
Abstract
The poly (ADP-ribose) polymerase (PARP) family of enzymes plays a critical role in the maintenance of DNA integrity as part of the base excision pathway of DNA repair. PARP1 is overexpressed in a variety of cancers, and its expression has been associated with overall prognosis in cancer, especially breast cancer. A series of new therapeutic agents that are potent inhibitors of the PARP1 and PARP2 isoforms have demonstrated important clinical activity in patients with breast or ovarian cancers that are caused by mutations in either the BRCA1 or 2 genes. Results from such studies may define a new therapeutic paradigm, wherein simultaneous loss of the capacity to repair DNA damage may have antitumor activity in itself, as well as enhance the antineoplastic potential of cytotoxic chemotherapeutic agents.
Similar content being viewed by others
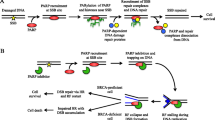
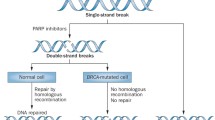

Background
Environmental exposures and cell replication result in DNA damage that is repaired by a variety of mechanisms, including base excision repair (BER), mismatch repair (MMR), nucleotide excision repair (NER), single strand annealing (SSA), homologous recombination (HR), and nonhomologous end joining (NHEJ) [1]. Poly (ADP-ribose) polymerases (PARPs) are a family of proteins involved in DNA repair that utilize the BER pathway [2] and share enzymatic and scaffolding properties. PARP1 and PARP2 are the best studied members of this family of enzymes. PARP1 has three domains that are responsible for DNA-binding, automodification, and catalysis. DNA cleavage results in the recruitment and binding of PARP1 to the site of damage, with an increase in its catalytic activity, and the formation of long, branched, poly (ADP-ribose) (PAR) chains. PAR has a net negative charge that promotes recruitment of DNA repair proteins involved in the BER pathway to the site of DNA damage, and facilitates removal of PARP1 from damage sites, allowing access to other repair proteins. Apart from its role in BER, PARP1 has been implicated in the HR and NHEJ pathways, suggesting a broader role for this enzyme family in the overall DNA repair process.
PARPs were initially identified in 1963; the potential for PARP inhibition to enhance DNA damage caused by cytotoxic chemotherapy was first considered in 1980 [3, 4]. PARP1 is overexpressed in a variety of cancers and its expression has been associated with overall prognosis in cancer, especially breast cancer [5]. PARP inhibitors in clinical development mimic the nicotinamide moiety of nicotinamide adenine dinucleotide, and bind to the enzyme's catalytic domain, inhibiting automodification and subsequent release of the enzyme from the site of DNA damage. In so doing, PARP inhibitors also prevent access of other repair proteins to the site of DNA cleavage.
Several PARP inhibitors are in clinical development (see Additional Files 1 and 2); as a whole, these agents have generated considerable interest because of their potential clinical activity for patients whose tumors harbor defects in the HR pathway [6–8]. Although several of these drugs have been shown to inhibit PARP in vivo, their spectrum of activity and effects on DNA repair pathways make them distinct. This review summarizes current insights into the mechanism of action, recent clinical trials, and potential next steps in the evaluation of this promising class of anti-cancer drugs.
Mechanism of action and pharmacology of PARP inhibitors
A number of PARP inhibitors are under clinical development: rucaparib (CO-338; AG014699, PF-0367338; oral/IV), iniparib (BSI-201), olaparib (AZD-2281; oral), veliparib (ABT-888; oral), MK-4827, BMN-673, CEP-9722 (oral) and E7016 (GPI 21016, oral). The loss of BER capacity produced by PARP inhibition has prompted the evaluation of these drugs as potential enhancers of DNA damaging cytotoxic chemotherapeutic agents such as alkylating agents (for example, platinum, cyclophosphamide) and topoisomerase 1 inhibitors (for example, camptothecin analogs) [9]. However, recent studies strongly suggest that, unlike the other drugs, the mechanism of action of iniparib is unclear and is probably not related to PARP inhibition per se [10].
PARP inhibition enhances the therapeutic index of cytotoxic chemotherapy only if DNA damage is selectively increased in tumor compared to normal tissues, such as the gastrointestinal mucosa or bone marrow. The opportunity to achieve selectivity in tumor cell killing with these agents would, therefore, be improved in tumors that already harbor DNA repair defects. Simultaneous dysfunction of two DNA damage repair (DDR) pathways, termed 'synthetic lethality', decreases the ability of tumor cells to withstand the DNA damage produced during normal cellular replication [8]. Duplication of this phenomenon pharmacologically is possible in tumors harboring somatic or germline defects in a non-BER pathway of DDR by treating with a PARP inhibitor so that BER and non-BER pathways are blocked simultaneously. Clinical development programs are testing this idea directly in settings where the HR pathway is compromised, for example, with PARP inhibitor monotherapy for tumors with BRCA1/2 defects. This could also be extended to include treatment of tumors with defects in other HR pathway proteins. For instance, PTEN-deficient cells have been shown to be sensitive to PARP inhibition, due to the role of PTEN in the expression of RAD51 [11]. One question for the further development of PARP inhibitors is whether they can effectively enhance DNA damage, in the presence of DNA damaging agents, in tumors that lack an intrinsic defect in DDR.
New data are emerging on the myriad of effects of PARP on DNA repair and other pathways (see Additional File 3). PARP has also been implicated in DNA repair by recruiting mitotic recombination 11 (MRE11) and ataxia telangiectasia-mutated (ATM) to DNA DSBs; effects on BRCA 1 and RAD 51 expression via repressive E2F4 and p130 complexes; interaction with the DNA protein kinase (DNA PK) complex [Ku70, Ku80, DNA PK] involved in NHEJ of DSBs; and the epigenetic regulation of chromatin structure [2, 12–15]. Recent reports evaluate the role of PARP in BER and its interaction with DNA single strand break intermediate forms. A differential effect of repair of SSBs has been demonstrated in the presence of PARP inhibitors as compared to PARP1 siRNA cells treated with the alkylating agent, dimethyl sulfate [16].
Pharmacodynamics of PARP inhibitors on PARP1 and PARP2
Assays have been developed to quantify drug-induced inhibition of PARP enzymatic activity in patient specimens. The primary effect of PARP inhibitors changes two parameters, each of which could be used as a pharmacodynamic endpoint: decreased PARP1/2-specific activity and decreased production of PARP1/2 reaction products, which are poly-ADP-ribosylated macromolecules ('PARylated substances'). However, a major concern with any ex vivo enzymatic assay is the dilution of the extract with sample processing and enzyme assay buffers. Diluting a tissue sample also dilutes the concentration of the competitive PARP inhibitor that was present at the time of sample collection. Under conditions of linear enzyme kinetics, the measured enzymatic activity may be more an indication of the resulting drug concentration in each diluted sample, rather than a measure of enzymatic activity originally present in the tissue in the face of actual tissue concentrations of the drug.
On the other hand, measurement of PARylated substances produced by PARP1/2 activity reflects the balance between degradation of those PARylated molecules by an enzyme called poly (ADP-ribose) glycohydrolase (PARG), and production by PARP1/2. A sandwich immunoassay (IA) was developed and validated at the US National Cancer Institute to quantify the level of poly-ADP-ribosylated macromolecules from a calibration curve of poly-ADP-ribose standard ('PAR antigen') [17]. This PAR-IA was designed for the first-in-human use of veliparib, where the measurement of PARylated substances would be the primary objective of a Phase '0' clinical trial. Sufficient assay sensitivity was required to distinguish a 30% decrease in the PARP1/2 reaction product PAR, with a lower limit of quantitation sufficient to quantify a 90% drop in PAR relative to baseline in approximately 85% of paired mononuclear cell samples. Prior to the clinical trial, a fit-for-purpose study was conducted in mice harboring human tumor xenografts to model the proposed use of the PAR IA. The results proved that veliparib significantly decreased PAR levels from baseline by four to seven hours after a single oral dose - the timeframe planned for the clinical biopsies. The results of the subsequent first-in-human clinical trial confirmed the findings of the fit-for-purpose animal modeling studies [18].
The PAR IA has been used to define a reproducible response of PARP1/2 to veliparib in tumor biopsies and mononuclear cell samples from treated patients. In addition, the PAR IA has been used to confirm pharmacodynamic effects, similar to those observed with veliparib, produced by olaparib and MK-4827 in human tumor xenografts and human tumor cell lines in vitro. However, iniparib and its two major metabolites failed to cause any change in the level of PARylated substances in the model systems, as measured by the PAR-IA [10]. The apparent lack of a pharmacodynamic effect by iniparib on PARP1/2 is very different from the responses elicited by veliparib, olaparib, and MK-4827. This may explain the recently reported lack of an effect of iniparib on the efficacy and toxicity of combination chemotherapy for triple negative breast cancer (TNBC) patients in a properly powered Phase III clinical trial [19].
Clinical experience with PARP inhibitors
PARP inhibitors have been evaluated in clinical trials either as single agents, with an emphasis on patients carrying BRCA mutations, or in combination with DNA damaging therapies (see Additional Files 1 and 2). Olaparib has demonstrated single agent activity in breast or ovarian cancer patients with germline mutations in BRCA1/2 [20–22]; an over 40% response rate has been reported in patients with BRCA mutant ovarian cancer, especially in patients with platinum sensitive disease [23].
The Cancer Genome Atlas Research Project recently reported on the molecular aberrations in high grade serous ovarian adenocarcinoma, demonstrating a defect in the HR pathway in half of the 489 tumors analyzed [24]. These results suggest that ovarian cancer patients with sporadic abnormalities in the HR pathway impairing DNA repair might benefit from treatment with PARP inhibitors. Similar abnormalities in DNA repair pathways have been reported in primary peritoneal cancers, and in patients with TNBC, forming the basis for recent clinical trials that have explored the use of PARP inhibitors in such patient populations [25–28]. Tumor types with defects in other DNA repair pathways, such as tumors with microsatellite instability, may also be susceptible to inhibition of the BER pathway [29].
Despite the evaluation of PARP inhibition in a number of clinical trials, the degree and duration of inhibition required for optimal clinical benefit has yet to be established [18]. This has resulted in the continuation of studies that have explored higher PARP inhibitor doses, well beyond those demonstrated to result in near-complete inhibition of PARP activity in clinical tumor samples; the results of some of these trials, such as the ICEBERG study, have suggested a dose response for deriving clinical benefit from PARP inhibitors [21, 22, 30].
Conclusions and outlook for the use of PARP inhibitors in the future
A major focus for the future clinical development of PARP inhibitors is to determine whether or not potentiating chemotherapy- or radiation-induced DNA damage in patients without known defects in DDR is either possible or fruitful. Enhancement of DNA damage by the addition of a PARP inhibitor to a topoisomerase I poison has been demonstrated in tumor biopsies and circulating tumor cells by measurement of γH2AX foci, a marker of DNA double strand breaks, in patients treated with veliparib and topotecan compared to those receiving topotecan alone [31]. However, the development of PARP inhibitors as chemopotentiating agents has been limited by an increase in observed toxicities, mainly myelosuppression, necessitating dose reductions of the cytotoxic chemotherapeutic agent and the PARP inhibitor [31, 32]. This raises the question of whether administering the combination is more efficacious than administering full doses of the chemotherapeutic agent alone, as well as the need to design clinical trial strategies to improve the therapeutic index of these combinations. It seems likely that to optimize the use of PARP inhibitors in the future will require the development of predictive assays to determine the presence of unsuspected defects in DDR pathways in tumors. It also presents an opportunity to rationally develop combinations of PARP inhibitors with new classes of DNA repair inhibitors that are on the horizon, and classical cytotoxic agents [33].
Abbreviations
- ATM:
-
ataxia telangiectasia-mutated
- BER:
-
base excision repair
- DDR:
-
DNA damage repair
- DNA PK:
-
DNA protein kinase
- DSBs:
-
double strand breaks
- HR:
-
homologous recombination
- MMR:
-
mismatch repair
- MRE11:
-
mitotic recombination 11
- NER:
-
nucleotide excision repair
- NHEJ:
-
non-homologous end joining
- PARG:
-
poly (ADP-ribose) glycohydrolase
- PARP:
-
poly (ADP-ribose) polymerase
- TNBC:
-
triple negative breast cancer
References
Sharova NP: How does a cell repair damaged DNA?. Biochemistry (Mosc). 2005, 70: 275-291. 10.1007/s10541-005-0113-4.
Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG: PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010, 10: 293-301. 10.1038/nrc2812.
Chambon P, Weill JD, Mandel P: Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963, 11: 39-43. 10.1016/0006-291X(63)90024-X.
Durkacz BW, Omidiji O, Gray DA, Shall S: (ADP-ribose)n participates in DNA excision repair. Nature. 1980, 283: 593-596. 10.1038/283593a0.
Rojo F, Garcia-Parra J, Zazo S, Tusquets I, Ferrer-Lozano J, Menendez S, Eroles P, Chamizo C, Servitja S, Ramirez-Merino N, Lobo F, Bellosillo B, Corominas JM, Yelamos J, Serrano S, Lluch A, Rovira A, Albanell J: Nuclear PARP-1 protein overexpression is associated with poor overall survival in early breast cancer. Ann Oncol. 2011.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T: Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005, 434: 913-917. 10.1038/nature03443.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A: Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005, 434: 917-921. 10.1038/nature03445.
Iglehart JD, Silver DP: Synthetic lethality-a new direction in cancer-drug development. N Engl J Med. 2009, 361: 189-191. 10.1056/NEJMe0903044.
Zhang YW, Regairaz M, Seiler JA, Agama KK, Doroshow JH, Pommier Y: Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011, 39: 3607-3620. 10.1093/nar/gkq1304.
Ji J, Lee MP, Kadota M, Zhang Y, Parchment RE, Tomaszewski JE, Doroshow JH: Pharmacodynamic and pathway analysis of three presumed inhibitors of poly (ADP-ribose) polymerase: ABT-888, AZD2281, and BSI201[abstract]. Proceedings of the AACR 102nd Annual Meeting. 2011, Orlando, FL, 52: 1080.
Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ, Ashworth A: Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009, 1: 315-322. 10.1002/emmm.200900041.
Caiafa P, Guastafierro T, Zampieri M: Epigenetics: poly(ADP-ribosyl)ation of PARP-1 regulates genomic methylation patterns. FASEB J. 2009, 23: 672-678. 10.1096/fj.08-123265.
Hegan DC, Lu Y, Stachelek GC, Crosby ME, Bindra RS, Glazer PM: Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc Natl Acad Sci USA. 2010, 107: 2201-2206. 10.1073/pnas.0904783107.
Patel AG, Sarkaria JN, Kaufmann SH: Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci USA. 2011, 108: 3406-3411. 10.1073/pnas.1013715108.
Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G: PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34: 6170-6182. 10.1093/nar/gkl840.
Strom CE, Johansson F, Uhlen M, Szigyarto CA, Erixon K, Helleday T: Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39: 3166-3175. 10.1093/nar/gkq1241.
Kinders RJ, Hollingshead M, Khin S, Rubinstein L, Tomaszewski JE, Doroshow JH, Parchment RE: Preclinical modeling of a phase 0 clinical trial: qualification of a pharmacodynamic assay of poly (ADP-ribose) polymerase in tumor biopsies of mouse xenografts. Clin Cancer Res. 2008, 14: 6877-6885. 10.1158/1078-0432.CCR-08-0214.
Kummar S, Kinders R, Gutierrez ME, Rubinstein L, Parchment RE, Phillips LR, Ji J, Monks A, Low JA, Chen A, Murgo AJ, Collins J, Steinberg SM, Eliopoulos H, Giranda VL, Gordon G, Helman L, Wiltrout R, Tomaszewski JE, Doroshow JH: Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol. 2009, 27: 2705-2711. 10.1200/JCO.2008.19.7681.
Domchek SM, Mitchell G, Lindeman GJ, Tung NM, Balmana J, Isakoff SJ, Schmutzler R, Audeh MW, Loman N, Scott C, Friedlander M, Kaufman B, Garber JE, Tutt A, Robson ME: Challenges to the Development of New Agents for Molecularly Defined Patient Subsets: Lessons From BRCA1/2-Associated Breast Cancer. J Clin Oncol. 2011, 29: 4224-4226. 10.1200/JCO.2011.36.8134.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009, 361: 123-134. 10.1056/NEJMoa0900212.
Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, Lu K, Schmutzler RK, Matulonis U, Wickens M, Tutt A: Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010, 376: 245-251. 10.1016/S0140-6736(10)60893-8.
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J: Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010, 376: 235-244. 10.1016/S0140-6736(10)60892-6.
Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De GJ, Lubinski J, Shanley S, Messiou C, A'Hern R, Tutt A, Ashworth A, Stone J, Carmichael J, Schellens JH, de Bono JS, Kaye SB: Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010, 28: 2512-2519. 10.1200/JCO.2009.26.9589.
The Cancer Genome Atlas Research Network: Integrated genomic analyses of ovarian carcinoma. Nature. 2011, 474: 609-615. 10.1038/nature10166.
Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J, Oza A: Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12: 852-861. 10.1016/S1470-2045(11)70214-5.
Dent RA, Lindeman GJ, Clemons M, Wildiers H, Chan A, McCarthy NJ, Singer CF, Lowe ES, Kemsley K, Carmichael J: Safety and efficacy of the oral PARP inhibitor olaparib (AZD 2281) in combination with paclitaxel for the first- or second-line treatment of patients with metastatic triple-negative breast cancer: results from the safety cohour of a phase I/II multicenter trial. [abstract]. J Clin Oncol. 2010, 28 (Suppl.1): 118s.
Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote IB, Rustin GJS, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Macpherson E, Watkins C, Carmichael J, Matulonis U: Phase II randomized placebo-controlled study of olaparib (AZD2281) in patients with palatinum-sensitive relapsed serous ovarian cancer (PSROC) [abstract]. J Clin Oncol. 2011, 29 (Suppl): 5003.
Sandu SK, Wenham RM, Wilding G, McFadden M, Sun L, Toniatti C, Stroh M, Carpenter CL, DeBono JS, Schelman WR: First-in-human trial of a poly(ADP-ribose) polymerase (PARP) inhibitor MK-4827 in advanced cancer patients (pts) with antitumor activity in BRCA-deficient and sporadic ovarian cancers [abstract]. J Clin Oncol. 2010, 28 (Suppl 1): 233.
Leichman LP, Cohen SJ, Hochster HS, Messersmith WA, Lenz H, Boman BM, Gold PJ, O'Neill BH, Berlin J, Carmichael J: A phase II trial to assess the single-agent efficacy and safety of the PARP inhibitor olaparib (O) in previously treated patients (pts) with metastatic, measurable colorectal cancer (mCRC) stratified by microsatellite status (MSs) [abstract]. Proceedings of the ASCO-NCI-EORTC Annual Meeting on Molecular Markers in Cancer. 2010, Abstract No. 118
Kaye S, Kaufman B, Lubinski J, Matulonis U, Gourley C, Karlan B, Taylor D, Wickens M, Carmichael J: Phase II study of the oral PARP inhibitor olaparib (AZD2281) versus liposomal doxorubicin in ovarian cancer patients with BRCA1 and/or BRCA2 mutations [abstract]. Ann Oncol. 2010, 21 (Suppl 8): vii304.
Kummar S, Chen A, Ji J, Zhang Y, Reid JM, Ames M, Jia L, Weil M, Speranza G, Murgo AJ, Kinders R, Wang L, Parchment RE, Carter J, Stotler H, Rubinstein L, Hollingshead M, Melillo G, Pommier Y, Bonner W, Tomaszewski JE, Doroshow JH: Phase I study of ABT-888, a PARP inhibitor, in combination with topotecan hydrochloride in adults with refractory solid tumors and lymphomas. Cancer Res. 2011, 71: 5626-5634. 10.1158/0008-5472.CAN-11-1227.
Giaccone G, Rajan A, Kelly RJ, Gutierrez M, Kummar S, Yancey M, Ji JJ, Zhang Y, Parchment RE, Doroshow JH: A phase I combination study of olaparib (AZD2281;KU-0059436) and cisplatin (C) plus gemcitabine (G) in adults with solid tumors [abstract]. J Clin Oncol. 2010, 28 (Suppl 15): 3027.
Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS: Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle. 2011, 10: 1192-1199. 10.4161/cc.10.8.15273.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1741-7015/10/25/prepub
Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SK, AC, REP, RK, and JHD jointly wrote the manuscript. All authors were involved in revising the manuscript, and gave final approval. All authors read and approved the final manuscript.
Electronic supplementary material
12916_2011_508_MOESM1_ESM.PDF
Additional File 1: Early phase clinical trials with PARP inhibitors. A table listing clinical trials of PARP inhibitors currently in development in early phase clinical trials. (PDF 247 KB)
12916_2011_508_MOESM2_ESM.PDF
Additional File 2: Clinical trials with PARP inhibitors in defined diseases. A table listing clinical trials of PARP inhibitors currently in development in specified cancers. (PDF 268 KB)
12916_2011_508_MOESM3_ESM.PDF
Additional File 3: Structural and functional characteristics of PARP1. A: Poly(ADP-ribose) polymerase 1 (PARP1) is shown with its DNA-binding (DBD), automodification (AD) and catalytic domains. The PARP signature sequence (yellow box within the catalytic domain) comprises the sequence most conserved among PARPs. Crucial residues for nicotinamide adenine dinucleotide (NAD+) binding (histidine; H and tyrosine; Y) and for polymerase activity (glutamic acid; E) are indicated. B: Consequences of PARP1 activation by DNA damage. Although not shown to simplify the scheme, PARP1 is active in a homodimeric form. PARP1 detects DNA damage through its DBD. This activates PARP1 to synthesize poly(ADP) ribose (pADPr; yellow beads) on acceptor proteins, including histones and PARP1. Owing to the dense negative charge of pADPr, PARP1 loses affinity for DNA, allowing the recruitment of repair proteins by pADPr to the damaged DNA (blue and purple circles). Poly(ADP-ribose) glycohydrolase (PARG) and possibly ADP-ribose hydrolase 3 (ARH3) hydrolyse pADPr into ADP-ribose molecules and free pADPr. ADP-ribose is further metabolized by the pyrophosphohydrolase NUDiX enzymes into AMP, raising AMP:ATP ratios, which in turn activate the metabolic sensor AMP-activated protein kinase (AMPK). NAD+ is replenished by the enzymatic conversion of nicotinamide into NAD+ at the expense of phosphoribosylpyrophosphate (PRPP) and ATP. Examples of proteins non-covalently (pADPr-binding proteins) or covalently poly(ADP-ribosyl)ated are shown with the functional consequences of modification. It is important to note that many potential protein acceptors of pADPr remain to be identified owing to the difficulty of purifying pADPr-binding proteins in vivo. PARP inhibitors prevent the synthesis of pADPr and hinder subsequent downstream repair processes, lengthening the lifetime of DNA lesions. ATM, ataxia telangiectasia-mutated; BER, base excision repair; BRCT, BRCA1 carboxy-terminal repeat motif; DNA-PKcs, DNA-protein kinase catalytic subunit; DSB, double-strand break; HR, homologous recombination; NHEJ, non-homologous end joining; NLS, nuclear localization signal; PPi, inorganic pyrophosphate; SSB, single-strand break; Zn, zinc finger. Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Cancer [2], copyright (2010). (PDF 249 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kummar, S., Chen, A., Parchment, R.E. et al. Advances in using PARP inhibitors to treat cancer. BMC Med 10, 25 (2012). https://doi.org/10.1186/1741-7015-10-25
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7015-10-25