
Abstract
Background
Traditional diagnoses of major depressive disorder (MDD) suggested that the presence or absence of stress prior to onset results in either ‘reactive’ or ‘endogenous’ subtypes of the disorder, respectively. Several lines of research suggest that the biological underpinnings of ‘reactive’ or ‘endogenous’ subtypes may also differ, resulting in differential response to treatment. We investigated this hypothesis by comparing the gene-expression profiles of three animal models of ‘reactive’ and ‘endogenous’ depression. We then translated these findings to clinical samples using a human post-mortem mRNA study.
Methods
Affymetrix mouse whole-genome oligonucleotide arrays were used to measure gene expression from hippocampal tissues of 144 mice from the Genome-based Therapeutic Drugs for Depression (GENDEP) project. The study used four inbred mouse strains and two depressogenic ‘stress’ protocols (maternal separation and Unpredictable Chronic Mild Stress) to model ‘reactive’ depression. Stress-related mRNA differences in mouse were compared with a parallel mRNA study using Flinders Sensitive and Resistant rat lines as a model of ‘endogenous’ depression. Convergent genes differentially expressed across the animal studies were used to inform candidate gene selection in a human mRNA post-mortem case control study from the Stanley Brain Consortium.
Results
In the mouse ‘reactive’ model, the expression of 350 genes changed in response to early stresses and 370 in response to late stresses. A minimal genetic overlap (less than 8.8%) was detected in response to both stress protocols, but 30% of these genes (21) were also differentially regulated in the ‘endogenous’ rat study. This overlap is significantly greater than expected by chance. The VAMP-2 gene, differentially expressed across the rodent studies, was also significantly altered in the human study after correcting for multiple testing.
Conclusions
Our results suggest that ‘endogenous’ and ‘reactive’ subtypes of depression are associated with largely distinct changes in gene-expression. However, they also suggest that the molecular signature of ‘reactive’ depression caused by early stressors differs considerably from that of ‘reactive’ depression caused by late stressors. A small set of genes was consistently dysregulated across each paradigm and in post-mortem brain tissue of depressed patients suggesting a final common pathway to the disorder. These genes included the VAMP-2 gene, which has previously been associated with Axis-I disorders including MDD, bipolar depression, schizophrenia and with antidepressant treatment response. We also discuss the implications of our findings for disease classification, personalized medicine and case-control studies of MDD.
Similar content being viewed by others


Background
Although antidepressants remain the first line treatment for major depressive disorder (MDD), antidepressant response varies considerably between individuals: fewer than half of all patients achieve remission following their first course of treatment [1]. The absence of robust predictors of treatment response means that the most effective antidepressant for a given patient is currently identified by trial and error. This is often a long and costly process which both delays recovery and has a negative effect on long-term outcome [2].
Clinicians have long intuited that heterogeneity in treatment response is the direct result of etiological heterogeneity in MDD [3]. Indeed, traditional diagnoses of major depression proposed that the presence or absence of stress prior to the onset of MDD results in two etiologically distinct subgroups of the disorder with different treatment recommendations. Early studies, which categorized these subtypes as ‘reactive’ (occurring as the result of a stressor) or ‘endogenous’ (occurring in the absence of stress), suggested that those with ‘endogenous’ depression responded more favorably to tricyclic antidepressants (TCAs) than selective serotonin reuptake inhibitors (SSRIs) [4]. While the validity of these subtypes remains unclear, reports continue to show that both distal stress (occurring early in life [5]) and proximal stress (occurring near the onset of a depressive episode [6]) are predictive of treatment response.
It remains unclear how the presence or absence of stress in the etiology of MDD affects response to treatment. However, it has been suggested that ‘endogenous’ and ‘reactive’ subtypes of depression are associated with largely distinct biological mechanisms, which respond differentially to treatment [3]. In line with this hypothesis, a recent animal study reported that the hippocampal gene-expression profile of a ‘reactive’ model of depression (induced by chronic restraint stress) differed considerably from that of an ‘endogenous’ model [7].
While this study suggests that the gene-expression profiles of ‘reactive’ depression caused by proximal stress may indeed differ from ‘endogenous’ depression, the role of distal early-life stress in this distinction remains unknown. Several studies have highlighted the importance of the timing of adversity and show that early and late stressors may have differential tissue-specific effects on gene-expression in the hippocampus [8–12]. The pathophysiological processes underlying MDD may therefore differ not only in the presence or absence of a stressor, but also by the timing of adversity (distal vs. proximal stress).
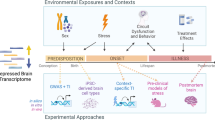
We investigated this hypothesis by exploring hippocampal gene-expression (mRNA) differences in three animal models of depression chosen to represent ‘reactive’ and ‘endogenous’ depression. In the ‘reactive’ depression model, mice were exposed to either distal stress (maternal separation) or proximal stress (unpredictable chronic mild stress). Flinders sensitive rats, which show congenital depression-like behavior, were used to model ‘endogenous’ depression.
Whole genome transcription profiles from disease relevant brain tissues in animals may provide valuable support and important information on the molecular mechanisms that may be relevant in humans. Nevertheless, the specific features of psychiatric illnesses means that molecular mechanisms uncovered in animal models are only suggestive and need to be validated in human studies [13, 14]. We therefore used findings from the animal models to inform probe set prioritization in a comparable human post-mortem case-control study of depression from the Stanley Brain Consortium. Specifically, we hypothesize that a set of genes that shows concordant expression differences in response to ‘reactive’ and ‘endogenous’ depression models in the rodent studies may represent a common final pathway to MDD. These same genes may therefore also be differentially regulated in the post-mortem brain tissue of humans with the disorder.
Methods
Design
Genome-wide expression profiling of the hippocampus (HIP) from two studies from the rodent arm of the Genome-based Therapeutic Drugs for Depression (GENDEP) study [15] was used to inform candidate gene selection in a comparable human post-mortem, case-control study on MDD from the Stanley Brain Consortium. The GENDEP project is a large-scale, multi-center human pharmacogenomics study that also includes a series of large-scale studies using animal models and in vitro experiments. The GENDEP project was designed to allow for integrative analysis of the results of the transcriptomics and proteomics on the samples from the human, the rodent and the in vitro studies, in order to gain further insight into the molecular mechanisms of MDD and identify biomarkers of antidepressant drugs (AD) treatment response. The mouse study used 144 animals from four strains of well-characterized inbred mice to model individual variation in humans. The mice were subjected to one of two stress protocols and a control condition (maternal separation (MS) - ‘early stress’, unpredictable chronic mild stress (UCMS) - ‘late stress’ - or the control condition (ENV)) to model ‘reactive’ depression. Litters of each strain were randomly allocated to the MS, UCMS or control group. Findings from the mouse study were cross validated in a parallel rat study that compared HIP mRNA differences between Flinders Sensitive and Flinders Resistant rat lines as models of ‘endogenous’ depression. Finally, genes differentially expressed in response to both stress protocols in the mouse study and in the rat study were used to inform probe set selection in comparable mRNA expression study in humans.
Animals
A total of 144 male and female mice (72 of each sex) from four different strains ((129S1/SvImJ, C57LB/6 J, DBA/2 J and FVB/NJ) were bred in the barrier unit at the Institute of Psychiatry, London, UK. Weaning took place when the animals were 21 to 28 days old. Animals were group-housed under standard conditions with a 12:12 h light:dark cycle, 22°C ± 11°C, food and water ad libitum. A total of 144 animals were sacrificed by cervical dislocation. Animals used for the transcriptomic study were not behaviorally tested. The hippocampus, liver and spleen were dissected following previously published protocols [16, 17]. All housing and experimental procedures were carried out in accordance with the UK Home Office Animals (Scientific Procedures) Act, 1986.
A total of 39 animals from two cohorts of Flinders Sensitive Lines and Flinders Resistant Lines (22 FRL and 17 FSL) were bred and maintained at Karolinska Institutet (Stockholm) and housed under standard room temperature (22 ± 1°C), relative humidity (45 to 55%) and a 12 h light:dark schedule (light on at 07:00 a.m.). Food and water were available ad libitum. The study was conducted as part of a parallel GENDEP investigation. The Stockholm's Ethical Committee for Protection of Animals approved the study and all procedures were conducted in conformity with the Karolinska Institutet's guidelines for the care and use of laboratory animals, which follows the European Communities Council Directive of 24 November 1986. Additional information on the rat study is available elsewhere [18].
UCMS (Unpredictable Chronic Mild Stress)
In mice, ‘reactive’ depression caused by proximal stress was modeled using an Unpredictable Chronic Mild Stress (UCMS) paradigm. A third of the 144 mice (48 male and female mice) were exposed to varying stressors on a daily basis for a period of two weeks. Exposure to UCMS commenced when the animals were 10 weeks of age. The UCSM protocols included exposure to different stressors each day in a pseudorandom order. The stressors in the UCMS regime were based on previously published protocols including two hours of home cage tilting at 45°, damp bedding for four hours, cage switching for two hours, flooded cage for 10 minutes, altered length and time of light-dark cycle and air-puff [19]. Animals were exposed to either one or two stressors each day for varying lengths of time (Figure 1). All UCMS-exposed mice were tested and maintained under standard laboratory conditions but were single-housed. Following the UCMS regimen, a set of animals was tested with a battery of behavioral tests including Porsolt as an index of UCMS-evoked depressive-behavior [19]. However, all animals used for this mRNA characterization were not behaviorally tested to control for the potential stressor effects of the tests.

This figure shows the stress administration regime for the unpredictable chronic mild stress paradigm. The duration of the stress regime was for two consecutive weeks and the order of the different stressors was randomized. This figure shows the stressors and time/duration of administration for each of the two weeks.
MS (Maternal Separation)
A maternal separation protocol was used to model ‘reactive’ depression caused by distal stress in a further 48 mice. A single 24-hour separation of the pup from the dam at postnatal day (PND) 9 protocol was chosen to elicit a sufficiently strong biological response. Day of birth was defined as PND 0 for that particular litter. On postnatal Day 9 the dam was removed from the litter for 24 hours. The litter was kept on a heating pad in their home cage at 33°C ± 2°C in a different room than the dam in order to avoid contact through vocalization. Separated pups did not have access to food or water during their separation period. Litters were always separated and reunited with the mother during the first half of the light phase. The first hour after reuniting the litter with the mother was videotaped. Litters were of different sizes and when possible each litter came from a different breeding pair. A more detailed description of the litters is published elsewhere [19].
‘Endogenous’ model of depression
Flinders Sensitive Lines (FSL) and Flinders Resistant Lines (FRL) rats represent an ‘endogenous’ model of depression [20–23]. Flinders Lines are strains originally obtained by selective breeding of out-bred Sprague-Dawley rats (SD), according to their resistance or sensitivity to anticholinesterase diisopropyl fluorophosphates (DFP) treatment [24]. FSL are congenitally more sensitive to DFP and cholinergic agonists than FRL, which is a neurobiological feature shared with depressed cases in humans [21]. They also show many behavioral similarities to human depressed patients, including decreased psychomotor activity and appetite, cholinergic hypersensitivity, immune and sleep abnormalities including delay in rapid eye movement (REM) sleep but preserved cognitive function and hedonic response [25]. Flinders rats remain a robust model of depression to date [26].
mRNA extraction and lab protocols
Mouse brains, livers and spleens were dissected from each animal and frozen on dry ice. Total RNA was extracted from frozen hippocampal tissue and 3-ug RNA was processed using the One Cycle Target Labelling kit (Affymetrix, Santa Clara, CA, USA) and hybridized to the mouse MOE430v2 Gene Expression Array (Affymetrix) following standard Affymetrix protocols. Hippocampal mRNA extraction from Flinders rats was performed by another participating group from the GENDEP project [18, 22]. Briefly, cRNA probes were obtained and hybridized to Affymetrix Rat Genome 230 2.0 using Affymetrix’s One-Cycle Eukaryotic Target Labelling Assay protocol. Protocols used for the human post-mortem mRNA extraction are described in detail in the paper by Iwamoto and colleagues [27]. Briefly, total RNA was extracted from 0.1 g of frozen prefrontal cortex tissues using Trizol (Invitrogen, Groningen, The Netherlands). A total of 8 to 10 mg of mRNA was reverse transcribed and synthesized into cDNA, hybridized onto Affymetrix HU95A oligonucleotide arrays and scanned using an HP GeneArray scanner (Hewlett-Packard, Palo Alto, CA, USA). Information on The Stanley Foundation brain collection and Neuropathology Consortium is found elsewhere [28].
Human samples
The human samples used in this study were donated to the Stanley Foundation Brain Collection at the Department of Psychiatry, University of the Health Sciences, Bethesda, MD, USA and have been made available to researchers world-wide. Human brain tissues were donated under standardized legislation according to the Uniform Anatomical Gift Act (USA). Information on Stanley Medical Research Institute (SMRI) and its research was offered to the next of kin at the time of the donation. Additional information is publically available from the Stanley Brain Consortium website [29]. The primary transcription-wide analysis was performed and described by Iwamoto and colleagues [27]. For consistency and quality assurance, the same subset has been used without additions or subtractions of cases. All data have been processed from raw files. The samples used consist of post-mortem prefrontal cortex from the Stanley Foundation Neuropathology Consortium from deceased patients affected with major depressive disorder and carefully matched controls. Exclusion criteria include poor mRNA quality and age (>65). A total of 26 samples, 11 cases and 15 controls, were used congruent with the primary data analysis (Table 1). Clinical diagnosis of MDD was made following Diagnostic and Statistical Manual of Mental Disorders – 4th Edition (DSM-IV) diagnostic guidelines and reviewed independently by a pathologist and psychiatrist. Additional information on the human sample can be found in the Iwamoto and colleagues paper [27].
Statistical analysis of microarray data
Probe intensity data from 144 Affymetrix mouse whole-genome oligonucleotide arrays (MOE 430 v2) were normalized and summarized using the Robust Multichip Average (RMA method) [30]. Probe sets that were systematically absent (based on the MAS 5.0 detection present/absent call) across all the arrays were removed leaving 37,231 out of the original 45,101 probe sets. A battery of quality control metrics and exploratory analysis on the 144 arrays identified 10 arrays that differed significantly in quality. These arrays were removed for the purpose of the subsequent analysis; further description on normalization methods is available elsewhere [12, 16].
In order to identify genes differentially expressed in response to early and late stress protocols we performed two sets of analyses. First, we compared normalized gene expression measurements between maternally separated animal (MS) and control (CON). Second, we compared normalized gene expression measurements between UCMS and CON. Differences were statistically evaluated using the non-parametric algorithms implemented in the RankProd package in the R environment [31, 32]. RankProd enabled us to combine datasets from four different strains using a meta-analysis approach with the RPadvance function. This allowed us to circumvent issues arising from the predominant strain effects by evaluating differences within each strain first. Genes differentially expressed in a single strain were analyzed using rank product (RP) function from the same package, using the ‘data from single origin’ option. The P-values were calculated with 1,000,000 permutations, and multiple testing was taken into account by using the percentage of false prediction at the very conservative threshold of PFP <0.001. A common method to control for the number of rejected hypothesis in ‘omics’ study is to compute and report the false discovery rate (FDR) as proposed by Benjamini and Hochberg. The RankProd package returns proportion of false positive (PFP), which is a method proposed by Fernando and colleagues. Contrary to FDR, PFP does not rely on the correlation between tests and the number of tests performed [33]. Although PFP and FDR are often equated, the two methods differ in that PFP controls the proportion of accumulated false positives while FDR controls the expected proportion of false positive. FDR is not the best method to use in cases where there is a relationship between variables, which in mRNA studies is generally driven by genetic regulatory pathways and cross hybridization. We therefore corrected using the PFP method across all studies where we use the RankProd algorithim. The genes significantly altered were identified by the PANTHER classification system [34]. Genes with PFP <0.001 were subsequently uploaded to the Ingenuity database for pathway analysis with the Ingenuity Pathway Analysis (IPA) software (QIAGEN’s Ingenuity® Pathway Analysis (IPA®, Redwood City, USA) [35].
Expression data from FSL and FRL animals have been made available on the Gene Expression Omnibus (GEO; accession number GS2088, [36]. Data have been processed from raw. CEL files to ensure consistency of data analysis across all animal studies. To control for potential batch effects we combined the rat datasets from two cohorts using the ComBat function built into the inSilicoMerging package for the R environment [37]. Probe sets were normalized and summarized using Robust Multichip Average (RMA method). Probe sets that were systematically absent (based on the MAS 5.0 absent/present detection call) were removed. Probe-set summaries from FSL and FRL were then compared using the RankProd non-parametric algorithm implemented in R using the PRadvance function and single origin option. P-values were evaluated using 1,000,000 permutations. A conservative false discovery rate (PFP) threshold of P <0.001 and a change fold >1.5 was used. Probe sets that met the statistical thresholds were subsequently annotated using PANTHER [34] to obtain a list of gene symbols. We then matched all genes differentially expressed across all rodent studies using scripts written in Python [38]. Convergent genes differentially expressed across all rodent studies were subsequently analyzed using IPA software. Lastly, all genes differentially expressed in response to both “reactive” and “endogenous” models of depression were used to inform probe set selection in the human study.
Raw scores from 26 Affymetrix human oligonucleotide arrays (HU95A) were normalized and summarized into probe sets using the RMA method, which returned log2 transformed intensities [30]. Intensity distributions, profile correlations and quality control metrics were applied. MAS 5.0 expression values were calculated based on scaling to a target intensity of 100, then transformed by Log2 and calls were computed using the MAS5.0 present/absent algorithm. Affymetrix HU95A incorporates over 12,000 probe sets, tagging the expression of over 5,000 well-characterized genes. Human genes, ortholog to genes differentially expressed across all three rodent studies, were obtained using the Mouse Genome Informatics orthology query [39]. The Affymetrix Netaffx tool [40] was used to identify probe sets on the HU95A chip (Affymetrix) tagging the expression of the human genes. Expression differences between human MDD cases and controls were evaluated using the RankProd non-parametric algorithm implemented in R using the single origin function. Candidate genes in humans informed by the results from the mouse study were considered differentially expressed at a stringent corrected significance threshold PFP <0.05 using permutation testing with 1,000,000 permutations.
Results
Gene expression profiles in ‘reactive’ depression models
The Rankprod method was used to identify the most robustly differentially expressed genes between ‘late’ (UCMS) stressed animals and control and between ‘early’ (MS) stressed animals and control. We considered only those genes that show consistency in the direction of change across all four strains. Inconsistency in the direction of change indicates Stress x Strain interaction effects, which are not specific to our research question. The results of this analysis uncovered 406 probe sets altered in response to UCMS across all four strains. These probes tag the expression of 370 known genes in mice. A summary of genes uncovered from this analysis with a previous association with stress response or MDD is presented in Table 1. The results reveal a number of genes previously associated with UCMS protocols and believed to play a role in the pathogenesis of MDD. The same analysis was repeated to compare the maternally separated animal (MS) and control. The results from this analysis revealed 396 probe sets differentially regulated in response to the maternal separation protocol. These probe sets could be mapped to 350 known genes in mice. A summary of the top genes differentially expressed in response to maternal separation protocols is presented in Table 2. We then explored the number of altered genes in response to either ‘early’ or ‘late’ stressors as well as the genetic overlap between the two conditions (Figure 2). There were remarkably few. Only 67 genes, less than 8.8% of significantly altered genes were in common between mice exposed to early and late stress paradigms.

Venn diagram showing the number of genes significantly altered in response each depressogenic protocol. A compelling finding is the limited number of overlapping genes (approximately 8.8%) suggesting that etiologically different molecular mechanisms underpin a congruent set of behaviors.
The minimal gene expression overlap suggests that the biological mechanisms underpinning ‘reactive’ depression caused by early and late stressors differs considerably. In order to gain further understanding into these differences, genes differentially expressed for each of the two models were analyzed using IPA [35]. This allowed us to uncover gene networks showing the molecular relationship between the genes and evaluate networks according to the fit of significant genes in each dataset [12]. First, we explored gene networks associated with ‘late’ UCMS protocols. A total of 350 genes from our reference list were found on the IPA database. The top two functional networks identified by IPA have a score >42, with 29 reference molecules included in the first network and 23 in the second. Both networks were significantly associated with stress signaling response. The most significant transcriptional regulators included ELK1/2/4 TFIIA, SMARCB, CREB1 and THRB (see Additional file 1: Figure S1 and Additional file 2: Figure S2). We repeated the pathway analysis with genes differentially expressed in response to ‘early’ (MS) stressors. A total of 347 genes from our reference list were found on the IPA knowledge database. IPA returned three networks with a score >40. The associated functions of the top networks include mRNA post-transcriptional modification, protein synthesis and cellular development. The networks are associated with developmental and neurological disorders, which is a good match to the “early” stress protocol used. The top-ranking network (see Additional file 3: Figure S3) includes 29 focus molecules from our reference gene set. The most prominent interacting genes within this network are with the Yhwaz and Yhwag. These genes are of particular interest as they have been systematically uncovered across several proteomic and transcriptomic studies from the GENDEP project in both mice and rats [12, 13, 17, 22]. These genes show a direct interaction with the STK25 kinase, which plays a role in stress response. The second network (Additional file 4: Figure S4) is composed of 27 molecules from our reference dataset. This network is centered on the NF-κB complex. The nuclear factor-κB (NF-κB) is a ubiquitous transcription factor involved in the regulation of gene expression and cell stress response and cell proliferation. Interestingly, NF-κB can be activated by different stimuli, including cytokines (such as TNF-α and IL-1): this finding is congruent with the inflammation hypothesis for MDD [41–45].
Gene expression profiles in ‘endogenous’ depression models
To gain an understanding of the similarities between stress-induced ‘reactive’ depression and a congenital ‘endogenous’ model of depression, we compared genes differentially regulated in response to early (MS) and late stress (UCMS) with mRNA differences between Flinders sensitive and resistant lines. Flinders lines are a genetic animal model of depression that allows us to cross-validate stress-altered genes within a parallel, independent mRNA study where depressive-behaviors occur in the absence of environmental stressors. The RankProd algorithm and conservative cut-offs described previously was used to evaluated mRNA differences between Flinders Sensitive and Flinders Resistant lines. The results revealed 715 down-regulated and 1,145 up-regulated probe sets. To obtain a list of gene names, probe sets obtained were subsequently annotated using PANTHER [34]. The probe sets tagged the expression of 501 down-regulated and 727 up-regulated genes. A total of 1,228 genes were used for cross-validation with the mouse study. First, we explored the genomic overlap between maternally deprived animals and Flinders line rats. From a total of 350 genes differentially regulated in maternally deprived mice, a total of 65 genes (19%) were also differentially regulated in rats. The same comparison was performed with genes differentially expressed in mice exposed to UCMS. A total of 52 genes (11%) were differentially expressed between ‘late’ stress animals and Flinders rats. A compelling finding is that 21 genes are differentially expressed in rats and in both early and late stressed mice (Figure 3). This is an important genetic overlap given that only 67 genes were commonly expressed between early and late stressed animals in the first place. Validations in an independent, methodologically different study using a genetic model of depression point to an important genetic overlap between stress-related and syndrome-related mechanisms. In order to gain further biological insight, genes significantly altered in response to both stresses in mice and between Flinders Sensitive and Resistant lines were carried forward for analysis using Ingenuity’s IPA system. All 21 genes were found in the Ingenuity reference database. A significant network with a score >40 consisting of over 55% of the reference molecules (12/21) was revealed (Figure 4). Among the genes in the pathway, four genes (Ywhaz, Ppm1a, Nkfb and Mapk1) are of particular interest as they have been previously reported across different “omics” GENDEP investigations [12, 17, 46–48].

Venn diagram showing the number of genes overlapping across all rodent studies. Only 67 genes were differentially regulated in response to both early (MS) and late stressors (UCMS) pointing at minimal genetic overlap. However, many of these genes (approximately 30%) were also differentially regulated in an endogenous rat model of depression. The replication of these genes in a different organism that shows congenital depression-like symptoms, points at molecular mechanisms that may be involved in the human pathology.

Network analysis performed on converging genes differentially regulated in response to different stresses in mouse and between different selected Flinders lines. The pathway comprises over half the reference molecules uploaded to the Ingenuity database system (12 out of the 21 reference molecules). The pathway implicates a number of genes previously associated with MDD and antidepressant treatment response, including Ppm1a, Ywhaz, NkFb and Mapk.
Translating findings to humans
From a total of 21 genes differentially expressed across all three rodent studies, 15 human orthologs were found. The expression of these 15 genes is tagged by 21 probe sets on the Affymetrix HU95A oligonucleotide array. The RankProd algorithm was used to evaluate expression between post-mortem cases and controls. Out of a total of 15 genes, the VAMP-2 is significantly down-regulated after correcting for the number of multiple non-independent tests using a prediction of false discovery rate of PFP <0.05 (Table 3). In our study, the Vesicle-Associated Membrane Protein 2 (VAMP-2: Synaptobrevin2) gene is significantly altered across all rodent studies and in the human study.
Discussion
The main objective of this study was to compare the genomic signatures of ‘reactive’ and ‘endogenous’ models of depression in three rodent studies and translate these findings in a human study. We found that all three animal models of depression had largely unique gene-expression profiles indicating divergent molecular mechanisms. Nevertheless, a small set of genes was consistently dysregulated across each paradigm and in the post-mortem brain tissue of depressed patients, which may represent a final common pathway to the disorder.
Gene-expression profiles of ‘endogenous’ and ‘reactive’ models of depression
Consistent with our hypothesis and with previous findings, the gene-expression profiles of both of our ‘reactive’ models of depression were largely distinct from our ‘endogenous’ model. Interestingly, this differed according to which ‘reactive’ depression paradigm was compared. For the early or stress ‘reactive’ model 19% of genes overlapped with the endogenous model, while for the late stress ‘reactive’ model the overlap was considerably lower at just 11%.
Surprisingly, the genomic signatures of our two ‘reactive’ models were more distinct from one another than the ‘endogenous’ model with fewer than 9% of genes shared between the two paradigms. This suggests that the two different models result in depressive-like behavior in mice through distinct biological mechanisms. Interestingly, gene pathway analysis returned plausible functional networks, with the more significant network for ‘early’ stressed animals associated with neurodevelopmental disorders and those of ‘late’ stressed animals associated with cell stress response and cell-signaling. Taken together, our results suggest that early exposure to stress modulates the expression of genes belonging to pathways associated with neurodevelopmental mechanisms. These changes may condition an individual’s exposure to later stresses and response to pharmacological and behavioral interventions later in life in yet unclear ways. Conversely, late onset stresses may act primarily on brain neurochemistry with neurostructural changes occurring via the cascading effects of neurochemically-related mechanisms, including neurogenesis and apoptosis [49].
Genes differentially regulated across all three paradigms
While each of our three animal models of depression showed largely distinct gene-expression profiles, a set of genes were differentially regulated across all three paradigms. Pathway analysis of these genes revealed a gene network which included Ppm1a, Ywhaz, Nkfb and Mapk.
All four genes have each been implicated in both the etiology of MDD and the response to treatment and may, therefore, represent a final common pathway to the disorder. We previously reported that the expression ppm1a was significantly modulated by the antidepressant nortriptyline. We have also shown that several single nucleotide polymorphisms in the human ortholog of this gene (PPM1A) predict a response to the same drug in a parallel human pharmacogenetic study [46]. Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein (Ywhaz) has systematically been uncovered across several GENDEP studies and plays a role in cell proliferation and neurogenesis, which is a current explanatory model of MDD [50–54]. Moreover, it interacts with IRS1 protein and the MAPK pathway by modulating the activation of JNK1 and p38 MAPK both of which have been systematically associated with depressive mechanisms [52–56]. Lastly, the Nfkb gene has extensively been associated with peripheral inflammation and is consistent with the inflammation hypothesis for MDD [57].
Convergent animal-human genes
Animal models are an attractive proposition for the study of mood disorders as they allow access to disease relevant brain tissues and to control for environmental conditions. However, given the nature and characteristic of psychiatric disorders, there are aspects of these illnesses that can only be studied in humans. We therefore attempted to translate our set of convergent genes emerging from the rodent studies in a matching human post-mortem mRNA study of depressed cases and controls. One gene, the VAMP-2 gene, remained significantly down-regulated after correcting for multiple testing in the human study. The vesicle-associated membrane protein (VAMP-2; synaptobrevin2) plays a role in the molecular regulation of transmitter release at the presynaptic plasma membrane. The expression of VAMP-2 has been found to be altered in both schizophrenia and bipolar disorder within a combined microarray analysis of the Stanley Foundation's brain collections [58]. Moreover, several other studies have implicated this gene in Axis-I psychiatric disorders and in antidepressant treatment response [59–62]. Previous studies have also shown that the VAMP2/synaptobrevin-2 gene is increased in rat frontal cortex after chronic antidepressant treatment and repeated electro-convulsive therapy (ECT), although the finding has not been consistently replicated [63, 64].
Implications
If replicated, the results of our study may have far reaching implications for both personalized medicine for MDD and case-control studies of the disorder.
Our findings suggest that etiological factors (such as proximal and distal stressors) could be used to indicate the molecular mechanisms at work in a given patient and, therefore, select the most effective treatment. Indeed, several studies have shown that proximal and distal stress predicts a response to antidepressants. Interestingly, while proximal stressors, such as divorce or job loss, have been linked with a good response [3], distal stressors, such as childhood maltreatment, are associated with a less favorable outcome [5]. Our results suggest that these contradictory findings may be explained by the divergent molecular mechanisms underlying ‘reactive’ depression caused by early versus late stressors. Nevertheless, further studies in clinical samples would be required to test this hypothesis.
Heterogeneity in the molecular mechanisms underlying depression could explain why, despite considerable efforts, genome-wide association studies (GWAS) of depression have yet to identify statistically significant associations with MDD [65]. This same heterogeneity may also explain the paucity of findings from pharmacogenetic studies of MDD, including the very large GWAS, NEWMEDS [66]. If the molecular mechanisms underlying MDD differ according to stress, it is plausible that different genetic variants would predict response to treatment in stressed and non-stressed individuals. In line with this, several studies have shown that genetic variants and stress have interdependent effects on antidepressant response [67].
While our findings highlight the heterogeneity of depression, they also suggest that a small set of genes may be involved in a final common pathway to the disorder. Replication of these findings in further transcriptomic studies of clinical samples is necessary before any firm conclusions can be drawn about the role of these genes in MDD. However, if they are successful, the existence of a final common pathway provides an exciting prospect for the development of novel antidepressants. If indeed the heterogeneity of MDD explains inter-individual variation in treatment response, it is plausible that antidepressants, which target this final common pathway, may prove to be effective for all patients, regardless of their etiological factors.
Limitations
Our findings should be considered in the context of several important limitations.
First, we used whole genome gene-expression data from four different samples in our study. While this approach allowed us to conduct integrative analyses across species and translate our findings from rodents to humans, it also meant that our analyses were subject to multiple testings. We used stringent thresholds both within and across analyses in order to protect against the risk of false positive findings. Nevertheless, in taking such an approach it is possible that we inflated the number of false negatives. Further replication of our results in larger independent samples is therefore necessary to confirm our findings.
Second, our rodent models focused exclusively on gene-expression in the hippocampus and did not include further brain structures implicated in the neurobiology of MDD, such as the amygdala. Moreover, limited public human depression hippocampal transcript array data were available at the time of analysis which meant that our human study used gene-expression data collected from a different, but still disease relevant, brain region (the prefrontal cortex). It is plausible, therefore, that the use of a different brain region resulted in false negatives in the human component of our study. Our findings, therefore, require confirmation in further brain regions in both animal and human samples.
Finally, it is important to note that rodents do not capture the complex characteristics of psychiatric illnesses that can only be fully investigated in human studies. Nevertheless, there are many advantages of animal models, which allow access to disease relevant brain tissues and control of environmental conditions. In the current study, we attempted to capture the full potential of both animal and human studies by conducting integrative analyses using several independent animal studies and translating results in a disease-relevant but different brain region in humans.
Conclusion
It is largely accepted that there are multiple causal pathways to MDD consisting of different combinations of genetic and environmental risk factors [67]. However, it remains unclear whether these factors converge on a unitary molecular mechanism underlying MDD or MDD consists of a heterogeneous group of disorders with multiple causal factors and distinct molecular mechanisms. Our findings provide support for both of these hypotheses. Using an animal model, we have shown that the presence and timing of stress determines distinct molecular processes underlying depressive behavior. However, we also identified a small set of genes which were consistently dysregulated across each stress paradigm and in post-mortem brain tissue of depressed patients suggestive of a final common pathway to the disorder. These genes included VAMP-2, a gene which has previously been associated with Axis-I disorders including MDD, bipolar depression, schizophrenia and with antidepressant treatment response.
Careful consideration of the etiological pathways to MDD may be key to dissecting the heterogeneity of the disorder and understanding and predicting response to treatment. Nevertheless, a final common pathway which unites the disparate etiologies of MDD may yet provide a target for novel treatments which are effective for all, rather than just subsets of patients.
Abbreviations
- CON:
-
Control
- DFP:
-
Diisopropyl fluorophosphates
- FDR:
-
False discovery rate
- FRL:
-
Flinders Resistant Line
- FSL:
-
Flinders Sensitive Line
- GWAS:
-
Genome Wide Association Study
- HIP:
-
Hippocampus
- IPA:
-
Ingenuity Pathway Analysis
- MDD:
-
Major Depressive Disorder
- MS:
-
Maternal Separation
- PFP:
-
Proportion of false positive
- PND:
-
Post Natal Day
- RMA:
-
Robust Multichip Averaging
- RP:
-
Rank Prod
- SSRIs:
-
Selective Serotonin Reuptake Inhibitors
- TCAs:
-
Tricyclic Antidepressants
- UCMS:
-
Unpredictable Chronic Mild Stress.
References
Thase ME, Entsuah AR, Rudolph RL: Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J Psychiatry. 2001, 178: 234-241.
Uher R, Huezo-Diaz P, Perroud N, Smith R, Rietschel M, Mors O, Hauser J, Maier W, Kozel D, Henigsberg N, Barreto M, Placentino A, Dernovsek MZ, Schulze TG, Kalember P, Zobel A, Czerski PM, Larsen ER, Souery D, Giovannini C, Gray JM, Lewis CM, Farmer A, Aitchison KJ, McGuffin P, Craig I: Genetic predictors of response to antidepressants in the GENDEP project. Pharmacogenomics J. 2009, 9: 225-233.
Keers R, Uher R: Gene-environment interaction in major depression and antidepressant treatment response. Curr Psychiatry Rep. 2012, 14: 129-137.
Paroxetine: a selective serotonin reuptake inhibitor showing better tolerance, but weaker antidepressant effect than clomipramine in a controlled multicenter study. Danish University Antidepressant Group. J Affect Disord. 1990, 18: 289-299.
Nanni V, Uher R, Danese A: Childhood maltreatment predicts unfavorable course of illness and treatment outcome in depression: a meta-analysis. Am J Psychiatry. 2012, 169: 141-151.
Chen JD, Liu F, Xun GL, Chen HF, Hu MR, Guo XF, Xiao CQ, Wooderson SC, Guo WB, Zhao JP: Early and late onset, first-episode, treatment-naive depression: same clinical symptoms, different regional neural activities. J Affect Disord. 2012, 143: 56-63.
Andrus BM, Blizinsky K, Vedell PT, Dennis K, Shukla PK, Schaffer DJ, Radulovic J, Churchill GA, Redei EE: Gene expression patterns in the hippocampus and amygdala of endogenous depression and chronic stress models. Mol Psychiatry. 2012, 17: 49-61.
Alfonso J, Frasch AC, Flugge G: Chronic stress, depression and antidepressants: effects on gene transcription in the hippocampus. Rev Neurosci. 2005, 16: 43-56.
Champagne DL, Bagot RC, van Hasselt F, Ramakers G, Meaney MJ, de Kloet ER, Joels M, Krugers H: Maternal care and hippocampal plasticity: evidence for experience-dependent structural plasticity, altered synaptic functioning, and differential responsiveness to glucocorticoids and stress. J Neurosci. 2008, 28: 6037-6045.
Gotlib IH, Joormann J, Minor KL, Hallmayer J: HPA axis reactivity: a mechanism underlying the associations among 5-HTTLPR, stress, and depression. Biol Psychiatry. 2008, 63: 847-851.
Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A, Sharma S, Pearson D, Plotsky PM, Meaney MJ: Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science. 1997, 277: 1659-1662.
Malki K, Lourdusamy A, Binder E, Payá-Cano J, Sluyter F, Craig I, Keers R, McGuffin P, Uher R, Schalkwyk LC: Antidepressant-dependent mRNA changes in mouse associated with hippocampal neurogenesis in a mouse model of depression. Pharmacogenet Genomics. 2012, 22: 765-776.
Carboni L, Becchi S, Piubelli C, Mallei A, Giambelli R, Razzoli M, Mathe AA, Popoli M, Domenici E: Early-life stress and antidepressants modulate peripheral biomarkers in a gene-environment rat model of depression. Prog Neuropsychopharmacol Biol Psychiatry. 2010, 34: 1037-1048.
Holmes PV: Rodent models of depression: reexamining validity without anthropomorphic inference. Crit Rev Neurobiol. 2003, 15: 143-174.
GENEDEP Official Site. [http://gendep.iop.kcl.ac.uk]
Malki K, Tosto MG, Jumabhoy I, Lourdusamy A, Sluyter F, Craig I, Uher R, McGuffin P, Schalkwyk LC: Integrative mouse and human mRNA studies using WGCNA nominates novel candidate genes involved in the pathogenesis of major depressive disorder. Pharmacogenomics. 2013, 14: 1979-1990.
Malki K, Campbell J, Davies M, Keers R, Uher R, Ward M, Paya-Cano J, Aitchinson KJ, Binder E, Sluyter F, Kuhn K, Selzer S, Craig I, McGuffin P, Schalkwyk LC: Pharmacoproteomic investigation into antidepressant response in two mouse inbred strains. Proteomics. 2012, 12: 2355-2365.
Piubelli C, Carboni L, Becchi S, Mathe AA, Domenici E: Regulation of cytoskeleton machinery, neurogenesis and energy metabolism pathways in a rat gene-environment model of depression revealed by proteomic analysis. Neuroscience. 2011, 176: 349-380.
Binder E, Malki K, Paya-Cano JL, Fernandes C, Aitchison KJ, Mathe AA, Sluyter F, Schalkwyk LC: Antidepressants and the resilience to early-life stress in inbred mouse strains. Pharmacogenet Genom. 2011, 21: 779-789.
Nishi K, Kanemaru K, Diksic M: A genetic rat model of depression, Flinders sensitive line, has a lower density of 5-HT1A receptors, but a higher density of 5-HT1B receptors, compared to control rats. Neurochem Int. 2009, 54: 299-307.
Osterlund MK, Overstreet DH, Hurd YL: The Flinders Sensitive Line rats, a genetic model of depression, show abnormal serotonin receptor mRNA expression in the brain that is reversed by 17 beta-estradiol. Mol Brain Res. 1999, 74: 158-166.
Blaveri E, Kelly F, Mallei A, Harris K, Taylor A, Reid J, Razzoli M, Carboni L, Piubelli C, Musazzi L, Racagni G, Mathé A, Popoli M, Domenici E, Bates S: Expression profiling of a genetic animal model of depression reveals novel molecular pathways underlying depressive-like behaviours. Plos One. 2010, 5: e12596.
Friedman EM, Becker KA, Overstreet DH, Lawrence DA: Reduced primary antibody responses in a genetic animal model of depression. Psychosom Med. 2002, 64: 267-273.
Overstreet DH: The Flinders sensitive line rats: a genetic animal model of depression. Neurosci Biobehav Rev. 1993, 17: 51-68.
Overstreet DH: Commentary: a behavioral, psychopharmacological, and neurochemical update on the Flinders Sensitive Line rat, a potential genetic animal model of depression. Behav Genet. 1991, 21: 67-74.
Overstreet DH, Wegener G: The Flinders sensitive line rat model of depression–25 years and still producing. Pharmacol Rev. 2013, 65: 143-155.
Iwamoto K, Kakiuchi C, Bundo M, Ikeda K, Kato T: Molecular characterization of bipolar disorder by comparing gene expression profiles of postmortem brains of major mental disorders. Mol Psychiatry. 2004, 9: 406-416.
Torrey EF, Webster M, Knable M, Johnston N, Yolken RH: The Stanley Foundation Brain Collection and Neuropathology Consortium. Schizophr Res. 2000, 44: 151-155.
The Stanley Medical Research Institute. [http://www.stanleyresearch.org]
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP: Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003, 4: 249-264.
The R Project for Statistical Computing. [http://www.r-project.org/]
Hong F, Breitling R, McEntee CW, Wittner BS, Nemhauser JL, Chory J: RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics. 2006, 22: 2825-2827.
Fernando RL, Nettleton D, Southey BR, Dekkers JC, Rothschild MF, Soller M: Controlling the proportion of false positives in multiple dependent tests. Genetics. 2004, 166: 611-619.
PANTHER Classification System. [http://www.pantherdb.org]
Ingenuity. [http://www.ingenuity.com/]
Gene Expression Omnibus. [http://www.ncbi.nlm.nih.gov/geo]
inSilicoMerging. http://www.bioconductor.org/packages/release/bioc/html/inSilicoMerging.html.
Python™. [http://www.python.org/]
Mouse Genome Informatics. [http://www.informatics.jax.org/]
Affymetrix: Biology for a better world. [http://www.affymetrix.com/analysis/index.affx]
Zunszain PA, Hepgul N, Pariante CM: Inflammation and depression. Curr Top Behav Neurosci. 2013, 14: 135-151.
Miller AH, Maletic V, Raison CL: Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009, 65: 732-741.
Gardner A, Boles RG: Beyond the serotonin hypothesis: mitochondria, inflammation and neurodegeneration in major depression and affective spectrum disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2011, 35: 730-743.
Maes M: The cytokine hypothesis of depression: inflammation, oxidative & nitrosative stress (IO&NS) and leaky gut as new targets for adjunctive treatments in depression. Neuro Endocrinol Lett. 2008, 29: 287-291.
Berk M, Williams LJ, Jacka FN, O'Neil A, Pasco JA, Moylan S, Allen NB, Stuart AL, Hayley AC, Byrne ML, Maes M: So depression is an inflammatory disease, but where does the inflammation come from?. BMC Med. 2013, 11: 200.
Malki K, Uher R, Paya-Cano J, Binder E, Rietschel M, Zobel A, Mors O, Hauser J, Henigsberg N, Jerman B, Souery D, Placentino A, Ng MY, Cohen-Woods S, Sluyter F, Farmer A, Aitchison KJ, Craig IW, Lewis CM, McGuffin P, Schalkwyk LC: Convergent animal and human evidence suggests a role of PPM1A gene in response to antidepressants. Biol Psychiatry. 2011, 69: 360-365.
Wegener G, Harvey BH, Bonefeld B, Muller HK, Volke V, Overstreet DH, Elfving B: Increased stress-evoked nitric oxide signalling in the Flinders sensitive line (FSL) rat: a genetic animal model of depression. Int J Neuropsychopharmacol. 2010, 13: 461-473.
Carboni L, Piubelli C, Pozzato C, Astner H, Arban R, Righetti PG, Hamdan M, Domenici E: Proteomic analysis of rat hippocampus after repeated psychosocial stress. Neuroscience. 2006, 137: 1237-1246.
Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS: Hippocampal volume reduction in major depression. Am J Psychiatry. 2000, 157: 115-118.
Choi JE, Hur W, Jung CK, Piao LS, Lyoo K, Hong SW, Kim SW, Yoon HY, Yoon SK: Silencing of 14-3-3zeta over-expression in hepatocellular carcinoma inhibits tumor growth and enhances chemosensitivity to cis-diammined dichloridoplatium. Cancer Lett. 2011, 303: 99-107.
Leivonen SK, Rokka A, Ostling P, Kohonen P, Corthals GL, Kallioniemi O, Perala M: Identification of miR-193b targets in breast cancer cells and systems biological analysis of their functional impact. Mol Cell Proteomics. 2011, 10: M110.005322.
Zhang Z, Luo X, Ding S, Chen J, Chen T, Chen X, Zha H, Yao L, He X, Peng H: MicroRNA-451 regulates p38 MAPK signaling by targeting of Ywhaz and suppresses the mesangial hypertrophy in early diabetic nephropathy. FEBS Lett. 2012, 586: 20-26.
Li XM, Li CC, Yu SS, Chen JT, Sabapathy K, Ruan DY: JNK1 contributes to metabotropic glutamate receptor-dependent long-term depression and short-term synaptic plasticity in the mice area hippocampal CA1. Eur J Neurosci. 2007, 25: 391-396.
Clarke M, Pentz R, Bobyn J, Hayley S: Stressor-like effects of c-Jun N-terminal kinase (JNK) inhibition. Plos One. 2012, 7: e44073.
Brust TB, Cayabyab FS, MacVicar BA: C-Jun N-terminal kinase regulates adenosine A1 receptor-mediated synaptic depression in the rat hippocampus. Neuropharmacology. 2007, 53: 906-917.
Miller AH, Raison CL: Cytokines, p38 MAP kinase and the pathophysiology of depression. Neuropsychopharmacology. 2006, 31: 2089-2090.
Maes M, Yirmyia R, Noraberg J, Brene S, Hibbeln J, Perini G, Kubera M, Bob P, Lerer B, Maj M: The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis. 2009, 24: 27-53.
Higgs BW, Elashoff M, Richman S, Barci B: An online database for brain disease research. BMC Genomics. 2006, 7: 70.
Chana G, Lucero G, Salaria S, Lozach J, Du P, Woelk C, Everall I: Upregulation of NRG-1 and VAMP-1 in human brain aggregates exposed to clozapine. Schizophr Res. 2009, 113: 273-276.
Fung SJ, Webster MJ, Weickert CS: Expression of VGluT1 and VGAT mRNAs in human dorsolateral prefrontal cortex during development and in schizophrenia. Brain Res. 2011, 1388: 22-31.
Munakata K, Iwamoto K, Bundo M, Kato T: Mitochondrial DNA 3243A > G mutation and increased expression of LARS2 gene in the brains of patients with bipolar disorder and schizophrenia. Biol Psychiatry. 2005, 57: 525-532.
Webster MJ, Elashoff M, Weickert CS: Molecular evidence that cortical synaptic growth predominates during the first decade of life in humans. Int J Dev Neurosci. 2011, 29: 225-236.
Yamada M, Takahashi K, Tsunoda M, Nishioka G, Kudo K, Ohata H, Kamijima K, Higuchi T, Momose K, Yamada M: Differential expression of VAMP2/synaptobrevin-2 after antidepressant and electroconvulsive treatment in rat frontal cortex. Pharmacogenomics J. 2002, 2: 377-382.
Saito S, Takahashi N, Ishihara R, Ikeda M, Suzuki T, Kitajima T, Yamanouchi Y, Iwata N, Yamada M, Yoshida K, Inada T, Ozaki N: Association study between vesicle-associated membrane protein 2 gene polymorphisms and fluvoxamine response in Japanese major depressive patients. Neuropsychobiology. 2006, 54: 226-230.
Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G, Byrne EM, Blackwood DH, Boomsma DI, Cichon S, Heath AC, Holsboer F, Lucae S, Madden PA, Martin NG, McGuffin P, Muglia P, Noethen MM, Penninx BP, Pergadia ML, Potash JB, Rietschel M, Lin D, Müller-Myhsok B, Shi J, Steinberg S, Grabe HJ, Lichtenstein P, Magnusson P, Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium, et al: A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013, 18: 497-511.
Tansey KE, Guipponi M, Perroud N, Bondolfi G, Domenici E, Evans D, Hall SK, Hauser J, Henigsberg N, Hu X, Jerman B, Maier W, Mors O, O'Donovan M, Peters TJ, Placentino A, Rietschel M, Souery D, Aitchison KJ, Craig I, Farmer A, Wendland JR, Malafosse A, Holmans P, Lewis G, Lewis CM, Stensbøl TB, Kapur S, McGuffin P, Uher R: Genetic predictors of response to serotonergic and noradrenergic antidepressants in major depressive disorder: a genome-wide analysis of individual-level data and a meta-analysis. PLoS Med. 2012, 9: e1001326.
Keers R, Uher R, Gupta B, Rietschel M, Schulze TG, Hauser J, Skibinska M, Henigsberg N, Kalember P, Maier W, Zobel A, Mors O, Kristensen AS, Kozel D, Giovannini C, Mendlewicz J, Kumar S, McGuffin P, Farmer AE, Aitchison KJ: Stressful life events, cognitive symptoms of depression and response to antidepressants in GENDEP. J Affect Disord. 2010, 127: 337-342.
Piva R, Belardo G, Santoro MG: NF-kappaB: a stress-regulated switch for cell survival. Antioxid Redox Signal. 2006, 8: 478-486.
Pace TW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH, Heim CM: Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry. 2006, 163: 1630-1633.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1741-7015/12/73/prepub
Acknowledgements
The Genome-Based Therapeutic Drugs for Depression study was funded by a European Commission Framework 6 grant, EC Contract Ref: LSHB-CT-2003–503428. The Biomedical Research Centre for Mental Health at the Institute of Psychiatry, King’s College London and South London, and Maudsley National Health Service Foundation Trust (funded by the National Institute for Health Research, Department of Health, United Kingdom) and GlaxoSmithKline contributed by funding add-on projects at the London Centre. Dr. Uher is supported by the Canada Research Chairs program (http://www.chairs-chaires.gc.ca/). RK is supported by an MRC Population Health Scientist Award (MR/K021281/1).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
Prof. Peter McGuffin, Dr. Enrico Domenici and Dr. Lucia Carboni have received consultancy fees and honoraria for participating in expert panels from pharmaceutical companies, including Roche and GlaxoSmithKline. All other authors report no biomedical financial interests or potential conflicts of interest.
Authors’ contributions
KM and RK analyzed the data and conceived the study. MGT and AL contributed to the analyses and methods section. ED and LC conducted the study on the Flinders rats and contributed to the revisions of the manuscript. RU contributed to the design. PM is the GENDEP PI and contributed to the revision and LS conducted the mouse study and revised the manuscript. All authors read and approved the final version of the manuscript.
Karim Malki, Robert Keers contributed equally to this work.
Electronic supplementary material
12916_2013_967_MOESM1_ESM.png
Additional file 1: Figure S1: Gene network obtained from genes differentially expressed in response to the UCMS protocol. The most significant network returned from the Ingenuity Pathway Analysis software for genes differentially expressed in response to Unpredictable Chronic Mild Stress. The network consists of 29 reference molecules and is significantly associated with cell stress response. (PNG 2 MB)
12916_2013_967_MOESM2_ESM.png
Additional file 2: Figure S2: Second gene network obtained from genes differentially expressed in response to the UCMS protocol. A second significant network with a score >42 returned by the Ingenuity Pathway Analysis software for genes differentially expressed in the mouse study in response to the Unpredictable Chronic Mild Stress protocol. The pathway includes 23 reference molecules and it is also associated with cell stress response. The pathway is centered on the ELK complex hub and is of particular interest as it shows the VAMP-2 complex and its association with the N-type calcium channel and potassium channel. (PNG 2 MB)
12916_2013_967_MOESM3_ESM.png
Additional file 3: Figure S3: Gene network obtained from genes differentially expressed in response to the Maternal Separation protocol. The most significant network returned from the Ingenuity Pathway Analysis software for genes differentially expressed in response to the maternal separation depressogenic protocol. Of particular interest is the presence of the Yhwaz reference molecule. The Yhwaz gene has been systematically uncovered across several animal studies and been shown to influence neurotransmission of dopamine by regulating exocytosis or phosphorylation of synaptic proteins. The pathway consists of 29 reference molecules and is associated with cell stress response. (PNG 2 MB)
12916_2013_967_MOESM4_ESM.png
Additional file 4: Figure S4: Second Gene network obtained from genes differentially expressed in response to the Maternal Separation protocol. Second pathway returned from the Ingenuity Pathway Analysis software for genes differentially expressed in response to the maternal separation protocol in mouse. This pathway includes 27 reference molecules and is centered on the NF-κB hub. The pathway is associated with cell proliferation and the NF-κB hub has been previously found to be associated with inflammation. Activation of the NF-κB transcription family, by nuclear translocation of cytoplasmic complexes, plays a central role in inflammation [68]. Human studies have shown that MDD patients with increased early life stress exhibit enhanced inflammatory responsiveness to psychosocial stress [69]. (PNG 2 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Malki, K., Keers, R., Tosto, M.G. et al. The endogenous and reactive depression subtypes revisited: integrative animal and human studies implicate multiple distinct molecular mechanisms underlying major depressive disorder. BMC Med 12, 73 (2014). https://doi.org/10.1186/1741-7015-12-73
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7015-12-73