: Microglial cell population dynamics in the injured adult central nervous system. Brain Res Rev 2005, 48: 196-206. 10.1016/j.brainresrev.2004.12.009
Minghetti L, Visentin S, Patrizio M, Franchini L, Antonietta M, Ajmone-Cat MA, Levi G: Multiple actions of the human immunodeficiency virus type-1 Tat protein on microglial cell functions. Neurochem Res 2004, 29: 965-978.
Hahn YK, Vo P, Fitting S, Block ML, Hauser KF, Knapp PE: beta-Chemokine production by neural and glial progenitor cells is enhanced by HIV-1 Tat: effects on microglial migration. J Neurochem 2010, 114: 97-109.
Kiebala M, Polesskaya O, Yao Z, Perry SW, Maggirwar SB: Nuclear factor-kappa B family member RelB inhibits human immunodeficiency virus-1 Tat-induced tumor necrosis factor-alpha production. Plos One 2010., 5:
Marker DF, Puccini JM, Mockus TE, Barbieri J, Lu SM, Gelbard HA: LRRK2 kinase inhibition prevents pathological microglial phagocytosis in response to HIV-1 Tat protein. J Neuroinflammation 2012, 9: 261. 10.1186/1742-2094-9-261
Jin J, Lam L, Sadic E, Fernandez F, Tan J, Giunta B: HIV-1 Tat-induced microglial activation and neuronal damage is inhibited via CD45 modulation: a potential new treatment target for HAND. Am J Transl Res 2012, 4: 302-315.
Turchan-Cholewo J, Dimayuga VM, Gupta S, Gorospe RMC, Keller JN, Bruce-Keller AJ: NADPH oxidase drives cytokine and neurotoxin release from microglia and macrophages in response to HIV-Tat. Antioxid Redox Signal 2009, 11: 193-204. 10.1089/ars.2008.2097
Gupta S, Knight AG, Gupta S, Knapp PE, Hauser KF, Keller JN, Bruce-Keller AJ: HIV-Tat elicits microglial glutamate release: role of NAPDH oxidase and the cystine-glutamate antiporter. Neurosci Lett 2010, 485: 233-236. 10.1016/j.neulet.2010.09.019
Kiebala M, Maggirwar SB: Ibudilast, a pharmacologic phosphodiesterase inhibitor, prevents human immunodeficiency virus-1 Tat-mediated activation of microglial cells. Plos One 2011., 6:
Mishra R, Chhatbar C, Singh SK: HIV-1 Tat C-mediated regulation of tumor necrosis factor receptor-associated factor-3 by microRNA 32 in human microglia. J Neuroinflammation 2012, 9: 131. 10.1186/1742-2094-9-131
Yao H, Duan M, Yang L, Buch S: Nonmuscle myosin light-chain kinase mediates microglial migration induced by HIV Tat: involvement of β1 integrins. FASEB J 2013, 27: 1532-1548. 10.1096/fj.12-219600
Kriegstein A, Alvarez-Buylla A: The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 2009, 32: 149-184. 10.1146/annurev.neuro.051508.135600
Ballabh P, Braun A, Nedergaard M: The blood–brain barrier: an overview - structure, regulation, and clinical implications. Neurobiol Dis 2004, 16: 1-13. 10.1016/j.nbd.2003.12.016
Dunfee R, Thomas ER, Gorry PR, Wang J, Ancuta P, Gabuzda D: Mechanisms of HIV-1 neurotropism. Curr HIV Res 2006, 4: 267-278. 10.2174/157016206777709500
Weiss JM, Nath A, Major EO, Berman JW: HIV-1 Tat induces monocyte chemoattractant protein-1-mediated monocyte transmigration across a model of the human blood–brain barrier and up-regulates CCR5 expression on human monocytes. J Immunol 1999, 163: 2953-2959.
Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, Estes ML, Thomas DM, Tuohy VK: Astrocyte expression of mesenger-RNA encoding cytokines IP-10 and JE/MCP-1 experimental autoimmune encephalomyelitis. FASEB J 1993, 7: 592-600.
Mahad DJ, Ransohoff RM: The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin Immunol 2003, 15: 23-32. 10.1016/S1044-5323(02)00125-2
Van der Voorn P, Tekstra J, Beelen RHJ, Tensen CP, Van der Valk P, De Groot CJ: Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am J Pathol 1999, 154: 45-51. 10.1016/S0002-9440(10)65249-2
Bethel-Brown C, Yao H, Hu G, Buch S: Platelet-derived growth factor (PDGF)-BB-mediated induction of monocyte chemoattractant protein 1 in human astrocytes: implications for HIV-associated neuroinflammation. J Neuroinflammation 2012, 9: 262. 10.1186/1742-2094-9-262
Bethel-Brown C, Yao H, Callen S, Lee YH, Dash PK, Kumar A, Buch S: HIV-1 Tat-mediated induction of platelet-derived growth factor in astrocytes: role of early growth response gene 1. J Immunol 2011, 186: 4119-4129. 10.4049/jimmunol.1002235
Fan Y, Zou W, Green LA, Kim BO, He JJ: Activation of Egr-1 expression in astrocytes by HIV-1 Tat: new insights into astrocyte-mediated Tat neurotoxicity. J Neuroimmune Pharmacol 2011, 6: 121-129. 10.1007/s11481-010-9217-8
Zou W, Wang Z, Liu Y, Fan Y, Zhou BY, Yang XF, He JJ: Involvement of p300 in constitutive and HIV-1 Tat-activated expression of glial fibrillary acidic protein in astrocytes. Glia 2010, 58: 1640-1648.
Blanco A, Alvarez S, Fresno M, Munoz-Fernandez MA: Extracellular HIV-tat induces cyclooxygenase-2 in glial cells through activation of nuclear factor of activated T cells. J Immunol 2008, 180: 530-540.
Sharma AHX, Napier TC, Al-Harthi L: Methamphetamine and HIV-1 Tat down regulate β-catenin signaling: implications for methampetamine abuse andHIV-1 co-morbidity. J Neuroimmune Pharmacol 2011, 6: 597-607. 10.1007/s11481-011-9295-2
Henderson LJ, Sharma A, Monaco MC, Major EO, Al-Harthi L: Human immunodeficiency virus type 1 (HIV-1) transactivator of transcription through its intact core and cysteine-rich domains inhibits Wnt/β-catenin signaling in astrocytes: relevance to HIV neuropathogenesis. J Neurosci 2012, 32: 16306-16313. 10.1523/JNEUROSCI.3145-12.2012
Song HY, Ju SM, Seo WY, Goh AR, Lee J-K, Bae YS, Choi SY, Park J: Nox2-based NADPH oxidase mediates HIV-1 Tat-induced up-regulation of VCAM-1/ICAM-1 and subsequent monocyte adhesion in human astrocytes. Free Radic Biol Med 2011, 50: 576-584. 10.1016/j.freeradbiomed.2010.12.019
Astarci ESA, Cimen I, Savaş B: The NF-κB target genes ICAM-1 and VCAM-1 are differentially regulated during spontaneous differentiation of Caco-2 cells. FEBS J 2012, 279: 2966-2986. 10.1111/j.1742-4658.2012.08677.x
El-Hage N, Podhaizer EM, Sturgill J, Hauser KF: Toll-like receptor expression and activation in astroglia: differential regulation by HIV-1 Tat, gp120, and morphine. Immunol Invest 2011, 40: 498-522. 10.3109/08820139.2011.561904
Amini S, Mameli G, Del Valle L, Skowronska A, Reiss K, Gelman BB, White MK, Khalili K, Sawaya BE: p73 interacts with human immunodeficiency virus type 1 Tat in astrocytic cells and prevents its acetylation on lysine 28. Mol Cell Biol 2005, 25: 8126-8138. 10.1128/MCB.25.18.8126-8138.2005
Saunders M, Eldeen MB, Del Valle L, Reiss K, Peruzzi F, Mameli G, Gelman BB, Khalili K, Amini S, Sawaya BE: p73 modulates HIV-1 Tat transcriptional and apoptotic activities in human astrocytes. Apoptosis 2005, 10: 1419-1431. 10.1007/s10495-005-2467-x
Clark E, Santiago F, Deng L, Chong S, de La Fuente C, Wang L, Fu P, Stein D, Denny T, Lanka V, Mozafari F, Okamoto T, Kashanchi F: Loss of G (1)/S checkpoint in human immunodeficiency virus type 1-infected cells is associated with a lack of cyclin-dependent kinase inhibitor p21/Waf1. J Virol 2000, 74: 5040-5052. 10.1128/JVI.74.11.5040-5052.2000
Longo F, Marchetti MA, Castagnoli L, Battaglia PA, Gigliani F: A novel approach to protein-protein interaction: complex formation between the p53 tumor suppressor and the HIV Tat proteins. Biochem Biophys Res Commun 1995, 206: 326-334. 10.1006/bbrc.1995.1045
Fitting SI-JB, Bull C, Skoff RP, Lichtman AH, Wise LE, Fox MA, Su J, Medina AE, Krahe TE, Knapp PE, Guido W, Hauser KF: Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol Psychiatry 2012, 3223: 852-859.
Hauser KF, Hahn YK, Adjan VV, Zou S, Buch SK, Nath A, Bruce-Keller AJ, Knapp PE: HIV-1 Tat and morphine have interactive effects on oligodendrocyte survival and morphology. Glia 2009, 57: 194-206. 10.1002/glia.20746
Aksamit AJ Jr: Progressive multifocal leukoencephalopathy. Continuum (Minneapolis, Minn) 2012, 18: 1374-1391.
Datta S, Wattal C, Sethi PK, Buxi T, Jain D: Use of John Cunningham virus polymerase chain reaction in the diagnosis of progressive multifocal leucoencephalopathy - a rare presenting manifestation in an HIV-positive patient. Indian J Med Microbiol 2012, 30: 239-241. 10.4103/0255-0857.96710
Adachi E, Koibuchi T, Imai K, Kikuchi T, Koga M, Nakamura H, Miura T, Iwamoto A, Fujii T: Favourable outcome of progressive multifocal leukoencephalopathy with mefloquine treatment in combination with antiretroviral therapy in an HIV-infected patient. Int J STD AIDS 2012, 23: 603-605. 10.1258/ijsa.2012.011305
Naito K, Ueno H, Sekine M, Kanemitsu M, Ohshita T, Nakamura T, Yamawaki T, Matsumoto M: Akinetic mutism caused by HIV-associated progressive multifocal leukoencephalopathy was successfully treated with mefloquine: a serial multimodal MRI study. Intern Med 2012, 51: 205-209. 10.2169/internalmedicine.51.6253
Biscione FM: Regarding “Persistence of neuropsychologic deficits despite long-term highly active antiretroviral therapy in patients with HIV-related neurocognitive impairment: prevalence and risk factors”. J Acquir Immune Defic Syndr 2007, 46: 510.
Giancola ML, Lorenzini P, Balestra P, Larussa D, Baldini F, Corpolongo A, Narciso P, Bellagamba R, Tozzi V, Antinori A: Neuroactive antiretroviral drugs do not influence neurocognitive performance in less advanced HIV-infected patients responding to highly active antiretroviral therapy. J Acquir Immune Defic Syndr 2006, 41: 332-337. 10.1097/01.qai.0000197077.64021.07
Volianskis ABN, Collett VJ, Irvine MW, Monaghan DT, Fitzjohn S, Jensen MS, Jane DE, Collingridge GL: Different NMDAR subtypes mediate induction of LTP and two forms of STP at CA1 synapses in the rat hippocampus in vitro. J Physiol 2013, 591: 955-972. 10.1113/jphysiol.2012.247296
Eugenin EA, King JE, Hazleton JE, Major EO, Bennett MVL, Zukin RS, Berman JW: Differences in NMDA receptor expression during human development determine the response of neurons to HIV-Tat-mediated neurotoxicity. Neurotox Res 2011, 19: 138-148. 10.1007/s12640-010-9150-x
King JE, Eugenin EA, Hazleton JE, Morgello S, Berman JW: Mechanisms of HIV-tat-induced phosphorylation of N-Methyl-D-Aspartate receptor subunit 2A in human primary neurons implications for NeuroAIDS pathogenesis. Am J Pathol 2010, 176: 2819-2830. 10.2353/ajpath.2010.090642
Silvers JM, Aksenova MV, Aksenov MY, Mactutus CF, Booze RM: Neurotoxicity of HIV-1 tat protein: Involvement of D1 dopamine receptor. Neurotoxicology 2007, 28: 1184-1190. 10.1016/j.neuro.2007.07.005
Aksenov MY, Aksenova MV, Silvers JM, Mactutus CF, Booze RM: Different effects of selective dopamine uptake inhibitors, GBR 12909 and WIN 35428, on HIV-1 Tat toxicity in rat fetal midbrain neurons. Neurotoxicology 2008, 29: 971-977. 10.1016/j.neuro.2008.06.003
Midde NM, Gomez AM, Zhu J: HIV-1 Tat protein decreases dopamine transporter cell surface expression and vesicular monoamine transporter-2 function in Rat striatal synaptosomes. J Neuroimmune Pharmacol 2012, 7: 629-639. 10.1007/s11481-012-9369-9
Perry SW, Barbieri J, Tong N, Polesskaya O, Pudasaini S, Stout A, Lu R, Kiebala M, Maggirwar SB, Gelbard HA: Human immunodeficiency virus-1 Tat activates calpain proteases via the ryanodine receptor to enhance surface dopamine transporter levels and increase transporter-specific uptake and V-max. J Neurosci 2010, 30: 14153-14164. 10.1523/JNEUROSCI.1042-10.2010
Fittipaldi A, Ferrari A, Zoppe M, Arcangeli C, Pellegrini V, Beltram F, Giacca M: Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. Mol Ther 2003, 278: 34141-34149.
Zhong Y, Hennig B, Toborek M: Intact lipid rafts regulate HIV-1 Tat protein-induced activation of the Rho signaling and upregulation of P-glycoprotein in brain endothelial cells. J Cereb Blood Flow Metab 2010, 30: 522-533. 10.1038/jcbfm.2009.214
Gutheil WG, Subramanyam M, Flentke GR, Sanford DG, Munoz E, Huber BT, Bachovchin WW: Human immunodeficiency virus 1 Tat binds to dipeptidyl aminopeptidase IV (CD26): a possible mechanism for Tat's immunosuppressive activity. Proc Natl Acad Sci U S A 1994, 91: 6594-6598. 10.1073/pnas.91.14.6594
Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT: Selective CXCR4 antagonism by Tat: Implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci U S A 2000, 97: 11466-11471. 10.1073/pnas.97.21.11466
Chattopadhyay N, Zastre J, Wong H-L, Wu XY, Bendayan R: Solid lipid nanoparticles enhance the delivery of the HIV protease inhibitor, atazanavir, by a human brain endothelial cell line. Pharm Res 2008, 25: 2262-2271. 10.1007/s11095-008-9615-2
Deshmane SL, Mukerjee R, Fan S, Sawaya BE: High-performance capillary electrophoresis for determining HIV-1 Tat protein in neurons. Plos One 2011., 6:
Nixon RA, Cataldo AM: The endosomal-lysosomal system of neurons-new roles. Trends Neurosci 1995, 18: 489-496. 10.1016/0166-2236(95)92772-I
Gelman BB, Soukup VM, Holzer CE, Fabian RH, Schuenke KW, Keherly MJ, Richey FJ, Lahart CJ: Potential role for white matter lysosome expansion in HIV-associated dementia. J Acquir Immune Defic Syndr 2005, 39: 422-425. 10.1097/01.qai.0000164250.41475.f2
Nixon RA, Cataldo AM: Lysosomal system pathways: genes to neurodegeneration in Alzheimer's disease. Journal of Alzheimer's disease: J Alzheimers Dis 2006, 9: 277-289.
Hui L, Chen X, Haughey NJ, Geiger JD: Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. ASN Neuro 2012, 4: 243-252. 10.1042/AN20120017
Zhang Y, Wang M, Li H, Zhang H, Shi Y, Wei F, Liu D, Liu K, Chen D: Accumulation of nuclear and mitochondrial DNA damage in the frontal cortex cells of patients with HIV-associated neurocognitive disorders. Brain Res 2012, 1458: 1-11.
Lecoeur H, Borgne-Sanchez A, Chaloin O, El-Khoury R, Brabant M, Langonne A, Porceddu M, Briere JJ, Buron N, Rebouillat D, et al.: HIV-1 Tat protein directly induces mitochondrial membrane permeabilization and inactivates cytochrome c oxidase. Cell Death Dis 2012, 3: 282. 10.1038/cddis.2012.21
Shin AH, Kim HJ, Thayer SA: Subtype selective NMDA receptor antagonists induce recovery of synapses lost following exposure to HIV-1 Tat. British J Pharmacol 2012, 166: 1002-1017. 10.1111/j.1476-5381.2011.01805.x
Lu S-M, Tremblay M-E, King IL, Qi J, Reynolds HM, Marker DF, Varrone JJP, Majewska AK, Dewhurst S, Gelbard HA: HIV-1 Tat-induced microgliosis and synaptic damage via interactions between peripheral and central myeloid cells. PLoS One 2011, 6: 23915. 10.1371/journal.pone.0023915
Kim HJ, Martemyanov KA, Thayer SA: Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci 2008, 28: 12604-12613. 10.1523/JNEUROSCI.2958-08.2008
Feligioni M, Raiteri L, Pattarini R, Grilli M, Bruzzone S, Cavazzani P, Raiteri M, Pittaluga A: The human immunodeficiency virus-1 protein Tat and its discrete fragments evoke selective release of acetylcholine from human and rat cerebrocortical terminals through species-specific mechanisms. J Neurosci 2003, 23: 6810-6818.
Ferris MJ, Frederick-Duus D, Fadel J, Mactutus CF, Booze RM: In vivo microdialysis in awake, freely moving rats demonstrates HIV-1 Tat-induced alterations in dopamine transmission. Synapse 2009, 63: 181-185. 10.1002/syn.20594
Musante V, Summa M, Neri E, Puliti A, Godowicz TT, Severi P, Battaglia G, Raiteri M, Pittaluga A: The HIV-1 viral protein Tat increases glutamate and decreases GABA exocytosis from human and mouse neocortical nerve endings. Cereb Cortex 2010, 20: 1974-1984. 10.1093/cercor/bhp274
Mukerjee R, Chang JR, Del Valle L, Bagashev A, Gayed MM, Lyde RB, Hawkins BJ, Brailoiu E, Cohen E, Power C, et al.: Deregulation of microRNAs by HIV-1 Vpr protein leads to the development of neurocognitive disorders. J Biol Chem 2011, 286: 34976-34985. 10.1074/jbc.M111.241547
Nelson PT, Wang W-X, Rajeev BW: MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol 2008, 18: 130-138. 10.1111/j.1750-3639.2007.00120.x
Wang W, Kwon EJ, Tsai L-H: MicroRNAs in learning, memory, and neurological diseases. Learn Mem 2012, 19: 359-368. 10.1101/lm.026492.112
Yao H, Peng F, Fan Y, Zhu X, Hu G, Buch SJ: TRPC channel-mediated neuroprotection by PDGF involves Pyk2/ERK/CREB pathway. Cell Death Differ 2009, 16: 1681-1693. 10.1038/cdd.2009.108
Gibellini D, De Crignis E, Ponti C, Borderi M, Clo A, Miserocchi A, Viale P, Re MC: HIV-1 Tat protein enhances RANKL/M-CSF-mediated osteoclast differentiation. Biochem Biophys Res Commun 2010, 401: 429-434. 10.1016/j.bbrc.2010.09.071
Doublier S, Zennaro C, Spatola T, Lupia E, Bottelli A, Deregibus MC, Carraro M, Conaldi PG, Camussi G: HIV-1 Tat reduces nephrin in human podocytes: a potential mechanism for enhanced glomerular permeability in HIV-associated nephropathy. AIDS 2007, 21: 423-432. 10.1097/QAD.0b013e328012c522
Mermis J, Gu H, Xue B, Li F, Tawfik O, Buch S, Bartolome S, O'Brien-Ladner A, Dhillon NK: Hypoxia-inducible factor-1 alpha/platelet derived growth factor axis in HIV-associated pulmonary vascular remodeling. Respi Res 2011, 12: 103. 10.1186/1465-9921-12-103
Wang J, Zhang W, Nardi MA, Li Z: HIV-1 Tat-induced platelet activation and release of CD154 contribute to HIV-1-associated autoimmune thrombocytopenia. J Thromb Haemost 2011, 9: 562-573. 10.1111/j.1538-7836.2010.04168.x
Nunnari G, Smith JA, Daniel R: HIV-1 Tat and AIDS-associated cancer: targeting the cellular anti-cancer barrier? J Exp Clin Cancer Res 2008, 27: 3. 10.1186/1756-9966-27-3
Huynh D, Vincan E, Mantamadiotis T, Purcell D, Chan C-K, Ramsay R: Oncogenic properties of HIV-Tat in colorectal cancer cells. Curr HIV Res 2007, 5: 403-409. 10.2174/157016207781023974
Louboutin J-P, Reyes BAS, Agrawal L, Van Bockstaele EJ, Strayer DS: HIV-1 gp120 upregulates matrix metalloproteinases and their inhibitors in a rat model of HIV encephalopathy. European J Neurosci 2011, 34: 2015-2023. 10.1111/j.1460-9568.2011.07908.x
Chompre G, Cruz E, Maldonado L, Rivera-Amill V, Porter JT, Noel RJ Jr: Astrocytic expression of HIV-1 Nef impairs spatial and recognition memory. Neurobiol Dis 2013, 49: 128-136.
Cheng X, Mukhtar M, Acheampong EA, Srinivasan A, Rafi M, Pomerantz RJ, Parveen Z: HIV-1 Vpr potently induces programmed cell death in the CNS in vivo. DNA Cell Biol 2007, 26: 116-131. 10.1089/dna.2006.0541
Kuiken C, Thakallapalli R, Esklid A, de Ronde A: Genetic analysis reveals epidemiologic patterns in the spread of human immunodeficiency virus. Am J Epidemiol 2000, 152: 814-822. 10.1093/aje/152.9.814
Robertson DL, Anderson JP, Bradac JA, Carr JK, Foley B, Funkhouser RK, Gao F, Hahn BH, Kalish ML, Kuiken C, Learn GH, Leitner T, et al.: HIV-1 nomenclature proposal. Science 2000, 288: 55-56.
Osmanov S, Pattou C, Walker N, Schwardländer B, Esparza J: Estimated global distribution and regional spread of HIV-1 genetic subtypes in the year 2000. J Acquir Immune Defic Syndr 2002, 29: 184-190.
Artenstein AW, Coppola J, Brown AE, Carr JK, Sanders-Buell E, Galbarini E, Mascola JR, VanCott TC, Schonbrood P, McCutchan FE, et al.: Multiple introductions of HIV-1 subtype E into the western hemisphere. Lancet 1995, 346: 1197-1198. 10.1016/S0140-6736(95)92900-2
Janssens W, Buvé A, Nkengasong JN: The puzzle of HIV-1 subtypes in Africa. AIDS 1997, 11: 705-712. 10.1097/00002030-199706000-00002
Paladin FJ, Monzon OT, Tsuchie H, Aplasca MR, Learn GH Jr, Kurimura T: Genetic subtypes of HIV-1 in the Philippines. AIDS 1998, 12: 291-300. 10.1097/00002030-199803000-00007
Piot P, Bartos M, Ghys PD, Walker N, Schwartländer B: The global impact of HIV/AIDS. Nature 2001, 410: 968-973. 10.1038/35073639
Quiñones-Mateu ME, Albright JL, Torre V, Reinis M, Vandasová J, Brucková M, Arts EJ: Molecular epidemiology of HIV type 1 isolates from the Czech Republic: identification of an env E subtype case. AIDS Res Hum Retroviruses 1999, 15: 85-89. 10.1089/088922299311763
Delaporte E, Janssens W, Peeters M, Buvé A, Dibanga G, Perret JL, Ditsambou V, Mba JR, Courbot MC, Georges A, et al.: Epidemiological and molecular characteristics of HIV infection in Gabon, 1986–1994. AIDS 1996, 10: 903-910. 10.1097/00002030-199607000-00014
Kalish ML, Baldwin A, Raktham S, Wasi C, Luo CC, Schochetman G, Mastro TD, Young N, Vanichseni S, Rübsamen-Waigmann H, et al.: The evolving molecular epidemiology of HIV-1 envelope subtypes in injecting drug users in Bangkok, Thailand: implications for HIV vaccine trials. AIDS 1995, 9: 851-857. 10.1097/00002030-199508000-00004
Takehisa J, Zekeng L, Ido E, Mboudjeka I, Moriyama H, Miura T, Yamashita M, Gürtler LG, Hayami M, Kaptué L: Various types of HIV mixed infections in Cameroon. Virology 1998, 245: 1-10. 10.1006/viro.1998.9141
Bikandou B, Takehisa J, Mboudjeka I, Ido E, Kuwata T, Miyazaki Y, Moriyama H, Harada Y, Taniguchi Y, Ichimura H, Ikeda M, et al.: Genetic subtypes of HIV type 1 in Republic of Congo. AIDS Res Hum Retroviruses 2000, 16: 613-619. 10.1089/088922200308837
Wainberg MA: HIV-1 subtype distribution and the problem of drug resistance. AIDS 2004,18(Suppl 3):63-68.
Kantor R, Katzenstein D: Polymorphism in HIV-1 non-subtype B protease and reverse transcriptase and its potential impact on drug susceptibility and drug resistance evolution. AIDS Rev 2003, 5: 25-35.
De Arellano ER, Soriano V, Holguín A: Genetic analysis of regulatory, promoter, and TAR regions of LTR sequences belonging to HIV type 1 Non-B subtypes. AIDS Res Hum Retroviruses 2005, 21: 949-954. 10.1089/aid.2005.21.949
Ramírez De Arellano E, Soriano V, Alcamil J, Holguín A: New findings on transcription regulation across different HIV-1 subtypes. AIDS Rev 2006, 8: 9-16.
Snoeck J, Kantor R, Shafer RW, Van Laethem K, Deforche K, Carvalho AP, et al.: Discordances between interpretation algorithms for genotypic resistance to protease and reverse transcriptase inhibitors of human immunodeficiency virus are subtype dependent. Antimicrob Agents Chemother 2006, 50: 694-701. 10.1128/AAC.50.2.694-701.2006
Rodriguez MA, Shen C, Ratner D, Paranjape RS, Kulkarni SS, Chatterjee R, Gupta P: Genetic and functional characterization of the LTR of HIV-1 subtypes A and C circulating in India. AIDS Res Hum Retroviruses 2007, 23: 1428-1433. 10.1089/aid.2007.0152
De Baar MP, Abebe A, Kliphuis A, Tesfaye G, Goudsmit J, Pollakis G: HIV type 1C and C’ subclusters based on long terminal repeat sequences in the Ethiopian type 1 subtype C epidemic. AIDS Res Hum Retroviruses 2003, 19: 917-922. 10.1089/088922203322493094
Montano MA, Nixon CP, Essex M: Dysregulation through the NF-kappaB enhancer and TATA box of the human immunodeficiency virus type 1 subtype E promoter. J Virol 1998, 72: 8446-8452.
Naghavi MH, Estable MC, Schwartz S, Roeder RG, Vahlne A: Upstream stimulating factor affects human immunodeficiency virus type 1 long terminal repeat-directed transcription in a cell-specific manner, independently of the HIV-1 subtype and the core-negative regulatory element. J Gen Virol 2001, 82: 547-559.
Roof P, Ricci M, Genin P, Montano MA, Essex M, Wainberg MA, Gatignol A, Hiscott J: Differential regulation of HIV-1 clade-specific B, C, and E long terminal repeats by NF-kappaB and the Tat transactivator. Virology 2002, 296: 77-83. 10.1006/viro.2001.1397
Jeeninga RE, Hoogenkamp M, Armand-Ugon M, de Baar M, Verhoef K, Berkhout B: Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J Virol 2000, 74: 3740-3751. 10.1128/JVI.74.8.3740-3751.2000
Montano MA, Nixon CP, Ndung'u T, Bussmann H, Novitsky VA, Dickman D, Essex M: Elevated tumor necrosis factor-alpha activation of human immunodeficiency virus type 1 subtype C in Southern Africa is associated with an NF-kappaB enhancer gain-of-function. J Infect Dis 2000, 181: 76-81. 10.1086/315185
Mukerjee R, Deshmane SL, Fan S, Del Valle L, White MK, Khalili K, Amini S, Sawaya BE: Involvement of the p53 and p73 transcription factors in neuroAIDS. Cell Cycle 2008, 7: 2682-2690. 10.4161/cc.7.17.6450
Desfosses Y, Solis M, Sun Q, Grandvaux N, Van Lint C, Burny A, Gatignol A, Wainberg MA, Lin R, Hiscott J: Regulation of human immunodeficiency virus type 1 gene expression by clade-specific Tat proteins. J Virol 2005, 79: 9180-9191. 10.1128/JVI.79.14.9180-9191.2005
Tikhonov I, Ruckwardt TJ, Hatfield GS, Pauza CD: Tat-neutralizing antibodies in vaccinated macaques. J Virol 2003, 77: 3157-3166. 10.1128/JVI.77.5.3157-3166.2003
Ramakrishna L, Anand KK, Mohankumar KM, Ranga U: Codon optimization of the tat antigen of human immunodeficiency virus type 1 generates strong immune responses in mice following genetic immunization. J Virol 2004, 78: 9174-9189. 10.1128/JVI.78.17.9174-9189.2004
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AB wrote the manuscript. BES modified parts of the manuscript. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Bagashev, A., Sawaya, B.E. Roles and functions of HIV-1 Tat protein in the CNS: an overview. Virol J 10, 358 (2013). https://doi.org/10.1186/1743-422X-10-358
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-10-358