
Abstract
Background
β1- and β2–adrenergic receptors (ARs) play distinct roles in the heart, e.g. β1AR is pro-contractile and pro-apoptotic but β2AR anti-apoptotic and only weakly pro-contractile. G protein coupled receptor kinase (GRK)-2 desensitizes and opposes βAR pro-contractile signaling by phosphorylating the receptor and inducing beta-arrestin (βarr) binding. We posited herein that GRK2 blockade might enhance the pro-contractile signaling of the β2AR subtype in the heart. We tested the effects of cardiac-targeted GRK2 inhibition in vivo exclusively on β2AR signaling under normal conditions and in heart failure (HF).
Results
We crossed β1AR knockout (B1KO) mice with cardiac-specific transgenic mice expressing the βARKct, a known GRK2 inhibitor, and studied the offspring under normal conditions and in post-myocardial infarction (MI). βARKct expression in vivo proved essential for β2AR-dependent contractile function, as β2AR stimulation with isoproterenol fails to increase contractility in either healthy or post-MI B1KO mice and it only does so in the presence of βARKct. The main underlying mechanism for this is blockade of the interaction of phosphodiesterase (PDE) type 4D with the cardiac β2AR, which is normally mediated by the actions of GRK2 and βarrs on the receptor. The molecular “brake” that PDE4D poses on β2AR signaling to contractility stimulation is thus “released”. Regarding the other beneficial functions of cardiac β2AR, βARKct increased overall survival of the post-MI B1KO mice progressing to HF, via a decrease in cardiac apoptosis and an increase in wound healing-associated inflammation early (at 24 hrs) post-MI. However, these effects disappear by 4 weeks post-MI, and, in their place, upregulation of the other major GRK in the heart, GRK5, is observed.
Conclusions
GRK2 inhibition in vivo with βARKct is absolutely essential for cardiac β2AR pro-contractile signaling and function. In addition, β2AR anti-apoptotic signaling in post-MI HF is augmented by βARKct, although this effect is short-lived.
Similar content being viewed by others

Background
Despite recent advances in prevention and management of heart disease, death due to post-myocardial infarction (MI) heart failure (HF) continues to rise and new treatments are needed [1]. β1- and β2–adrenergic receptors (ARs) are the main stimulatory receptors of cardiac function but are now known to play clearly distinct roles in cardiac physiology and pathology [2–5]. For instance, cardiomyocyte contraction is readily stimulated by β1ARs but not β2ARs and β1AR signaling is generally considered pro-apoptotic whereas β2AR signaling anti-apoptotic in the heart [2–5]. These differences might be explained by differences in the signaling complexes assembled by activation of these two βARs: β1AR forms a complex with phosphodiesterase (PDE) 4D8 directly when inactive, and agonist binding dissociates it [6, 7]. Additionally, β1AR does not readily bind the receptor adapter proteins beta-arrestins (βarrs) following its agonist-promoted phosphorylation by G protein coupled receptor kinases (GRKs), the most prominent members of which in the heart are GRK2 and −5 [8–11]. In contrast, β2AR recruits another PDE variant upon its agonist activation, PDE4D5, via its interaction with βarrs following its GRK-dependent phosphorylation [6, 7, 12–16]. PDE recruitment to the receptor’s signaling complex plays a crucial role in compartmentalizing the cyclic adenosine monophosphate (cAMP) signal and thereby tightly regulating βAR-stimulated contractility [7]. It has been postulated that this PDE4D5 recruitment to the agonist-activated cardiac β2AR poses a “brake” on the β2AR cAMP signaling’s ability to stimulate contractility [6, 7, 13, 15]. By contrast, agonist-promoted dissociation of PDE from the β1AR underlies the more “diffuse” and more powerful at stimulating contraction signaling of this βAR subtype [6, 7]. Of note, cardiac β2AR signaling has been reported to become more “diffuse” and decompartmentalized, i.e. to adopt a β1AR-like signaling pattern, in a rat model of HF, which might underlie cardiac β2AR dysfunction in HF [17].
On the other hand, cardiac β2AR can switch its signaling from Gs protein-mediated to Gi/o protein-mediated, which is believed to underlie its anti-apoptotic effects and is a feature cardiac β1ARs lack [18–24]. Finally, the interactions of the β2AR with the βarrs, which require prior receptor phosphorylation by GRKs, can have pleiotropic effects in cardiac myocytes, such as inhibition of apoptosis/promotion of survival by promoting extracellular signal-regulated kinase (ERK) signaling [25] and inhibition of inflammation by blocking the pro-inflammatory transcription factor nuclear factor-kappaB (NF-κB) [26, 27], a crucial mediator of major pro-inflammatory cytokine expression, such as tumor necrosis factor-alpha (TNF-α) and interleukin-1 and −6 (IL-1 & -6) [28–30]. These βarr-dependent signaling effects may also play some part in the well known and described anti-apoptotic and other beneficial in post-MI HF effects of cardiac β2AR.
Cardiac GRK2 is a major negative regulator of βAR pro-contractile signaling [8–11]. By desensitizing both β1- and β2ARs, i.e. terminating their G protein-mediated signaling through cAMP, it dramatically reduces cardiac inotropic and adrenergic reserves, and since it is markedly elevated in HF, its blockade represents an attractive therapeutic strategy for heart disease treatment [8–11, 31–35]. Given that GRK2 can block the pro-contractile and other beneficial signaling of cardiac β2AR in HF, and also that its action on β2AR induces recruitment of βarrs with all their aforementioned myriad effects on this receptor’s signaling, we hypothesized, in the present study, that cardiac GRK2 blockade in vivo might enhance β2AR signaling post-MI. In order to study the effects of GRK2 blockade specifically on this subtype’s signaling, without any interference by the qualitatively different β1AR signaling, we utilized the β1AR knockout (B1KO) mice [36], which we crossed with mice overexpressing the known GRK2 inhibitor mini-gene βARKct (or GRK2ct) specifically in cardiac myocytes [32]. After breeding the offspring to homozygosity, we studied them both under normal conditions (i.e. healthy, sham-operated animals) and after surgically induced MI to induce HF. We found that GRK2 inhibition in vivo is absolutely necessary for the β2AR to be capable of increasing contractility. In addition, β2AR anti-apoptotic signaling post-MI is augmented by βARKct, but only acutely.
Results
βARKct restores cardiac β2AR-dependent pro-contractile signaling by reducing the interaction of PDE4D with the receptor
We developed the hybrid transgenic line βARKct/B1KO by crossing the B1KO mice with the βARKct transgenic mice, which express βARKct only in cardiomyocytes. The βARKct/B1KO’s breed normally, without any gross abnormalities and present no overt cardiovascular or other phenotype (data not shown). Three-month-old male mice were chosen to undergo surgical MI in order to induce HF and were studied alongside age-matched male homozygous B1KO’s (without βARKct expression), which served as the control group.
Since GRK2 is a major (negative) regulator of cardiac βAR-dependent contractility in vivo, and the β2AR stimulates contractility only very weakly, we first examined the cardiac function parameters of these mice, both in sham and post-MI groups. Echocardiography revealed that the B1KO mice display significantly decreased ejection fraction compared to control wild type (WT) mice, both under normal conditions (sham groups) and at 4 weeks post-MI, as expected since the β1AR is the major βAR subtype in the heart stimulating contractility (Table 1) [8]. Notably, βARKct overexpression led to significant augmentation of the ejection fraction of the B1KO mice, up to the levels of WT mice, again both in normal and in 4 week post-MI mice (Table 1), while, as already known from our studies in the past [10, 11, 32, 33], βARKct significantly augments contractility of the WT mice, as well (Table 1). Importantly, when the mice underwent in vivo cardiac catheterization to measure their hemodynamic responses to isoproterenol stimulation (a standard βAR full agonist), B1KO mice, remarkably, completely failed to show any increase in contractility (as measured by the +dP/dtmax LV pressure elevation parameter), even at the highest concentration of isoproterenol challenge (Max. Iso, Table 1). In contrast, the hybrid βARKct/B1KO mice showed very good contractile responses to isoproterenol, both in the sham (healthy) conditions and in post-MI HF (Table 1). As expected, the other two mouse lines, i.e. WT and βARKct, were responsive to βAR stimulation, with the HF animals in these groups showing somewhat reduced responses compared to their sham counterparts and the βARKct line displaying much more robust responses compared to the WT group (Table 1). These results strongly indicate that cardiac GRK2 is a major opposing force for the β2AR pro-contractile function and only when its activity is blocked (e.g. in the presence of βARKct) is the cardiac β2AR capable of promoting contractility in response to agonist stimulation.
To identify the main signaling mechanism underlying this dramatic effect of βARKct on cardiac β2AR-dependent contractiity, we examined the levels of PDE4D interaction with the β2AR in cardiac membranes of these mice in vivo. As shown in Figures 1A and 1B, the interaction of cardiac β2AR with both the PDE4D3 and -D5 isoforms is significantly reduced in βARKct/B1KO mouse hearts compared to control B1KO hearts, an effect that might enable βARKct to enhance cardiac β2AR-dependent pro-contractile signaling in vivo.

β2AR-PDE4D interaction in the heart. Co-immunoprecipitation (co-IP) followed by western blotting in cardiac extracts from normal (sham) B1KO and βARKct/B1KO (CT/B1KO) mice to measure the β2AR-PDE4D interaction in the heart. Representative immunoblots are shown in (A), and the amounts of the co-immunoprecipitated PDE4D isoforms, as measured by densitometry and normalized with the amount of β2AR pulled down in the co-IP, are shown in (B). *, p<0.05, vs. B1KO; n=4 independent experiments (i.e. performed on 4 different hearts from each mouse line). IP: Immunoprecipitation, IB: Immunoblotting.
βARKct and cardiac β2AR-dependent anti-apoptotic/inflammatory signaling
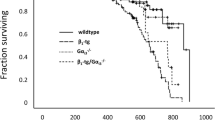
Next, we examined the impact of βARKct expression on the other major aspect of cardiac β2AR signaling, anti-apoptosis/cardiac survival. Post-MI βARKct/B1KO mice display markedly better survival post-MI compared to their B1KO counterparts (Figure 2A). Kaplan-Meier survival curves indicated that at 4 weeks post-MI, a significant (~70%) of βARKct/B1KO’s are still alive, compared to only ~40% of B1KO’s at the same time point post-MI (Figure 2A). In addition, cardiac apoptosis is found significantly decreased very early (at 24 hrs) post-MI in the βARKct/B1KO hearts compared to control B1KO hearts (Figure 2B) but similar between the two groups at 4 weeks post-MI (Figure 2B), indicating that this reduction in post-MI apoptosis induced by βARKct is short-lived. As for post-MI cardiac inflammation in the two animal groups, levels of the major pro-inflammatory cytokines TNFα(Figure 2C), IL-6 (Figure 2D) and IL-1β (Figure 2E) are significantly increased in the hearts of βARKct/B1KO mice, compared to control B1KO hearts at 24 hrs post-MI, indicating increased wound (infarct) healing-associated inflammation. By 4 weeks post-MI however, levels of all these three cytokines (TNFα, IL-6, IL-1β) in βARKct/B1KO hearts have returned to the levels of 24-hour post-MI B1KO hearts (data not shown), indicating that also the effect of βARKct on post-MI inflammation is short-lived.

Survival, cardiac apoptosis and inflammation post-MI. (A) Kaplan-Meier survival curves of the 4 groups of mice of the study: sham-operated (Sham) and post-MI (MI) B1KO and βARKct/B1KO mice. p=0.011 between MI B1KO and MI βARKct/B1KO; n=15 mice/group for sham, 37 mice/group for MI mice. (B) Apoptotic cell death at 24 hrs and at 4 weeks (wks) post-MI in the two transgenic (B1KO and CT/B1KO) lines, as measured by TUNEL performed in the border zone of the infarct. No difference in rate of apoptosis in the remote zone at either post-MI time point was found (data not shown). **, p<0.05, vs. B1KO 24 hrs, n=6 mice/group. (C-E) Levels of pro-inflammatory cytokines TNFα (C), IL-6 (D), and IL-1β (E), measured via ELISA in serum of intra-cardiac blood from B1KO and βARKct/B1KO (CT/B1KO) mice at 24 hrs post-MI. *, p<0.05, n=5 mice/group.
To identify potential signaling mechanisms underlying these effects of βARKct on apoptosis and inflammation in post-MI B1KO hearts, we examined protein levels of the major anti-apoptotic mediator Bcl-2 [37] and levels of activation of the crucial pro-inflammatory transcription factor NFκB. Bcl-2 was found significantly up-regulated in βARKct/B1KO hearts compared to control B1KO hearts at 24 hrs post-MI (Figures 3A and 3B), indicating enhanced cellular survival/inhibition of apoptosis. However, at 4 weeks post-MI, Bcl-2 protein was virtually undetectable in the hearts of both mouse lines (Figure 3C), which is consistent with the phenotypic finding of the short-lived inhibition of apoptosis in the heart by βARKct (Figure 2B). In addition, NFκB activation appears also markedly elevated in βARKct/B1KO hearts compared to control B1KO hearts at 24 hrs post-MI (Figures 3A and 3B), indicating enhanced cardiac inflammation. For NFκB to get activated, its inhibitory IκBα subunit must be phosphorylated and subsequently targeted for proteasomal degradation to release the transcriptionally active subunits of the complex [28–30]. Thus, increased phosphorylation of IκBα and decreased levels (increased degradation) of total IκBα in the hybrid transgenic hearts at 24 hrs post-MI (Figures 3A and 3B) suggest increased NFκB activation compared to B1KO hearts. Finally, blotting for βARKct in these hearts confirmed the robust expression of this GRK2 inhibitor in the hearts of βARKct/B1KO’s, which, of course, was absent from the hearts of B1KO mice (Figure 3A).

Levels of cardiac Bcl-2, NFκB activity and GRK5 post-MI. (A-B) Western blotting in total cardiac extracts from 24 hr post-MI B1KO and βARKct/B1KO (CT/B1KO) mice for βARKct, Bcl-2, phospho-IκBα (pIκBα), and total IκBα. Representative blots are shown in (A), including blots for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as loading control for each protein tested, and densitometric quantitation, normalized with GAPDH as control and expressed as % of B1KO levels, of 4 independent experiments done in duplicate, is shown in (B). *, p<0.05, vs. B1KO. (C) Western blotting for Bcl-2 protein in total cardiac extracts from 4 week post-MI B1KO and βARKct/B1KO (CT/B1KO) mice. Representative blots from 4 independent experiments done in duplicate are shown, including GAPDH as loading control and h9c2 cell extract as positive control (input) for Bcl-2. Bcl-2 was virtually undetectable in either group at 4 weeks post-MI. (D) Western blotting for GRK5 in total cardiac extracts from B1KO and βARKct/B1KO (CT/KO) mice at 24 hrs and at 4 weeks (wks) post-MI. Representative blots of 4 independent experiments done in duplicate, with GAPDH as loading control, are shown on top, and densitometric quantitation on bottom. *, p<0.05, vs. all other groups.
GRK5 and cardiac β2AR-dependent signaling
Apart from GRK2, the other major cardiac GRK that can phosphorylate and desensitize β2ARs, and thus oppose β2AR pro-contractile and anti-apoptotic signaling, is GRK5 [8–10]. As shown in Figure 3D, cardiac post-MI GRK5 levels are initially (at 24 hrs post-MI) similar between the two groups, as is the case also in the healthy, sham-operated groups (data not shown). By 4 weeks post-MI however, a significant up-regulation (~2-fold increase) of GRK5 is observed in βARKct/B1KO hearts compared to control B1KO hearts (Figure 3D), indicating that GRK2 inhibition with βARKct leads to a compensatory up-regulation of GRK5 over time.
Discussion
The present study reports for the first time, to our knowledge, that cardiac GRK2 is an endogenous “stumbling block” that normally prevents β2AR signaling from stimulating contractility, mainly because it promotes association of this βAR subtype with PDE4D in the heart, a major molecular “brake” on cardiac β2AR-dependent contractility [6, 7, 15]. Thus, only when cardiac GRK2 is blocked (e.g. with βARKct) is the β2AR capable of promoting cardiac contractility. Obviously, several signaling mechanisms/pathways are at play, the present study has identified the following two: 1) βARKct, by blocking GRK2, reduces the uncoupling of β2AR with the classical pro-contractile Gs protein-adenylyl cyclase-cAMP-PKA signaling pathway (Figure 4), and 2) GRK2 blockade reduces the interaction of β2AR with βarrs, which scaffold on themselves various isoforms of PDE4D (mainly PDE4D3 and PDE4D5) (Figure 4). PDE4D causes degradation of the local cAMP signals produced by activated β2AR, which are essential for stimulation of contractility, and thus it weakens these pro-contractile signals hampering β2AR-stimulated contractility [6, 7, 15]. By indirectly reducing the βarr-PDE4D interaction with the cardiac β2AR, βARKct releases the “brake” PDE4D poses on this receptor’s pro-contractile signaling and enhances its capacity to stimulate contractility (Figure 4).

Schematic illustration of the signaling pathways discussed in the present study that are elicited by β 2 AR activation in cardiac myocytes and are affected by GRK2 inhibition with βARKct. CA: Catecholamine; AC: Adenylyl cyclase; ATP: adenosine triphosphate. See text for details and all other molecular acronym descriptions.
Since the other major beneficial effect of cardiac β2AR signaling in vivo is inhibition of apoptosis (promotion of survival), we also tested the effects of cardiac GRK2 blockade in vivo on this aspect of β2AR signaling in the context of post-MI HF progression. We found that early on after MI, cardiac GRK2 blockade with βARKct also dramatically augments β2AR anti-apoptotic signaling, as well as its pro-infarct healing inflammatory signaling, in the heart. This results in significant reduction in all-cause mortality (marked increase in animal survival in the first 4 weeks post-MI), and reduced cellular apoptosis in the post-MI heart, compared to B1KO mice with unopposed cardiac GRK2 activity. Thus, βARKct enhances not only cardiac contractility, but also cardiac survival stimulated by the β2AR, which further reinforces its validity as an attractive therapeutic strategy to potentiate cardiac β2AR signaling and function in post-MI HF. Of course, enhancement of the anti-apoptotic signaling of other cardio-protective receptors that are also GRK2 substrates by βARKct cannot be ruled out and is, in fact, quite likely to have contributed to the observed cardiac apoptosis phenotype of βARKct/B1KO mice. However, βARKct’s cardio-protective and anti-apoptotic effects have been shown to be β2AR-dependent, since selective blockade of this receptor in cardiac myocytes abolishes βARKct-mediated anti-apoptotic effects [38]. On the other hand, effects of βARKct on β2AR-dependent pro-angiogenetic signaling, which plays an important role in peri-infarct HF progression [39], cannot be ruled out either. Nevertheless, it becomes quite clear from our current data that βARKct augments β2AR contractile function without negatively affecting its anti-apoptotic one, but rather actually preserving and further enhancing this β2AR function, as well.
However, this augmentation of anti-apoptotic signaling is short-lived: by 4 weeks post-MI, cardiac cellular apoptosis has returned to the 24-hour post-MI B1KO heart levels. This might be related to the nature of cardiac β2AR pro-survival signaling; β2AR can have remarkably different effects in the heart depending on its expression levels and on time [40, 41]. Cardiac β2AR is known to be beneficial (i.e. promoting survival) at low levels of overexpression and in the first few months of life in mice, but when overexpressed at extremely high levels in murine hearts or later on in the mouse’s life, these animals do not survive and die of severe cardiac complications [40]. Mechanistically, cardiac β2AR anti-apoptotic signaling is known to proceed mainly through the Gi/o protein signaling pathway [20–23], to which it is capable of switching following its phosphorylation by PKA [42]. GRK2 blockade by βARKct can increase this signaling by a) decreasing the pathway’s βarr-mediated desensitization, i.e. increasing the coupling of Gi/o proteins with the β2AR, and b) by increasing the PKA-dependent switching of β2AR signaling from Gs to Gi/o proteins thanks to the increase of β2AR signaling via the Gs protein-cAMP-PKA (the pro-contractile) pathway it also causes, discussed above (Figure 4). With regards to the pro-inflammatory signaling of cardiac β2AR, βarrs are known to scaffold and stabilize the inhibitory IκBα subunit of NFκB, thereby prohibiting NFκB activation [26, 27]. GRK2 blockade with βARKct decreases βarr interaction with the β2AR thereby “releasing” the inhibitory effect of βarrs on NFκB activation (Figure 4). Thus, NFκB activation and the subsequent pro-inflammatory cytokine production are enhanced (Figure 4). Indeed, NFκB activation and inflammatory cytokine levels were found significantly elevated in βARKct/B1KO hearts compared to B1KO hearts without GRK2 inhibition at 24 hrs post-MI.
Meanwhile however, βARKct also causes upregulation of the other major cardiac GRK, GRK5, in the first few weeks post-MI. This is also probably due to the enhanced NFκB activation (Figure 4), since NFκB can cause upregulation of GRK5 in cardiomyocytes [43]. Importantly, and given that GRK5 elevation is generally considered detrimental for the heart [8–10], this finding might explain, at least in part, the aforementioned switching of cardiac β2AR signaling from beneficial (anti-apoptotic) early in life of transgenic mice or at low levels of receptor expression to detrimental (pro-apoptotic) later on in the lifespan of these mice or at very high levels of cardiac β2AR overexpression [41]. Of note, GRK5 has also been reported to bind (via its non-catalytic, N-terminal domain) to, and stabilize IκBα, thereby inhibiting NFκB activity in several tissues, including the heart [44]. Therefore, our present findings strongly indicate that a negative feedback loop might exist in the heart, in which NFκB induces GRK5 expression, and GRK5 can subsequently suppress NFκB activation (Figure 4).
Conclusions
In summary, the present study reports that cardiac GRK2 inhibition with βARKct in vivo is absolutely essential for the cardiac β2AR subtype’s pro-contractile function, all the while preserving and augmenting this receptor’s beneficial anti-apoptotic/pro-survival and pro-infarct healing signaling pathways post-MI, early on after the cardiac insult. However, the effects of βARKct on the latter signaling modalities are transient due (in part) to compensatory elevation of cardiac GRK5 over time.
Methods
Experimental animals and surgical procedures
The animals in this study were handled according to animal welfare regulations and protocols approved by the authors’ Institutional Review Boards. Genetically engineered, 8- to 12-wk-old β1AR KO (B1KO) (on C57/B6 background) [36] and the offspring of their cross with Mini-27 mice, expressing the βARKct (or GRK2ct) transgene under the alpha-myosin heavy chain gene promoter [32], were used for this study. Mice were anesthetized with a mixture of ketamine (100 mg/kg) and xylazine (2.5 mg/kg). Animals were placed in the supine position on a heated operation board and a midline cervical incision was made to expose the trachea. Following successful endotracheal intubation, the cannula was connected to a rodent ventilator. The entire left ventricle (i.e. both infarct and non-infarct zones) were used for subsequent histological and biochemical assays. Myocardial infarction was performed as previously described [35].
Echocardiography & in vivo hemodynamics
Transthoracic echocardiography was performed with a linear 30-MHz transducer (VeVo 770 High Resolution Imaging System, VisualSonics, Toronto, ON, Canada), as described [35]. In vivo hemodynamic analysis was also performed as previously described [35].
In situ TUNEL staining
Heart specimens were fixed with 10% neutral buffered formalin, embedded in paraffin, and sectioned at 5-μm thickness. DNA fragmentation was detected in situ in deparaffinized sections using the ApopTag Kit (Intergene) and according to manufacturer’s instructions, as described previously [45]. The total number of nuclei was determined by manual counting of DAPI-stained nuclei in six random fields per section. All terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL)-positive nuclei were counted in each section.
Co-immunoprecipitation and western blotting
Cardiac extracts were prepared in 20 mM Tris pH 7.4, 137 mM NaCl, 1% Nonidet P-40, 20% glycerol, 10 mM PMSF, 1 mM Na3VO4, 10 mM NaF, 2.5 μg/ml aprotinin, and 2.5 μg/ml leupeptin. Protein concentration was then determined and equal amounts of protein per sample were loaded on SDS-PAGE gels for electrophoretic separation, as described previously [46]. For β2AR-PDE4D co-immunoprecipitation experiments, β2AR was immunoprecipitated with an anti-mouse β2AR antibody (sc-9042, Santa Cruz), immobilized on Protein A-sepharose beads (Invitrogen), prior to SDS-PAGE/western blotting. Total IκBα and phospho-IκBα were detected by using anti-IκBα (sc-1643, Santa Cruz) and anti-phosphoIκBα at Ser-32 (sc-7977, Santa Cruz) antibodies, Bcl-2, GRK5, GRK2/βARKct and GAPDH, with antibodies sc-492, sc-565, sc-562, and sc-25778, respectively (all from Santa Cruz), and PDE4D (various isoforms) with the PD4-401AP antibody (FabGennix). Immunoblots were revealed by enhanced chemiluminescence (ECL, Amersham Biosciences) and visualized in the FluorChem E Digital Darkroom (Cell Biosciences). Densitometry was performed with the AlphaView software (Cell Biosciences) in the linear range of signal detection (on non-saturated bands).
Cytokine measurements via ELISA
Pro-inflammatory cytokines TNFα, IL-6 and IL-1β were measured in serum obtained from left ventricular blood, immediately prior to heart excision and animal euthanizing, via multiplexed ELISA, as described [47, 48]. The assay was performed using the Mouse Cytokine ELISA Profiling Kit (EA-1091, Signosis), according to the manufacturer’s instructions.
Statistical analyses
Data are generally expressed as mean ± SEM. Unpaired 2-tailed Student’s t test and one- or two-way ANOVA with Bonferroni test were generally performed for statistical comparisons, unless otherwise indicated. For all tests, a p value of <0.05 was generally considered to be significant.
Abbreviations
- βAR:
-
Beta-adrenergic receptor
- B1KO:
-
β1AR knockout
- GRK:
-
G protein-coupled receptor kinase
- βARKct:
-
Beta-adrenergic receptor kinase (GRK2) carboxyl terminal fragment
- βarr:
-
Beta-arrestin
- PDE:
-
Phosphodiesterase
- MI:
-
Myocardial infarction
- HF:
-
Heart failure
- NFκB:
-
Nuclear factor-kappaB
- IκBα:
-
Inhibitor of nuclear factor-kappaB alpha subunit
- cAMP:
-
3′-5′ adenosine monophosphate (cyclic adenosine monophosphate)
- WT:
-
Wild type
- PKA:
-
Protein kinase A
- Gs:
-
Stimulatory G protein
- Gi/o:
-
Inhibitory or other G protein
- TNFα:
-
Tumor necrosis factor alpha
- IL:
-
Interleukin
- TUNEL:
-
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- Bcl-2:
-
B-cell lymphoma 2
- LV:
-
Left ventricular
- ELISA:
-
Enzyme-linked immunosorbent assay.
References
Tamargo J, López-Sendón J: Novel therapeutic targets for the treatment of heart failure. Nat Rev Drug Disc. 2011, 10: 536-555. 10.1038/nrd3431.
Xiang Y, Kobilka BK: Myocyte adrenoceptor signaling pathways. Science. 2003, 300: 1530-1532. 10.1126/science.1079206.
Xiao RP, Zhu W, Zheng M, Chakir K, Bond R, Lakatta EG, Cheng H: Subtype-specific beta-adrenoceptor signaling pathways in the heart and their potential clinical implications. Trends Pharmacol Sci. 2004, 25: 358-365. 10.1016/j.tips.2004.05.007.
Devic E, Xiang Y, Gould D, Kobilka B: Beta-adrenergic receptor subtype-specific signaling in cardiac myocytes from beta(1) and beta(2) adrenoceptor knockout mice. Mol Pharmacol. 2001, 60: 577-583.
Bernstein D, Fajardo G, Zhao M, Urashima T, Powers J, Berry G, Kobilka BK: Differential cardioprotective/cardiotoxic effects mediated by beta-adrenergic receptor subtypes. Am J Physiol Heart Circ Physiol. 2005, 289: H2441-H2449. 10.1152/ajpheart.00005.2005.
Xiang Y, Naro F, Zoudilova M, Jin SL, Conti M, Kobilka B: Phosphodiesterase 4D is required for beta2 adrenoceptor subtype-specific signaling in cardiac myocytes. Proc Natl Acad Sci USA. 2005, 102: 909-914. 10.1073/pnas.0405263102.
Richter W, Day P, Agrawal R, Bruss MD, Granier S, Wang YL, Rasmussen SG, Horner K, Wang P, Lei T, Patterson AJ, Kobilka B, Conti M: Signaling from beta1- and beta2-adrenergic receptors is defined by differential interactions with PDE4. EMBO J. 2008, 27: 384-393. 10.1038/sj.emboj.7601968.
Rockman HA, Koch WJ, Lefkowitz RJ: Seven-transmembrane-spanning receptors and heart function. Nature. 2002, 415: 206-212. 10.1038/415206a.
Rengo G, Lymperopoulos A, Koch WJ: Future g protein-coupled receptor targets for treatment of heart failure. Curr Treat Options Cardiovasc Med. 2009, 11: 328-338. 10.1007/s11936-009-0033-5.
Belmonte SL, Blaxall BC: G protein coupled receptor kinases as therapeutic targets in cardiovascular disease. Circ Res. 2011, 109: 309-319. 10.1161/CIRCRESAHA.110.231233.
Rengo G, Lymperopoulos A, Leosco D, Koch WJ: GRK2 as a novel gene therapy target in heart failure. J Mol Cell Cardiol. 2011, 50: 785-792. 10.1016/j.yjmcc.2010.08.014.
Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD: beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci USA. 2003, 100: 940-945. 10.1073/pnas.262787199.
Houslay MD, Adams DR: PDE4 cAMP phosphodiesterases: modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem J. 2003, 370: 1-18. 10.1042/BJ20021698.
De Arcangelis V, Liu R, Soto D, Xiang Y: Differential association of phosphodiesterase 4D isoforms with beta2-adrenoceptor in cardiac myocytes. J Biol Chem. 2009, 284: 33824-33832. 10.1074/jbc.M109.020388.
Fischmeister R, Castro LR, Abi-Gerges A, Rochais F, Jurevicius J, Leroy J, Vandecasteele G: Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res. 2006, 99: 816-828. 10.1161/01.RES.0000246118.98832.04.
Conti M, Beavo J: Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007, 76: 481-511. 10.1146/annurev.biochem.76.060305.150444.
Nikolaev VO, Moshkov A, Lyon AR, Miragoli M, Novak P, Paur H, Lohse MJ, Korchev YE, Harding SE, Gorelik J: Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010, 327: 1653-1657. 10.1126/science.1185988.
Chesley A, Lundberg MS, Asai T, Xiao RP, Ohtani S, Lakatta EG, Crow MT: The beta(2)-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through G(i)-dependent coupling to phosphatidylinositol 3′-kinase. Circ Res. 2000, 87: 1172-1179. 10.1161/01.RES.87.12.1172.
Patterson AJ, Zhu W, Chow A, Agrawal R, Kosek J, Xiao RP, Kobilka B: Protecting the myocardium: a role for the beta2 adrenergic receptor in the heart. Crit Care Med. 2004, 32: 1041-1048. 10.1097/01.CCM.0000120049.43113.90.
Xiao RP, Avdonin P, Zhou YY, Cheng H, Akhter SA, Eschenhagen T, Lefkowitz RJ, Koch WJ, Lakatta EG: Coupling of beta2-adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. Circ Res. 1999, 84: 43-52. 10.1161/01.RES.84.1.43.
Zhu W, Zeng X, Zheng M, Xiao RP: The enigma of beta2-adrenergic receptor Gi signaling in the heart: the good, the bad, and the ugly. Circ Res. 2005, 97: 507-509. 10.1161/01.RES.0000184615.56822.bd.
Hasseldine AR, Harper EA, Black JW: Cardiac-specific overexpression of human beta2 adrenoceptors in mice exposes coupling to both Gs and Gi proteins. Br J Pharmacol. 2003, 138: 1358-1366. 10.1038/sj.bjp.0705191.
Communal C, Singh K, Sawyer DB, Colucci WS: Opposing effects of beta(1)- and beta(2)-adrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin-sensitive G protein. Circulation. 1999, 100: 2210-2212. 10.1161/01.CIR.100.22.2210.
Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao RP: Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci USA. 2001, 98: 1607-1612. 10.1073/pnas.98.4.1607.
DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK: Beta-arrestins and cell signaling. Annu Rev Physiol. 2007, 69: 483-510. 10.1146/annurev.physiol.69.022405.154749.
Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ: beta-Arrestin inhibits NF-kappaB activity by means of its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc Natl Acad Sci USA. 2004, 101: 8603-8607. 10.1073/pnas.0402851101.
Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, Pei G: Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol Cell. 2004, 14: 303-317. 10.1016/S1097-2765(04)00216-3.
Valen G, Yan ZQ, Hansson GK: Nuclear factor kappa-B and the heart. J Am Coll Cardiol. 2001, 38: 307-314. 10.1016/S0735-1097(01)01377-8.
Valen G: Signal transduction through nuclear factor kappa B in ischemia-reperfusion and heart failure. Basic Res Cardiol. 2004, 99: 1-7. 10.1007/s00395-003-0442-7.
Rothwarf DM, Karin M: The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE. 1999, 1999: RE1-
Ungerer M, Böhm M, Elce JS, Erdmann E, Lohse MJ: Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation. 1993, 87: 454-463. 10.1161/01.CIR.87.2.454.
Koch WJ, Rockman HA, Samama P, Hamilton RA, Bond RA, Milano CA, Lefkowitz RJ: Cardiac function in mice overexpressing the β-adrenergic receptor kinase or a βARK inhibitor. Science. 1995, 268: 1350-1353. 10.1126/science.7761854.
Rockman HA, Choi DJ, Akhter SA, Jaber M, Giros B, Lefkowitz RJ, Caron MG, Koch WJ: Control of myocardial contractile function by the level of β-adrenergic receptor kinase 1 in gene-targeted mice. J Biol Chem. 1998, 273: 18180-18184. 10.1074/jbc.273.29.18180.
Matkovich SJ, Diwan A, Klanke JL, Hammer DJ, Marreez Y, Odley AM, Brunskill EW, Koch WJ, Schwartz RJ, Dorn GW: Cardiac-specific ablation of G-protein receptor kinase 2 redefines its roles in heart development and β-adrenergic signaling. Circ Res. 2006, 99: 966-1003.
Raake PW, Vinge LE, Gao E, Boucher M, Rengo G, Chen X, DeGeorge BR Jr, Matkovich S, Houser SR, Most P, Eckhart AD, Dorn GW, Koch WJ: G protein- coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ Res. 2008, 103: 413-422. 10.1161/CIRCRESAHA.107.168336.
Rohrer DK, Desai KH, Jasper JR, Stevens ME, Regula DP Jr, Barsh GS, Bernstein D, Kobilka BK: Targeted disruption of the mouse beta1-adrenergic receptor gene: developmental and cardiovascular effects. Proc Natl Acad Sci USA. 1996, 93: 7375-7380. 10.1073/pnas.93.14.7375.
Whelan RS, Kaplinskiy V, Kitsis RN: Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010, 72: 19-44. 10.1146/annurev.physiol.010908.163111.
Brinks H, Boucher M, Gao E, Chuprun JK, Pesant S, Raake PW, Huang ZM, Wang X, Qiu G, Gumpert A, Harris DM, Eckhart AD, Most P, Koch WJ: Level of G protein-coupled receptor kinase-2 determines myocardial ischemia/reperfusion injury via pro- and anti-apoptotic mechanisms. Circ Res. 2010, 107: 1140-1149. 10.1161/CIRCRESAHA.110.221010.
Santulli G, Cipolletta E, Sorriento D, Del Giudice C, Anastasio A, Monaco S, Maione AS, Condorelli G, Puca A, Trimarco B, Illario M, Iaccarino G: CaMK4 gene deletion induces hypertension. J Am Heart Assoc. 2012, 1: e001081-10.1161/JAHA.112.001081.
Dorn GW, Tepe NM, Lorenz JN, Koch WJ, Liggett SB: Low- and high- level transgenic expression of beta2-adrenergic receptors differentially affect cardiac hypertrophy and function in Galphaq-overexpressing mice. Proc Natl Acad Sci USA. 1999, 96: 6400-6405. 10.1073/pnas.96.11.6400.
Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, Yatani A, Dorn GW: Early and delayed consequences of beta(2)-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation. 2000, 101: 1707-1714. 10.1161/01.CIR.101.14.1707.
Daaka Y, Luttrell LM, Lefkowitz RJ: Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997, 390: 88-91. 10.1038/36362.
Islam KN, Koch WJ: Involvement of nuclear factor κB (NF-κB) signaling pathway in regulation of cardiac G protein-coupled receptor kinase 5 (GRK5) expression. J Biol Chem. 2012, 287: 12771-12778. 10.1074/jbc.M111.324566.
Sorriento D, Santulli G, Fusco A, Anastasio A, Trimarco B, Iaccarino G: Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-kappaB-dependent hypertrophic gene expression. Hypertension. 2010, 56: 696-704. 10.1161/HYPERTENSIONAHA.110.155960.
Yoo B, Lemaire A, Mangmool S, Wolf MJ, Curcio A, Mao L, Rockman HA: Beta1-adrenergic receptors stimulate cardiac contractility and CaMKII activation in vivo and enhance cardiac dysfunction following myocardial infarction. Am J Physiol Heart Circ Physiol. 2009, 297: H1377-H13786. 10.1152/ajpheart.00504.2009.
Lymperopoulos A, Rengo G, Funakoshi H, Eckhart AD, Koch WJ: Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat Med. 2007, 13: 315-323. 10.1038/nm1553.
Scott NJ, Cameron VA, Raudsepp S, Lewis LK, Simpson ER, Richards AM, Ellmers LJ: Generation and characterization of a mouse model of the metabolic syndrome: apolipoprotein E and aromatase double knockout mice. Am J Physiol Endocrinol Metab. 2012, 302: E576-E584. 10.1152/ajpendo.00222.2011.
Hu Y, Zhang H, Lu Y, Bai H, Xu Y, Zhu X, Zhou R, Ben J, Xu Y, Chen Q: Class A scavenger receptor attenuates myocardial infarction-induced cardiomyocyte necrosis through suppressing M1 macrophage subset polarization. Basic Res Cardiol. 2011, 106: 1311-1328. 10.1007/s00395-011-0204-x.
Acknowledgments
This work was supported in part by a Scientist Development Grant from the American Heart Association (AHA #09SDG2010138, National Center) and a Nova Southeastern University’s Health Professions Division (HPD) Research Grant (to AL), and NIH grants R37 HL061690, R01 HL085503, P01 HL075443 (Project 2) and P01 HL091799 (to WJK).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NCS, XV, AS, GR, AC, DLic, CDL, and EG performed research. DLeos assisted with writing of the paper. WJK supervised the project, provided funding for the research and assisted with writing of the paper. AL conceived and supervised the project, designed research, provided funding for it, and wrote the paper. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Salazar, N.C., Vallejos, X., Siryk, A. et al. GRK2 blockade with βARKct is essential for cardiac β2-adrenergic receptor signaling towards increased contractility. Cell Commun Signal 11, 64 (2013). https://doi.org/10.1186/1478-811X-11-64
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1478-811X-11-64