
Abstract
Intracerebral hemorrhage (ICH) is a common type of fatal stroke, accounting for about 15% to 20% of all strokes. Hemorrhagic strokes are associated with high mortality and morbidity, and increasing evidence shows that innate immune responses and inflammatory injury play a critical role in ICH-induced neurological deficits. However, the signaling pathways involved in ICH-induced inflammatory responses remain elusive. Toll-like receptor 4 (TLR4) belongs to a large family of pattern recognition receptors that play a key role in innate immunity and inflammatory responses. In this review, we summarize recent findings concerning the involvement of TLR4 signaling in ICH-induced inflammation and brain injury. We discuss the key mechanisms associated with TLR4 signaling in ICH and explore the potential for therapeutic intervention by targeting TLR4 signaling.
Similar content being viewed by others

Introduction
Intracerebral hemorrhage (ICH) is the least treatable type of stroke and has devastating consequences [1]. Hemorrhagic strokes account for 15% to 20% of all strokes and are associated with high mortality and morbidity [2, 3]. Primary damage caused by ICH occurs within the first few hours after the onset of bleeding and is mainly due to formation of hematomas, which compress adjacent tissues, thus destroying them [4]. Many patients with ICH deteriorate progressively with no sign of hematoma expansion, suggesting that secondary damage following ICH plays a critical role in neurological deterioration [5, 6]. Several lines of evidence show that secondary damage involves inflammation, cerebral edema, and cellular apoptosis, ultimately leading to blood–brain barrier disruption and massive brain cell death [4, 7–10]. The molecular mechanisms of secondary damage after ICH have not been well-established, but they may represent novel therapeutic targets to prevent further brain injury.
Secondary damage following ICH is triggered by the presence of intraparenchymal blood, which subsequently activates cytotoxic, oxidative and inflammatory pathways [4, 10]. The toxic effects of extravasated blood result mainly from blood components, including red blood cells (RBCs), coagulation factors, complement components and immunoglobulins [4, 5, 10]. Thrombin, a serine protease produced rapidly after ICH onset, contributes to edema formation and blood–brain barrier damage in early brain injury, and activates the cytotoxic, excitotoxic and inflammatory pathways that are involved in secondary injury following ICH [5, 10, 11]. Hemoglobin (Hb) released from RBC lysis is a potent cytotoxic chemical that generates free radicals and oxidative damage, causing death of surrounding cells [5, 10, 12, 13]. Hemin, the oxidative form of heme, plays a critical role in Hb-induced brain injury following ICH [5]. Hemin exerts its neurotoxic effects via release of excessive iron, depletion of glutathione and production of free radicals [14]. In addition, an inflammatory response occurs after ICH, which aggravates ICH-induced brain injury, leading to further tissue damage, blood–brain barrier disruption and edema [4, 15, 16]. The inflammatory mechanisms involved in progression of ICH-induced brain injury include activation of microglial cells, infiltration of inflammatory cells and production of cytokines and chemokines. Neutrophils are believed to contribute to brain injury after ICH [17, 18]. Depletion of neutrophils reduced blood–brain barrier disruption, axon injury and inflammation in a rat model of ICH [18] and was found to prevent tissue plasminogen activator (tPA)-induced ICH in a rat model of cerebral ischemia [17]. Neutrophils may damage brain tissues by producing reactive oxygen species (ROS) and releasing proinflammatory cytokines and matrix metalloproteinases (MMPs) [19, 20]. Several lines of evidence show that activation of innate immunity and inflammatory responses contributes to the pathogenesis of secondary injury after ICH [4, 5, 10, 16].
Toll-like receptors (TLRs) belong to a large family of pattern recognition receptors that play a key role in innate immunity and inflammatory responses [21, 22]. Thirteen mammalian TLRs have been identified in mice, eleven of which are also found in humans. They recognize distinct pathogen-associated molecular patterns from diverse organisms, including viruses, bacteria, mycobacteria, fungi and parasites [23, 24]. In addition, TLRs also recognize damage-associated molecular patterns and mediate host inflammatory responses to injury [22]. TLR1, TLR2, TLR4, TLR5, TLR6 and TLR10 are distributed on the cell surface, and other TLRs (TLR3, TLR7, TLR8, and TLR9) are expressed in the intracellular endosomes [22, 25]. TLR consists of leucine-rich repeats in the extracellular ectodomains that bind various ligands, as well as intracellular Toll-interleukin 1 receptor (TIR) domains that recruit intracellular adaptor proteins, including myeloid differentiation factor 88 (MyD88), TIR domain-containing adaptor-inducing interferons (TRIFs), TIR domain-containing adaptor protein (TIRAP) or TRIF-related adaptor molecule (TRAM). TLR signaling pathways include at least a MyD88-dependent pathway common to all TLRs except TLR3, as well as a MyD88-independent pathway selective to TLR3 and TLR4 [22, 26, 27]. In the MyD88-dependent pathway, MyD88 activates signal transduction molecules, including interleukin (IL)-1R-associated kinases (IRAKs), tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) and transforming growth factor (TGF)-β-activated kinase (TAK1), ultimately leading to activation of nuclear factorκB (NF-κB) and expression of proinflammatory cytokines. In the MyD88-independent pathway, TRIF activates signal transduction molecules including TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3), ultimately leading to expression of interferonβ (IFN-β) [26]. Our recent study shows that both the MyD88 and TRIF signaling pathways are involved in TLR4-mediated inflammatory responses after ICH [28].
It has been reported that TLR4 is upregulated in a rat model of ICH [29, 30] and that its signaling pathway contributes to poor outcome after ICH [31]. TLR4 is activated by many endogenous ligands, such as heme and fibrinogen [32, 33], which are produced in the brain after ICH. Our recent in vivo study shows that activation of TLR4 by heme causes ICH-induced inflammatory injury via the MyD88/TRIF signaling pathway and that effective blockade of TLR4 by its antibody suppresses ICH-induced inflammation [28]. Thus, the TLR4 signaling pathway could be a promising therapeutic target for ICH treatment.
TLR4 is expressed in microglia, the resident macrophages of the brain. Microglia are activated within minutes after ICH [34, 35] and subsequently release chemotactic factors to recruit hematogenous phagocytes to the hemorrhagic areas. Timely clearance of the extravasated RBCs by activated microglia/macrophages can provide protection from local damage resulting from RBC lysis. Successful removal of injured cells can reduce secondary damage by preventing discharge of injurious proinflammatory cell contents. Resolution of hematoma and inhibition of inflammation are considered potential targets for ICH treatment [5, 10, 36, 37]. In this review, we highlight the roles of TLR signaling pathways in ICH and discuss their potential as therapeutic targets.
Innate immunity and inflammation in the pathogenesis of ICH
Microglial cells are activated within minutes after the onset of ICH [34, 35]. Activated microglial cells undergo morphological and functional changes that include enlargement and thickening of processes, upregulation of proinflammatory proteins, and behavioral changes, including proliferation, migration and phagocytosis [10, 20]. The primary neuroprotective role of activated microglia is to clear the hematoma and damaged cell debris through phagocytosis, providing a nurturing environment for tissue recovery. However, accumulating evidence has shown that microglial activation contributes to ICH-induced secondary brain injury by releasing a variety of cytokines, chemokines, free radicals, nitric oxide and other potentially toxic chemicals [16, 34, 38, 39]. In addition, several studies have shown that inhibition of microglial activation reduces brain damages in animal models of ICH [39–41]. Microglial inhibitors, such as minocycline and microglia/macrophage inhibitory factors (tuftsin fragment 1–3), reduce ICH-induced brain injury and improve neurological function in rodents [40–45]. Clearly, microglial activation mediates ICH-mediated brain injury.
Besides microglia, other blood-derived inflammatory cells, such as leukocytes and macrophages, are also activated after ICH and contribute to ICH-induced brain injury [16]. Neutrophil infiltration occurs less than 1 day after the onset of ICH, and the infiltrating neutrophils die by apoptosis within 2 days [35, 46]. Dying leukocytes can cause further brain injury by stimulating microglia/macrophages to release proinflammatory factors [16]. Activated macrophages are indistinguishable from resident microglia in morphology and function [20]. Similar to activated microglia, activated leukocytes and macrophages release a variety of cytokines, chemokines, free radicals and other potentially toxic chemicals [16, 20, 34].
Cytokines are well-known to be associated with inflammation and immune activation [47]. Although cytokines are released by many cells, including microglia/macrophages, astrocytes and neurons, the major sources of cytokines are activated microglia/macrophages [48]. Many studies have shown that two major proinflammatory cytokines, TNF-α and interleukin1β (IL-1β), exacerbate ICH-induced brain injury. After ICH, TNF-α is significantly increased both in vivo and in vitro[16, 28, 34, 49], which may contribute to brain edema formation and brain injury in animal models of ICH [49, 50]. Consistent with animal studies, clinical studies support the proposition that TNF-α contributes to ICH-induced brain injury. Plasma TNF-α has been shown to correlate with the magnitude of the perihematomal brain edema in patients with ICH [51]. Single-nucleotide polymorphisms in the TNF-α gene promoter are associated with spontaneous deep ICH [52]. Similarly, IL-1β has been found to be upregulated after ICH in an animal model and to produce detrimental effects, including brain edema and blood–brain barrier disruption [28, 53, 54].
NF-κB, a transcription factor involved in inflammatory responses, is also activated after ICH, leading to upregulation of gene expression that contributes to brain injury [34, 55]. Activation of NF-κB occurs within minutes and lasts for at least 1 week after the onset of ICH [56]. NF-κB is a key regulator of many proinflammatory cytokines, such as TNF-α and IL-1β, in various pathological conditions, including ICH [16, 28, 55, 57]. The activity of NF-κB correlates with perilesional cell death after ICH in rats [58]. Activation of NF-κB is positively associated with the progression of apoptotic cell death in patients with ICH [59]. Therefore, understanding the signaling mechanisms underlying ICH-induced NF-κB activation may facilitate identification of therapeutic targets.
Several lines of evidence have shown that NF-κB is activated by RBCs and plasma via signaling pathways involving free radicals, cytokines and glutamate receptors [55]. It is well-known that TLR signaling pathways can lead to NF-κB activation, resulting in production of proinflammatory cytokines [27, 60, 61]. Increasing evidence has shown that TLR signaling pathways play an essential role in sterile inflammatory diseases in the central nervous system [62–64]. Herein we review recent advances in TLR4 signaling pathways in ICH-induced inflammatory brain injury.
TLR4 signaling in ICH-induced inflammatory brain injury
TLRs, especially TLR4, are involved in the inflammatory responses and neuronal damage associated with cerebral ischemia [22, 65]. Expression of TLR4 is upregulated in a mouse model of transient cerebral ischemia [66]. In ischemic brain injury, TLR4-deficient mice show significant suppression of inflammatory cytokine expression, including IRF1, inducible nitric oxide synthase, cyclooxygenase2, MMP9, and IFN-β [67]. TLR4-knockout mice have significantly smaller infarct volumes and better neurological function than wild-type (WT) mice [66, 67]. Similar to cerebral ischemia, TLR4 expression is significantly increased after ICH, and TLR4-knockout mice demonstrate improved neurological function after ICH [28–31]. These animal studies agree with recent clinical findings that increased expression of TLR4 is associated with poorer functional outcome and greater residual volume in patients with ICH [68]. However, although TLR4 participates in both cerebral ischemia- and ICH-induced brain injuries, the signaling pathways involved are different. Both the MyD88 and TRIF pathways are involved in TLR4-mediated brain injury in ICH [28], whereas only MyD88 is involved in ischemia [69]. Therefore, the roles of TLR4 in ICH and cerebral ischemia may be different.
The roles of TLR4 in inflammation and neurological impairment following ICH have been studied recently, using TLR4-knockout mice, by our research group and others [28, 31]. Compared to WT mice, after ICH, TLR4-knockout mice exhibited significantly decreased brain water content [28] and neurological deficits [28, 31]. Furthermore, the perihematomal region in TLR4-knockout mice had lower levels of infiltrating inflammatory cells, including macrophages [28], leukocytes and monocytes [31], as well as lower levels of inflammatory cytokines, such as TNF-α, IL-1β and IL-6, and decreased NF-κB activity [28]. In addition, Sansing et al. [31] reported increased gene expression of CD36, CSF2 and CX3CL1 in TLR4-knockout mice compared to WT mice. Taken together, these studies demonstrate that TLR4 activation is involved in ICH-induced neurological deficits and contributes to the detrimental inflammatory response.
TLR4 is expressed in various cell types in the central nervous system, including microglia, neurons and astrocytes, as well as in peripheral blood cells, such as leukocytes, macrophages and platelets [22, 28, 30, 33, 70, 71]. After ICH, TLR4 mRNA and protein expression is significantly increased by approximately 2 to 6 hours, peaks at day 3, declines somewhat at day 5, but remains elevated relative to baseline even at day 7 [28–30]. Though ICH induces upregulation of TLR4 expression in neurons, astrocytes and microglia, TLR4 is predominantly expressed in CD11b-positive microglial cells in mice [28]. In addition, TLR4 contributes to reduced recruitment of neutrophils and monocytes in the perihematomal area after ICH in TLR4-deficient mice [31]. Therefore, resident microglia in the brain as well as peripheral infiltrating leukocytes and monocytes are likely involved in the roles of TLR4 in ICH-induced brain injury.
Microglial activation in response to ICH contributes to ICH-induced brain injury by releasing cytokines, and inhibition of microglial activation has been shown to improve neurological function in animal models of ICH [38–41]. Recently, we investigated the role of TLR4 in microglial activation following ICH [28]. Exogenous hemin treatment of cultured microglia increases expression of TLR4, as well as proinflammatory cytokines, such as TNF-α, IL-1β and IL-6. This effect is completely abolished by knockout of TLR4 or treatment with anti-TLR4 antibodies, suggesting that TLR4 mediates hemin-stimulated microglial activation [28]. In addition, hemin-induced expression of proinflammatory cytokines TNF-α, IL-1β and IL-6 is completely blocked in TLR4-knockout mice [28]. These data demonstrate that TLR4 signaling mediates heme-induced inflammatory injury, possibly by activating microglial cells and subsequently releasing proinflammatory cytokines. However, Sansing et al. [31] reported that upregulation of IL-1β and IL-6 genes in the perihematomal region did not significantly differ between TLR4-knockout and WT mice, suggesting that TLR4 signaling may not be involved in the transcriptional regulation of proinflammatory cytokines in perihematomal inflammation. Alternately, it may not be possible to detect upregulation of proinflammatory cytokine genes in certain cell types, such as microglia, when the entire perihematomal region is used [31].
Using blood transfer experiments in which TLR4-deficient blood was injected into the brains of WT mice and WT blood into brains of TLR4-deficient mice, Sansing et al. [31] found that TLR4 signaling within the hemorrhage mediated the inflammatory response and contributed to ICH-induced neurological injury. Activation of TLR4 on leukocytes or platelets within the hemorrhage, but not on resident cells, promoted inflammation and resulted in poor functional outcomes. This study clearly demonstrated that TLR4 signaling on peripheral blood cells plays a critical role in ICH-induced brain injury.
Therefore, TLR4 signaling on resident microglia and on blood cells within the hemorrhage is critical for ICH-induced inflammatory injury. Because TLR4 is expressed on resident microglia which are activated within minutes after ICH [34, 35], the TLR4 signaling on microglia probably initiates ICH-induced inflammatory injury, causing release of inflammatory cytokines and infiltration of neutrophils. Significant upregulation of TLR4 expression occurs at approximately 2 to 6 hours after ICH [28–30], accompanied by the appearance of infiltrating neutrophils in the hematoma (at approximately 4 hours after ICH) [46]. TLR4 signaling in the neutrophils may further mediate release of proinflammatory cytokines, which contributes to the detrimental inflammatory response [17, 18]. There are abundant TLR4 activators in the perihematomal region, including heme [32], fibrinogen [33] and myeloid-related proteins 8 and 14, which are released from degranulating neutrophils [72]. These endogenous activators can stimulate TLR4 on leukocytes, leading to inflammatory injury.
TLR4 signaling pathways in ICH
TLRs are a group of Class I transmembrane proteins that consist of ectodomains, transmembrane domains, and intracellular TIR domains. TLRs recognize and bind various ligands via leucin-rich repeats in the extracellular ectodomains. The receptor-ligand binding results in conformational changes of the receptor, subsequently leading to recruitment of intracellular adaptor proteins, including MyD88, TRIF, TIRAP, or TRAM [22]. Once recruited, these adaptors initiate downstream signaling events, which ultimately lead to activation of transcription factors, such as NF-κB, and subsequent expression of various proinflammatory cytokines, such as IL-6, TNF-α and IL-1 [22, 27].
TLR4 interacts with two distinct adaptor proteins (MyD88 and TRIF) and therefore activates two parallel signaling pathways to initiate the activation of transcription factors that regulate expression of proinflammatory cytokine genes [22, 26, 73]. The MyD88-dependent pathway, common to all TLRs, is essential for activation of NF-κB and production of inflammatory cytokines [26, 74]. TRIF is essential for the TLR3- and TLR4-mediated MyD88-independent pathway, which involves the activation of IRF-3, leading to expression of IFN-β [26, 27]. Recently, we found that both MyD88 and TRIF signaling pathways are involved in TLR4-mediated inflammatory responses after ICH [28]. Deletion of MyD88 or TRIF in transgenic mice leads to improved neurological function and reduced cytokine release and macrophage infiltration following ICH. After ICH in TLR4-knockout mice, MyD88 and TRIF expression are reduced, further demonstrating the role of these factors in TLR4-mediated ICH sequelae. Furthermore, the ICH-induced increase in NF-κB activity is significantly lower in TLR4-knockout mice than that in WT mice, suggesting that TLR4 mediates ICH-induced inflammation via activation of NF-κB to regulate expression of inflammatory cytokines.
The MyD88 pathway of TLR4 signaling activates not only the NF-κB pathway but also the activating protein1 (AP-1) pathway. AP-1 is a dimeric protein composed of members of the Jun, Fos, and α-fetoprotein families of proteins [75]. AP-1 activation leads to the expression of many proinflammatory mediators, such as MMPs, proteases, and cytokines such as IL-1 and IFN [76, 77]. It is well-documented that these proinflammatory mediators are increased after ICH [16, 20]. For example, MMPs, including MMP2, MMP3, MMP9, and MMP12, have been reported to be upregulated after ICH as a result of their activation by proteases such as plasmin and tPA [20, 78, 79]. However, the role of AP-1 in ICH has not been explored yet. AP-1 activation in TLR4 signaling is mainly mediated by mitogen-activated protein kinases (MAPKs), including c-Jun N-terminal kinase (JNK), p38 and ERK [76]. JNK, a stress-activated kinase that mediates apoptosis in neurons and microglia, is activated after ICH, and inhibition of the JNK pathway results in a significant decrease in edema and hematoma volume in mice after ICH [80]. Therefore, proapoptotic signals may play a role in ICH-induced inflammatory injury. However, it remains unknown whether ILR4 can mediate ICH-induced inflammatory injury via AP-1 activation.
Interestingly, lively and Schlichter showed that the expression of inflammatory mediators after ICH is age-dependent [81]. They examined 27 genes, including TLR4, and found that 18 of the 27 genes were different in expression levels or timing between young adult and aged rats [81]. The delayed expression of TLR4 was observed in aged rats, accompanied by a delayed and/or decreased expression of many proinflammatory cytokines such as IL-1β and IL-6 [81]. The TLR4 signaling pathways involved in age-related expression of proinflammatory cytokines is not well-understood. It has been reported that lipopolysaccharide (LPS)-induced production of TLR4-mediated proinflammatory cytokines is age-dependently decreased, accompanied by a decrease in the expression of MAPKs, but not the surface expression of TLR4 [82]. Decreased TLR4/MPAK signaling pathway may be responsible for age-related decrease in the production of inflammatory cytokines.
Because the brain is a sterile organ without any pathogens derived from bacteria or viruses, the endogenous molecules released after ICH become TLR stimulators. Several endogenous molecules have been reported as TLR4 ligands, including heme, fibrinogen, high-mobility group protein B1 (HMGB1), heat shock proteins, hyaluronan, oxidized low-density lipoprotein and amyloid β [32, 33, 69, 83–87]. However, the molecular mechanisms underlying activation of TLR4 signaling pathways by these different ligands are not completely understood. Clearly, different TLR4 ligands can activate distinct signaling pathways. For example, in vivo animal studies show that heme triggers a TLR4 signaling pathway involving both MyD88 and TRIF [28], whereas HMBG1 initiates only the MyD88 pathway [69], and monophosphoryl lipid A, a low-toxicity derivative of LPS, activates only the TRIF pathway [88]. It remains unclear how different TLR4 ligands selectively activate distinct signaling pathways. However, different TLR4 receptor conformations induced by binding of different TLR4 ligands may contribute to the pathway-specific activation [89]. The ligand-biased signaling is well-known for G protein-coupled receptors, such as β-adrenergic receptors [25, 90]. Understanding the mechanisms of biased signaling can provide leads for designing more specific drugs.
Many endogenous TLR4 ligands are known to be released during ICH. Some TLR4 ligands are crucial for activating TLR4 to trigger ICH-induced inflammation and inflammatory cytokine expression [28, 31, 91, 92]. Heme, released from RBC lysis after ICH, is essential for TLR4-mediated inflammation because it potentiates microglial activation and increases cytokine expression [28]. Fibrinogen within the clots after ICH is also critical for activation of TLR4 on platelets or leukocytes within the hemorrhage, which contributes to poor outcome after ICH [31]. HMGB1, known to be essential for ischemic brain injury [65], is reported to be upregulated in the microglia after subarachnoid hemorrhage [91], and the HMGB1 inhibitor glycyrrhizin attenuates ICH-induced brain injury [92]. Additional studies of the roles of endogenous TLR4 ligands in ICH are warranted.
A deeper understanding of TLR4 signaling pathways should enable development of potential therapeutic targets for prevention and treatment of ICH. There are two ways to block TLR signaling: direct blockade by removal of TLR ligands (e.g., heme) and inhibition of TLRs and their downstream signaling pathways. Effective removal of deposited blood and disintegrated cells by promoting hematoma clearance has been demonstrated to reduce ICH-induced neurological deficits in a mouse model of ICH [36, 37]. Hematoma resolution could promote clearance of hemin, thus reducing hemin-mediated activation of TLR4 and subsequent inflammatory responses. In addition, effective blockade of TLR4 receptor using an antibody disrupted TLR4 signaling and has neuroprotective effects in a mouse model of ICH [28]. Therefore, effective blockade of TLR4 signaling pathway could be a potential therapeutic strategy for prevention and treatment of ICH.
Blockade of TLR4 signaling as a potential target in the treatment of ICH
As TLR4 signaling plays an important role in ICH-induced inflammatory injury, TLR4 inhibition should be beneficial. TLR antagonists have been developed for a number of inflammatory and autoimmune diseases [93–95] and include anti-TLR antibodies, small-molecule antagonists screened from compound libraries, and antagonists derived from medicinal plants. However, the efficacy of TLR antagonists in ICH has not been well-studied to date.
We have found that a specific antibody (Mts50) blocked TLR4 signaling in a mouse model of ICH [28]. TLR4 antibody treatment significantly reduced cerebral water content and improved neurological function after ICH, similar to effects observed in TLR4-knockout mice, suggesting that blockade of the TLR4 receptor is a potential therapeutic approach in the treatment of ICH. The neuroprotective effects of the TLR4 antibody in ICH may be associated with inhibition of cytokine expression and macrophage infiltration. However, the effectiveness of the antibody on hemin-induced TLR4 activation in macrophages is controversial. We found that the TLR4 antibody effectively suppressed hemin-induced microglial activation in mice [28]. However, Figueiredo et al. reported that this antibody blocked only LPS-induced, but not hemin-induced, TLR4 activation in macrophages, suggesting that different TLR4 receptor conformations exist in response to different ligands [32]. The discrepancy between the two studies may be due to use of different reagents and animal models, and further studies are needed to evaluate the efficacy of the TLR4 antibody for ICH.
Many TLR4 antagonists have been reported to produce anti-inflammatory effects [93–95]. Some antagonists, such as curumin, 6-shogaol, isoliquiritigenin, and OSL07 (4-oxo-4-(2-oxo-oxazolidin-3-yl)-but-2-enoic acid ethyl ester), block TLR4 signaling by inhibiting homodimerization of TLR4 [96–99]. Other agents, such as sparstolonin B, auranofin, TAK-242, and M62812, have also been reported to block TLR4 signaling pathways selectively [100–103]. However, most of these agents were tested in an animal model of LPS-induced sepsis. The effects of these agents on ICH-induced inflammation have not yet been explored.
In addition to these antagonists, many agents with multiple pharmacological mechanisms have been found to have neuroprotective effects in animal models of ICH via inhibition of TLR4 signaling. For example, oxymatrine, which has anti-inflammatory, antioxidative, and antiapoptotic activities, suppresses TLR4 and NF-κB gene expression and decreases production of proinflammatory cytokines, such as IL-6, TNF-α, and IL-1β [104]. Ginkgolide B, a specific platelet-activating factor receptor antagonist, reduces neuronal cell apoptosis after traumatic brain injury in rats, possibly via inhibition of TLR4 signaling pathways [105]. Progesterone treatment inhibits TLR4 signaling pathways and reduces brain edema and blood–brain barrier impairment after subarachnoid hemorrhage in rats [106]. These agents are not specific TLR4 receptor blockers, but they can inhibit TLR4 signaling to reduce ICH-induced inflammatory injury.
Paradoxically, TLR4 activation with low doses of LPS prior to ischemic brain injury protects against subsequent severe ischemic injury [107–109]. The mechanisms of preconditioning by TLR4 activation in ischemic brain injury are not fully understood. Recent studies have shown that LPS preconditioning redirects TLR4 singling through the TRIF-IRF3 pathway, but not through the MyD88 pathway [109, 110]. Enhanced IRF3 activity and increased anti-inflammatory IFN gene expression contribute to the beneficial effects of LPS preconditioning [109]. Though the suppression of NF-κB activity is suppressed in LPS preconditioning mice following ischemic injury, proinflammatory cytokine production does not change, suggesting that besides the TLR4 signaling pathway, other signaling cascades and transcription factors are involved in proinflammatory cytokine production during ischemic injury [109]. However, there have been relatively few studies examining the effects of preconditioning by TLR4 activation on ICH. It remains to be determined whether the TRIF/IRF3 pathway and enhanced anti-inflammatory IFN production are preferentially involved in preconditioning by TLR4 activation in ICH. It has been reported that progesterone inhibits TLR4/NF-κB signaling pathway and decrease proinflammatory cytokine production in rats following subarachnoid hemorrhage [106], suggesting that suppressed proinflammatory signaling may contribute to preconditioning by TLR4 activation in ICH. Therefore, suppressed proinflammatory signaling and/or enhanced anti-inflammatory IFN signaling may be associated with preconditioning by TLR4 activation in ICH. Additional studies of the mechanisms of preconditioning by TLR4 activation in ICH are warranted.
Heme removal and hematoma resolution as potential targets in the treatment of ICH
RBC lysis occurs at approximately 24 hours after the onset of ICH and continues for the next several days, leading to release of cytotoxic hemoglobin [111]. Hemoglobin then degrades to hemin, the oxidative form of heme [14]. Hemin is gradually cleared by hematogenous phagocytes and resident microglia [10]. Once inside these cells, hemin is degraded by heme oxygenase (HO) into biliverdin and carbon monoxide, releasing cytotoxic iron. The toxic effects of hemoglobin/hemin include release of redox-active iron, depletion of glutathione, and production of free radicals [14]. To avoid the toxicity of hemoglobin/hemin, these substances are cleared from the extracellular space via binding to haptoglobin and hemopexin or via phagocytosis by microglia/macrophages.
Haptoglobin, an abundant protein in blood plasma, has the ability to bind hemoglobin. In the brain, it is produced and released by oligodendrocytes, thereby protecting the brain against extravascular hemoglobin toxicity [112]. Expression of haptoglobin is increased around the hematoma in animal models of ICH. Haptoglobin-deficient mice are more vulnerable to ICH-induced brain injury, and mice with haptoglobin overexpression are less susceptible to injury [112]. Sulforaphane, an NF-E2-related factor2 (Nrf2) activator, increases haptoglobin in the brain and reduces brain injury following ICH [112]. In addition, sulforaphane treatment increases expression of Nrf2-mediated antioxidant genes, such as catalase, superoxide dismutase, and glutathioneS-transferase, in the brain after ICH [113]. The antioxidative effects of sulforphane are correlated with reduction of brain damage, measured by brain edema, blood–brain barrier impairment, cortical apoptosis, and motor deficits [114].
Peroxisome proliferator-activated receptorγ (PPARγ) plays an important role in augmenting phagocytosis and promoting hematoma absorption [36, 37]. PPARγ is a ligand-dependent transcription factor that regulates the expression of several target genes, such as scavenger receptor CD36 [36, 37]. CD36, a class B scavenger receptor, is important for phagocytic activity [115, 116]. Treatment with PPARγ agonists, such as rosiglitazone and 15d-PGJ2, increases expression of CD36 and promotes phagocytosis of RBCs by microglia/phagocytes in both in vitro and in vivo models of ICH [36, 37], and anti-CD36 antibodies prevent PPARγ agonist-induced increases in phagocytosis [37]. In addition, in an animal model, treatment of ICH with PPARγ agonists accelerated hematoma resolution and reduced neurological deficits [36, 37].
ROS are produced after ICH and contribute to ICH pathogenesis [4, 5, 13, 34]. In addition, phagocytosis generates a large amount of ROS that can damage macrophages and neurons. PPARγ also plays an important role in protecting microglia/macrophages from oxidative damage via upregulation of the antioxidant catalase [36, 37]. PPARγ agonists upregulate catalase expression in microglia in vitro and in vivo after ICH, enhance phagocytosis in vitro and increase hematoma absorption in vivo[37]. The PPARγ-mediated upregulation of catalase reduces oxidative stress, as demonstrated by a significant reduction of extracellular hydrogen peroxide in cultured microglia [37]. Phagocytosis of RBCs by microglia is also enhanced by upregulation of catalane, as demonstrated by the finding that addition of exogenous catalane to the culture media promotes phagocytosis [37].
PPARγ can also induce neuroprotection after ICH via anti-inflammatory effects. In both in vitro and in vivo experiments, PPARγ activators reduced expression of proinflammatory genes, including TNF-α, IL-1β,MMP9, and inducible nitric oxide synthase [37]. PPARγ is known to inhibit DNA binding of NF-κB [34, 55], which controls expression of proinflammatory cytokines and enzymes, suggesting that the anti-inflammatory effect of PPARγ probably results from inhibition of NF-κB [37, 117].
TLR4 signaling in hematoma resolution
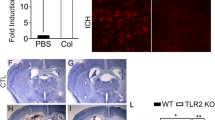
Microglia/macrophages express the scavenger receptor CD36, which has been reported to assist in phagocytosis-mediated removal of RBCs after ICH [37]. PPARγ agonists can promote phagocytosis of RBCs by microglia/phagocytes through upregulation of CD36 [36, 37]. In addition, PPARγ agonists suppress the subarachnoid hemorrhage-induced inflammatory response by inhibiting TLR4 signaling [118]. Knockout of TLR4 results in upregulation of CD36 in the perihematomal region [31], suggesting that the TLR4 signaling pathway could play a role in hematoma resolution. In agreement with this hypothesis, we found that hematoma resolved significantly faster in TLR4-knockout mice than in WT mice, as shown in Figure 1. Specifically, hematoma started to resolve at 3 days after ICH and almost completely resolved by 5 days. These data demonstrate that the TLR4 signaling pathway is involved in hematoma resolution after ICH, and the underlying mechanisms are currently under investigation.

TLR4 knockout results in faster hematoma resolution. (A) Representative brain sections from WT and TLR4-knockout mice at 1, 3, and 5 days after intrastriatal injection of blood. (B) Compared with WT mice, TLR4-knockout mice had significantly smaller hemotomas at 3 and 5 days after ICH. Hematoma volume was measured on coronal slices (2 mm thick) using image analysis software. Scale bar: 1 mm. **P < 0.05 versus WT mice.
Conclusions and perspectives
Increasing evidence has shown that TLR4 signaling plays important roles in ICH-induced inflammatory brain injury by stimulating activation of microglial cells, infiltration of leukocytes, and production of cytokines and chemokines [28, 31]. The TLR4 signaling pathway involved in ICH-induced inflammatory injury includes ligands (e.g., heme), TLR4 itself, and its downstream pathways, including adaptor proteins (MyD88 and TRIF) and transcription factors, such as NF-κB [28]. Therefore, TLR4 and its signaling pathways are potential targets for developing effective medical treatment of ICH.
There are many challenges to be overcome before inhibition of TLR4 signaling can be used in the prevention and treatment of ICH. Though inhibition of TLR4 signaling by anti-TLR4 antibodies or deletion of TLR4 genes can effectively reduce ICH-induced neurological deficits in mice [28], specific TLR4 antagonists that inhibit TLR4 signaling have not been investigated in models of ICH. In addition, a diverse range of endogenous ligands may activate TLR4 signaling to trigger the inflammatory response that is critical in ICH-induced brain injury. However, it remains unclear how these ligands activate TLR4 signaling, which ligands are the most critical, how TLR4 receptors recruit adaptor proteins and activate transcription factors, and whether TLR4 signaling pathways are similar across cell types (e.g., microglia versus leukocytes). Whether TLR4 interacts with other TLRs in ICH-induced inflammatory injury requires further investigation. Better understanding of the roles of TLR4 signaling in ICH will facilitate development of ICH treatments.
Brain injury following ICH is triggered by the presence of intraparenchymal blood. The mechanisms of ICH-induced brain injury are numerous, including cytotoxic, oxidative, and inflammatory pathways. It would be beneficial to develop a medical treatment that promotes hematoma resolution and inhibits cytotoxic, oxidative, and inflammatory insults following ICH. For example, PPARγ activators reduce ICH-induced brain injury by improving hematoma resolution and reducing oxidative injury [36, 37]. Another promising intervention is inhibition of TLR4 signaling, which also promotes hematoma resolution (see Figure 1) and inhibits ICH-induced inflammation [28]. However, the molecular mechanisms underlying hematoma resolution and cytotoxic, oxidative, and inflammatory injury following ICH remain elusive. Further research into the complex mechanisms involved in ICH pathogenesis will facilitate identification of novel therapeutic targets.
Abbreviations
- Hb:
-
Hemoglobin
- HMGB1:
-
High-mobility group box 1 protein
- HO:
-
Heme oxygenase
- ICH:
-
Intracerebral hemorrhage
- IFN-β:
-
Interferon β
- IL-1β:
-
Interleukin-1β
- LPS:
-
Lipopolysaccharide
- MyD88:
-
Myeloid differentiation factor 88
- NF-κB:
-
Nuclear factorκB
- Nrf2:
-
NF-E2-related factor2
- PPARγ:
-
Peroxisome proliferator-activated receptorγ
- RBC:
-
Red blood cell
- TIR:
-
Intracellular Tollinterleukin 1 receptor
- TIRAP:
-
TIR domain-containing adaptor protein
- TLR:
-
Toll-like receptor
- TNF-α:
-
Tumor necrosis factorα
- TRAM:
-
TRIF-related adaptor molecule
- TRIF:
-
TIR domain-containing adaptor-inducing interferon
- WT:
-
Wild type.
References
Hwang BY, Appelboom G, Ayer A, Kellner CP, Kotchetkov IS, Gigante PR, Haque R, Kellner M, Connolly ES: Advances in neuroprotective strategies: potential therapies for intracerebral hemorrhage. Cerebrovasc Dis 2011,31(3):211–222.
Flower O, Smith M: The acute management of intracerebral hemorrhage. Curr Opin Crit Care 2011,17(2):106–114.
Mayer SA, Rincon F: Treatment of intracerebral haemorrhage. Lancet Neurol 2005,4(10):662–672.
Xi G, Keep RF, Hoff JT: Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol 2006,5(1):53–63.
Babu R, Bagley JH, Di C, Friedman AH, Adamson C: Thrombin and hemin as central factors in the mechanisms of intracerebral hemorrhage-induced secondary brain injury and as potential targets for intervention. Neurosurg Focus 2012,32(4):E8.
Elliott J, Smith M: The acute management of intracerebral hemorrhage: a clinical review. Anesth Analg 2010,110(5):1419–1427.
Felberg RA, Grotta JC, Shirzadi AL, Strong R, Narayana P, Hill-Felberg SJ, Aronowski J: Cell death in experimental intracerebral hemorrhage: the “black hole” model of hemorrhagic damage. Ann Neurol 2002,51(4):517–524.
Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT: Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg 2002,96(2):287–293.
Lee KR, Kawai N, Kim S, Sagher O, Hoff JT: Mechanisms of edema formation after intracerebral hemorrhage: effects of thrombin on cerebral blood flow, blood–brain barrier permeability, and cell survival in a rat model. J Neurosurg 1997,86(2):272–278.
Aronowski J, Zhao X: Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke 2011,42(6):1781–1786.
Sharp F, Liu DZ, Zhan X, Ander BP: Intracerebral hemorrhage injury mechanisms: glutamate neurotoxicity, thrombin, and Src. Acta Neurochir Suppl 2008, 105:43–46.
Wagner KR, Packard BA, Hall CL, Smulian AG, Linke MJ, De Courten-Myers GM, Packard LM, Hall NC: Protein oxidation and heme oxygenase-1 induction in porcine white matter following intracerebral infusions of whole blood or plasma. Dev Neurosci 2002,24(2–3):154–160.
Nakamura T, Keep RF, Hua Y, Hoff JT, Xi G: Oxidative DNA injury after experimental intracerebral hemorrhage. Brain Res 2005,1039(1–2):30–36.
Robinson SR, Dang TN, Dringen R, Bishop GM: Hemin toxicity: a preventable source of brain damage following hemorrhagic stroke. Redox Rep 2009,14(6):228–235.
Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG: Microglia potentiate damage to blood–brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke 2006,37(4):1087–1093.
Wang J, Dore S: Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab 2007,27(5):894–908.
Gautier S, Ouk T, Petrault O, Caron J, Bordet R: Neutrophils contribute to intracerebral haemorrhages after treatment with recombinant tissue plasminogen activator following cerebral ischaemia. Br J Pharmacol 2009,156(4):673–679.
Moxon-Emre I, Schlichter LC: Neutrophil depletion reduces blood–brain barrier breakdown, axon injury, and inflammation after intracerebral hemorrhage. J Neuropathol Exp Neurol 2011,70(3):218–235.
Nguyen HX, O’Barr TJ, Anderson AJ: Polymorphonuclear leukocytes promote neurotoxicity through release of matrix metalloproteinases, reactive oxygen species, and TNF-α. J Neurochem 2007,102(3):900–912.
Wang J: Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol 2010,92(4):463–477.
Akira S, Uematsu S, Takeuchi O: Pathogen recognition and innate immunity. Cell 2006,124(4):783–801.
Kong Y, Le Y: Toll-like receptors in inflammation of the central nervous system. Int Immunopharmacol 2011,11(10):1407–1414.
Beutler BA: TLRs and innate immunity. Blood 2009,113(7):1399–1407.
Zhu J, Mohan C: Toll-like receptor signaling pathways: therapeutic opportunities. Mediators Inflamm 2010, 2010:781235.
Akira S, Takeda K: Toll-like receptor signalling. Nat Rev Immunol 2004,4(7):499–511.
Takeda K, Akira S: TLR signaling pathways. Semin Immunol 2004,16(1):3–9.
Lin Q, Li M, Fang D, Fang J, Su SB: The essential roles of Toll-like receptor signaling pathways in sterile inflammatory diseases. Int Immunopharmacol 2011,11(10):1422–1432.
Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, Wang JZ, Su BY, Yang QW: Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflammation 2012, 9:46.
Teng W, Wang L, Xue W, Guan C: Activation of TLR4-mediated NF-κB signaling in hemorrhagic brain in rats. Mediators Inflamm 2009, 2009:473276.
Ma CX, Yin WN, Cai BW, Wu J, Wang JY, He M, Sun H, Ding JL, You C: Toll-like receptor 4/nuclear factor-κ B signaling detected in brain after early subarachnoid hemorrhage. Chin Med J (Engl) 2009,122(13):1575–1581.
Sansing LH, Harris TH, Welsh FA, Kasner SE, Hunter CA, Kariko K: Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann Neurol 2011,70(4):646–656.
Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graça-Souza AV, Bozza MT: Characterization of heme as activator of Toll-like receptor 4. J Biol Chem 2007,282(28):20221–20229.
Smiley ST, King JA, Hancock WW: Fibrinogen stimulates macrophage chemokine secretion through Toll-like receptor 4. J Immunol 2001,167(5):2887–2894.
Aronowski J, Hall CE: New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res 2005,27(3):268–279.
Xue M, Del Bigio MR: Intracerebral injection of autologous whole blood in rats: time course of inflammation and cell death. Neurosci Lett 2000,283(3):230–232.
Zhao X, Grotta J, Gonzales N, Aronowski J: Hematoma resolution as a therapeutic target: the role of microglia/macrophages. Stroke 2009,40(3 Suppl):S92-S94.
Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J: Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor γ in microglia/macrophages. Ann Neurol 2007,61(4):352–362.
Wang J, Tsirka SE: Contribution of extracellular proteolysis and microglia to intracerebral hemorrhage. Neurocrit Care 2005,3(1):77–85.
Gao Z, Wang J, Thiex R, Rogove AD, Heppner FL, Tsirka SE: Microglial activation and intracerebral hemorrhage. Acta Neurochir Suppl 2008, 105:51–53.
Wang J, Rogove AD, Tsirka AE, Tsirka SE: Protective role of tuftsin fragment 1–3 in an animal model of intracerebral hemorrhage. Ann Neurol 2003,54(5):655–664.
Wang J, Tsirka SE: Tuftsin fragment 1–3 is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke 2005,36(3):613–618.
Wu J, Yang S, Xi G, Fu G, Keep RF, Hua Y: Minocycline reduces intracerebral hemorrhage-induced brain injury. Neurol Res 2009,31(2):183–188.
Wu J, Yang S, Hua Y, Liu W, Keep RF, Xi G: Minocycline attenuates brain edema, brain atrophy and neurological deficits after intracerebral hemorrhage. Acta Neurochir Suppl 2010, 106:147–150.
Zhao F, Hua Y, He Y, Keep RF, Xi G: Minocycline-induced attenuation of iron overload and brain injury after experimental intracerebral hemorrhage. Stroke 2011,42(12):3587–3593.
Wasserman JK, Schlichter LC: Minocycline protects the blood–brain barrier and reduces edema following intracerebral hemorrhage in the rat. Exp Neurol 2007,207(2):227–237.
Wang J, Dore S: Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain 2007,130(Pt 6):1643–1652.
Turrin NP, Plata-Salaman CR: Cytokine-cytokine interactions and the brain. Brain Res Bull 2000,51(1):3–9.
Emsley HC, Tyrrell PJ: Inflammation and infection in clinical stroke. J Cereb Blood Flow Metab 2002,22(12):1399–1419.
Hua Y, Wu J, Keep RF, Nakamura T, Hoff JT, Xi G: Tumor necrosis factor-α increases in the brain after intracerebral hemorrhage and thrombin stimulation. Neurosurgery 2006,58(3):542–550. discussion 542–550
Zhang X, Li H, Hu S, Zhang L, Liu C, Zhu C, Liu R, Li C: Brain edema after intracerebral hemorrhage in rats: the role of inflammation. Neurol India 2006,54(4):402–407.
Castillo J, Dávalos A, Alvarez-Sabín J, Pumar JM, Leira R, Silva Y, Montaner J, Kase CS: Molecular signatures of brain injury after intracerebral hemorrhage. Neurology 2002,58(4):624–629.
Chen YC, Hu FJ, Chen P, Wu YR, Wu HC, Chen ST, Lee-Chen GJ, Chen CM: Association of TNF-α gene with spontaneous deep intracerebral hemorrhage in the Taiwan population: a case control study. BMC Neurol 2010, 10:41.
Wu J, Yang S, Xi G, Song S, Fu G, Keep RF, Hua Y: Microglial activation and brain injury after intracerebral hemorrhage. Acta Neurochir Suppl 2008, 105:59–65.
Holmin S, Mathiesen T: Intracerebral administration of interleukin-1β and induction of inflammation, apoptosis, and vasogenic edema. J Neurosurg 2000,92(1):108–120.
Wagner KR: Modeling intracerebral hemorrhage: glutamate, nuclear factor-κB signaling and cytokines. Stroke 2007,38(2 Suppl):753–758.
Zhao X, Zhang Y, Strong R, Zhang J, Grotta JC, Aronowski J: Distinct patterns of intracerebral hemorrhage-induced alterations in NF-κB subunit, iNOS, and COX-2 expression. J Neurochem 2007,101(3):652–663.
Allan SM, Rothwell NJ: Inflammation in central nervous system injury. Philos Trans R Soc Lond B Biol Sci 2003,358(1438):1669–1677.
Hickenbottom SL, Grotta JC, Strong R, Denner LA, Aronowski J: Nuclear factor-κB and cell death after experimental intracerebral hemorrhage in rats. Stroke 1999,30(11):2472–2477. discussion 2477–2478
Wang YX, Yan A, Ma ZH, Wang Z, Zhang B, Ping JL, Zhu JS, Zhou Y, Dai L: Nuclear factor-κB and apoptosis in patients with intracerebral hemorrhage. J Clin Neurosci 2011,18(10):1392–1395.
Barton GM, Medzhitov R: Control of adaptive immune responses by Toll-like receptors. Curr Opin Immunol 2002,14(3):380–383.
Brown J, Wang H, Hajishengallis GN, Martin M: TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res 2011,90(4):417–427.
Shi XQ, Zekki H, Zhang J: The role of TLR2 in nerve injury-induced neuropathic pain is essentially mediated through macrophages in peripheral inflammatory response. Glia 2011,59(2):231–241.
Wu FX, Bian JJ, Miao XR, Huang SD, Xu XW, Gong DJ, Sun YM, Lu ZJ, Yu WF: Intrathecal siRNA against Toll-like receptor 4 reduces nociception in a rat model of neuropathic pain. Int J Med Sci 2010,7(5):251–259.
Hanamsagar R, Hanke ML, Kielian T: Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol 2012,33(7):333–342.
Shichita T, Sakaguchi R, Suzuki M, Yoshimura A: Post-ischemic inflammation in the brain. Front Immunol 2012, 3:132.
Hyakkoku K, Hamanaka J, Tsuruma K, Shimazawa M, Tanaka H, Uematsu S, Akira S, Inagaki N, Nagai H, Hara H: Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 2010,171(1):258–267.
Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I: Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation 2007,115(12):1599–1608.
Rodríguez-Yáñez M, Brea D, Arias S, Blanco M, Pumar JM, Castillo J, Sobrino T: Increased expression of Toll-like receptors 2 and 4 is associated with poor outcome in intracerebral hemorrhage. J Neuroimmunol 2012,247(1–2):75–80.
Yang QW, Lu FL, Zhou Y, Wang L, Zhong Q, Lin S, Xiang J, Li JC, Fang CQ, Wang JZ: HMBG1 mediates ischemia-reperfusion injury by TRIF-adaptor independent Toll-like receptor 4 signaling. J Cereb Blood Flow Metab 2011,31(2):593–605.
Carty M, Bowie AG: Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem Pharmacol 2011,81(7):825–837.
Aravalli RN, Peterson PK, Lokensgard JR: Toll-like receptors in defense and damage of the central nervous system. J Neuroimmune Pharmacol 2007,2(4):297–312.
Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MAD, Nacken W, Foell D, van der Poll T, Sorg C, Roth J: Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 2007,13(9):1042–1049.
Buchanan MM, Hutchinson M, Watkins LR, Yin H: Toll-like receptor 4 in CNS pathologies. J Neurochem 2010,114(1):13–27.
O’Neill LA: How Toll-like receptors signal: what we know and what we don’t know. Curr Opin Immunol 2006,18(1):3–9.
Jochum W, Passegue E, Wagner EF: AP-1 in mouse development and tumorigenesis. Oncogene 2001,20(19):2401–2412.
Kawai T, Akira S: TLR signaling. Cell Death Differ 2006,13(5):816–825.
Vincenti MP, Brinckerhoff CE: Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res Ther 2002,4(3):157–164.
Wasserman JK, Zhu X, Schlichter LC: Evolution of the inflammatory response in the brain following intracerebral hemorrhage and effects of delayed minocycline treatment. Brain Res 2007, 1180:140–154.
Xue M, Fan Y, Liu S, Zygun DA, Demchuk A, Yong VW: Contributions of multiple proteases to neurotoxicity in a mouse model of intracerebral haemorrhage. Brain 2009, 132:26–36.
Michel-Monigadon D, Bonny C, Hirt L: c-Jun N-terminal kinase pathway inhibition in intracerebral hemorrhage. Cerebrovasc Dis 2010,29(6):564–570.
Lively S, Schlichter LC: Age-related comparisons of evolution of the inflammatory response after intracerebral hemorrhage in rats. Transl Stroke Res 2012,3(Suppl 1):132–146.
Boehmer ED, Goral J, Faunce DE, Kovacs EJ: Age-dependent decrease in Toll-like receptor 4-mediated proinflammatory cytokine production and mitogen-activated protein kinase expression. J Leukoc Biol 2004,75(2):342–349.
Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ: CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 2010,11(2):155–161.
Ohashi K, Burkart V, Flohe S, Kolb H: Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol 2000,164(2):558–561.
Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H: HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem 2002,277(17):15107–15112.
Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC: Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med 2002,195(1):99–111.
Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL: Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem 2004,279(17):17079–17084.
Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC: The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 2007,316(5831):1628–1632.
Gangloff M: Different dimerisation mode for TLR4 upon endosomal acidification? Trends Biochem Sci 2012,37(3):92–98.
Schmidt P, Krook H, Goto M, Korsgren O: MyD88-dependent toll-like receptor signalling is not a requirement for fetal islet xenograft rejection in mice. Xenotransplantation 2004,11(4):347–352.
Murakami K, Koide M, Dumont TM, Russell SR, Tranmer BI, Wellman GC: Subarachnoid hemorrhage induces gliosis and increased expression of the pro-inflammatory cytokine high mobility group box 1 protein. Transl Stroke Res 2011,2(1):72–79.
Ohnishi M, Katsuki H, Fukutomi C, Takahashi M, Motomura M, Fukunaga M, Matsuoka Y, Isohama Y, Izumi Y, Kume T, Inoue A, Akaike A: HMGB1 inhibitor glycyrrhizin attenuates intracerebral hemorrhage-induced injury in rats. Neuropharmacology 2011,61(5–6):975–980.
Kanzler H, Barrat FJ, Hessel EM, Coffman RL: Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med 2007,13(5):552–559.
Liu X, Zheng J, Zhou H: TLRs as pharmacological targets for plant-derived compounds in infectious and inflammatory diseases. Int Immunopharmacol 2011,11(10):1451–1456.
Gearing AJ: Targeting toll-like receptors for drug development: a summary of commercial approaches. Immunol Cell Biol 2007,85(6):490–494.
Park SJ, Kang SH, Kang YK, Eom YB, Koh KO, Kim DY, Youn HS: Inhibition of homodimerization of toll-like receptor 4 by 4-oxo-4-(2-oxo-oxazolidin-3-yl)-but-2-enoic acid ethyl ester. Int Immunopharmacol 2011,11(1):19–22.
Youn HS, Saitoh SI, Miyake K, Hwang DH: Inhibition of homodimerization of Toll-like receptor 4 by curcumin. Biochem Pharmacol 2006,72(1):62–69.
Ahn SI, Lee JK, Youn HS: Inhibition of homodimerization of toll-like receptor 4 by 6-shogaol. Mol Cells 2009,27(2):211–215.
Park SJ, Youn HS: Suppression of homodimerization of Toll-like receptor 4 by isoliquiritigenin. Phytochemistry 2010,71(14–15):1736–1740.
Liang Q, Wu Q, Jiang J, Duan J, Wang C, Smith MD, Lu H, Wang Q, Nagarkatti P, Fan D: Characterization of sparstolonin B, a Chinese herb-derived compound, as a selective Toll-like receptor antagonist with potent anti-inflammatory properties. J Biol Chem 2011,286(30):26470–26479.
Park SJ, Lee AN, Youn HS: TBK1-targeted suppression of TRIF-dependent signaling pathway of toll-like receptor 3 by auranofin. Arch Pharm Res 2010,33(6):939–945.
Kuno M, Nemoto K, Ninomiya N, Inagaki E, Kubota M, Matsumoto T, Yokota H: The novel selective toll-like receptor 4 signal transduction inhibitor tak-242 prevents endotoxaemia in conscious guinea-pigs. Clin Exp Pharmacol Physiol 2009,36(5–6):589–593.
Nakamura M, Shimizu Y, Sato Y, Miyazaki Y, Satoh T, Mizuno M, Kato Y, Hosaka Y, Furusako S: Toll-like receptor 4 signal transduction inhibitor, M62812, suppresses endothelial cell and leukocyte activation and prevents lethal septic shock in mice. Eur J Pharmacol 2007,569(3):237–243.
Huang M, Hu YY, Dong XQ, Xu QP, Yu WH, Zhang ZY: The protective role of oxymatrine on neuronal cell apoptosis in the hemorrhagic rat brain. J Ethnopharmacol 2012,143(1):228–235.
Yu WH, Dong XQ, Hu YY, Huang M, Zhang ZY: Ginkgolide B reduces neuronal cell apoptosis in the traumatic rat brain: possible involvement of Toll-like receptor 4 and nuclear factor κB pathway. Phytother Res 2012,26(12):1838–1844.
Wang Z, Zuo G, Shi XY, Zhang J, Fang Q, Chen G: Progesterone administration modulates cortical TLR4/NF-κB signaling pathway after subarachnoid hemorrhage in male rats. Mediators Inflamm 2011, 2011:848309.
Tasaki K, Ruetzler CA, Ohtsuki T, Martin D, Nawashiro H, Hallenbeck JM: Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res 1997,748(1–2):267–270.
Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP: Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke 2004,35(11):2576–2581.
Vartanian KB, Stevens SL, Marsh BJ, Williams-Karnesky R, Lessov NS, Stenzel-Poore MP: LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J Neuroinflammation 2011, 8:140.
Stevens SL, Leung PY, Vartanian KB, Gopalan B, Yang T, Simon RP, Stenzel-Poore MP: Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J Neurosci 2011,31(23):8456–8463.
Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF: Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab 2003,23(6):629–652.
Zhao X, Song S, Sun G, Strong R, Zhang J, Grotta JC, Aronowski J: Neuroprotective role of haptoglobin after intracerebral hemorrhage. J Neurosci 2009,29(50):15819–15827.
Zhao X, Sun G, Zhang J, Strong R, Dash PK, Kan YW, Grotta JC, Aronowski J: Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke 2007,38(12):3280–3286.
Chen G, Fang Q, Zhang J, Zhou D, Wang Z: Role of the Nrf2-ARE pathway in early brain injury after experimental subarachnoid hemorrhage. J Neurosci Res 2011,89(4):515–523.
Fadok VA, Warner ML, Bratton DL, Henson PM: CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (αvvβ3). J Immunol 1998,161(11):6250–6257.
Ren Y, Silverstein RL, Allen J, Savill J: CD36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. J Exp Med 1995,181(5):1857–1862.
Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J: 15d-prostaglandin J2 activates peroxisome proliferator-activated receptor-γ, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab 2006,26(6):811–820.
Beutler B: Inferences, questions and possibilities in Toll-like receptor signalling. Nature 2004,430(6996):257–263.
Acknowledgements
This work was supported in part by a grant from the National Natural Science Foundation of China (Nos. 81070932and 81271283).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors read and approved the final manuscript. FH collected literatures and reviewed the literatures. WPF reviewed the literatures. WYC proofreaded and corrected the manuscript. ZY revised the manuscript. YQW wrote the manuscript and approved the final version of the manuscript.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Fang, H., Wang, PF., Zhou, Y. et al. Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J Neuroinflammation 10, 794 (2013). https://doi.org/10.1186/1742-2094-10-27
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-10-27