
Abstract
Autoimmune phenomena were identified in many different cases of hematological diseases and solid tumors, which may be due to alterations in the expression and function of the NF-κB signaling pathway. Recently, a number of studies have shown that the deletion or mutation of A20, a negative regulator of NF-κB, is frequently found in lymphomas, suggesting that it may be a linker between the altered immune response and leukemogenesis. The aim of this review is to summarize current findings of the A20 biological functions and its molecular mechanism as a tumor suppressor and immune regulator. The identification of A20 mutations and deletions in lymphocytic malignancy and the predictive significance of these aberrations are also reviewed.
Similar content being viewed by others


Introduction
Autoimmune phenomena were identified in many different cases of hematological diseases and solid tumors in which abnormal A20 expression was characterized. Recently, a number of studies have shown that A20 deletions and mutations are frequently found in lymphomas, suggesting that it may be involved in pathogenesis[1].
A20 is also known as tumor necrosis factor-α (TNFα)-induced protein 3 (TNFAIP3), which was first discovered in 1990 by Dixit and colleagues as a cytokine-induced gene in human umbilical vein endothelial cells[2, 3]. This protein was originally identified as a key regulator of inflammation signaling pathways and a negative regulator of the nuclear factor kappa B (NF-κB) activation pathway, which could attenuate the NF-κB activity triggered by signaling from several surface receptors including tumor necrosis factors receptor (TNFR), Toll-like receptor (TLR), and B cell receptor (BCR)[4].
The A20 structure and expression features
The A20 gene is located on chromosome 6q23.3, and the cDNA sequence is 4,440 bp long with an open reading frame of 2,370 nucleotides that encodes a protein predicted to contain 790 amino acids. A20 contains seven zinc finger (ZnF) domains in its C-terminus including one that functions as an E3 ligase, while an OUT (ovarian tumor) domain is embedded in its N-terminus[2, 3, 5–9]. Structural studies have revealed that the fourth zinc finger (ZnF4) interacts with mono-ubiquitins and K63-linked polyubiquitin chains[9].
A20 expression may be induced in multiple organs by various stimuli including interleukin-1β(IL-1β), TNF-α, and Epstein-Barr virus (EBV) - latent membrane protein1(LMP1). Although A20 is inducible by proinflammatory cytokines in most cell types, the regulation of A20 differs in lymphocytes. Temporally restricted and tissue-specific A20 expression patterns were observed with strikingly high levels in lymphoid organs including the thymus, spleen, and gut-associated lymphoid tissue. A20 is constitutively expressed in immature and mature thymocyte subpopulations and resting peripheral T cells. T cell activation leads to the down-regulation of A20 expression in mature thymocytes and peripheral T cells. TNF does not induce A20 expression in activated T cells even though these cells express TNF receptors. Thus, although A20 has been frequently described as an inducible immediate early gene in T cells, constitutive A20 expression is the prevailing pattern in these cells. Interestingly, constitutive A20 expression is not found in resting B cells, indicating the differential regulation of this gene among the B and T lineages[4, 10].
The biological functions of A20
A20 has been reported to be a ubiquitin-editing enzyme with several functions. Although A20 was initially described as an inhibitor of TNF-induced cell death[11], subsequent studies demonstrated that A20 overexpression inhibited NF-κB activation in response to different stimuli[12, 13]. Stable overexpression of A20 in a number of cell lines, such as the human breast carcinoma MCF7 cells and murine fibrosarcoma WEHI164 cells, was shown to result in partial resistance to TNF-induced apoptosis. It should be noted that inhibition of A20-mediated apoptosis has not been observed in all cell lines studied. For example, A20 overexpression in the human cervical carcinoma HeLa, lung epithelial A549, or human hepatoma HepG2 cells had no effect on apoptosis induced by the Fas receptor, lymphokine-activated killer cells, serum depletion, or oxidative stress[10]. The reason why some cell lines are protected by A20 and others are not is still unclear. However, recent studies have indicated that the most important A20 function is its function as a crucial tumor suppressor, and its deletion is closely associated with lymphomas[6].
NF-κB inhibitory factor
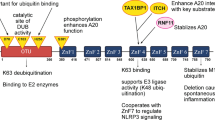
The cloning and characterization of the A20 promoter revealed two NF-κB DNA binding elements, which are recognition sequences for NF-κB transcription factors. It was also found that multiple NF-κB activating stimuli induce A20 expression via NF-κB sites in the A20 promoter[14]. Therefore, A20 has been demonstrated to downregulate its own expression, and it has been proposed that A20 participates in a negative feedback loop to attenuate TNFα-induced inflammatory responses. Subsequent studies have demonstrated that A20 is also induced in many other cell types by a wide variety of other stimuli including activation of the B cell surface receptor CD40 and overexpression of HTLV-I Tax and EBV-LMP1[15–19]. A20 overexpression was subsequently demonstrated to block the NF-κB activation mediated by TNF, IL-1, lipopolysaccharide (LPS), phorbol esters, and hydrogen peroxide in different cell types[12, 13, 20–23]. This blockage is most likely due to the inhibition of NF-κB activation in endothelial cells in response to proinflammatory stimuli and to an antiproliferative effect on smooth muscle cells as has been observed upon A20 overexpression in vitro. All of these findings suggest that A20 attenuates the activity of proximal signaling complexes at pro-inflammatory receptors[6, 10]. The regulation of NF‐κB signaling as it relates to A20-mediated ubiquitylation in T cells is diagramed in Figure1[6, 24–27].

Regulation of the NF‐κB signaling related A20 protein by ubiquitylation in T cells.
Regulation in innate and adaptive immunity
A20 regulates innate and adaptive immunity. This protein plays a pivotal role in the regulation of the immune response and prevents excessive NF-κB activation in response to a variety of external stimuli[28]. A20 has recently been suggested to suppress T cell activation. A20 regulates the strength and duration of the IκB kinase (IKK)/NF-κB response upon TCR/CD28 costimulation. By catalyzing the removal of K63-linked ubiquitin chains from mucosa associated lymphoid tissue 1 (MALT1), A20 prevents a sustained interaction between ubiquitinated MALT1 and the IKK complex, thus serving as a negative regulator of inducible IKK activity. Upon T cell stimulation, A20 is rapidly removed, and the paracaspase activity of MALT1, which is known to be recruited to lipid rafts after TCR stimulation, has been suggested to cleave A20[29].
A20 loss in B cells lowers their activation threshold and enhances their proliferation and survival in a gene dose-dependent fashion. Through the expression of proinflammatory cytokines, most notably IL-6, A20-deficient B cells trigger a progressive inflammatory reaction in naïve mice, which is characterized by the expansion of myeloid cells and effector-type T and regulatory T cells[30].
Moreover, A20 also serve as a negative regulator of the Toll-like receptor, which was found to play a crucial role in controlling the maturation, cytokine production and immunostimulatory potency of dendritic cells (DCs). A20-silenced DCs demonstrated spontaneous and enhanced expression of costimulatory molecules and proinflammatory cytokines and had different effects on T cell subsets i.e., they inhibited Treg cells and hyperactivated tumor-infiltrating cytotoxic T and T helper cells, which produce IL-6 and TNF-α, and were refractory to Treg cell–mediated suppression. Hence, A20 serves as an antigen presentation attenuator for the control of the antitumor immune responses during the priming and effector phases and provides a strategy for overcoming Treg cell–mediated suppression in an antigen-specific manner, reducing the need for directly targeting Treg cells[31].
Tumor suppressor
There are two different A20 expression patterns in cancer cells. First, A20 overexpression was found in undifferentiated nasopharyngeal carcinoma, poorly differentiated head and neck squamous cell carcinomas (SCCs) of the skin, estrogen receptor (ER)-negative breast cancer cell lines, tamoxifen resistant ER-positive tumors, and hepatitis B virus-related hepatocellular carcinoma[32]. Our recent study also found A20 overexpression in B-ALL samples that lack an A20 mutation (unpublished), suggesting that A20 plays a role in the pathogenesis of these malignancies. Moreover, A20 overexpression has been associated with poor prognosis in ER and progesterone receptor (PR)-negative breast tumor cells; thus, it may be thought of as a new cancer biomarker[6, 10, 27]. Second, and more importantly, A20 is inactivated in a substantial number of lymphomas by deletion, promoter methylation, frameshift mutations, and/or nonsense mutations that result in A20 truncations or point mutations. Notably, both A20 alleles are often affected, which is a hallmark of tumor suppressor genes[6].
Increasing data support the notion that A20 is a tumor suppressor. When re-expressed in a lymphoma-derived cell line with no functional A20 alleles, wildtype but not mutant A20 resulted in cell growth suppression and apoptosis induction accompanied by the downregulation of NF-κB activation. In A20-deficient cells, the suppression of cell growth and NF-κB activity due to A20 re-expression depended, at least in part, on cell surface-receptor signaling including that of the tumor necrosis factor receptor. These findings indicate that uncontrolled NF-κB signaling caused by the loss of A20 function is involved in the pathogenesis of B cell lymphoma subsets, and they strongly suggest a tumor suppressor role for A20, the loss of which may contribute to B cell lymphoma pathogenesis by causing supra-physiological NF-κB activation, which, in turn, has oncogenic properties including inhibiting apoptosis and promoting cell proliferation[28, 33].
Molecular mechanisms of the A20 biological functions
A20 functions as a ubiquitin-editing enzyme in the TNFR pathway by first cleaving K63-linked polyubiquitin chains on RIP1 (receptor interacting protein 1) followed by the catalysis of K48-linked chains via ZnF4, which triggers RIP1 proteasomal degradation[34]. In vitro studies conducted with recombinant A20 have indicated that A20 does not efficiently hydrolyze K63-linked polyubiquitin chains but instead has a preference for K48-linked polyubiquitin chains[35]. A20 cleaves Lys63-linked polyubiquitin chains and promotes Lys48-linked polyubiquitin chains, leading to negative regulation of TLR-mediated NF-κB and/or IRF3 (interferon regulatory factor 3) activation by regulating their substrates including TRAF6 (TNFR-Associated Factor 6), TRAF3, and RIP1[36]. Therefore, A20 may rely on accessory proteins to provide specificity for different pathways. One such protein is TAX1BP1 (Tax1-binding protein 1), which is also known as TXBP151. TAX1BP1 is a TRAF6-binding molecule that negatively regulates TRAF6-induced NF-κB activation by cooperating with the function of A20 and the E3 ubiquitin ligase Itch, suggesting that it cooperates with A20 to inhibit cell death[37, 38]. In addition, A20 associates with TXBP151 through the A20 C-terminal zinc finger-containing domain[10].
Regulation of A20 expression
A20 is regulated by the CARMA1–Bcl-10–MALT1(CBM) upstream signaling pathway complex, which bridges T cell antigen receptor (TCR) signaling with the canonical IKK/NF-κB pathway (Figure1)[6, 24–27]. TCR stimulation induced the recruitment of A20 and the Bcl-10 adaptor protein into the MALT1 complex, leading to MALT1-mediated A20 processing. Similarly, API2-MALT1 expression resulted in A20 cleavage. MALT1 cleaved human A20 at arginine 439 and impaired its NF-κB inhibitory function. Therefore, A20 was identified as a MALT1 substrate, emphasizing the importance of MALT1 proteolytic activity in ‘fine tuning’ T cell antigen receptor signaling[29].
Moreover, microRNA dysfunction can contribute to the constitutive activation of different signaling pathways such as the NF-κB pathway. miR-125a and miR-125b overexpression has been linked with dysfunctional hematopoiesis and aberrant immune cell responses[39–43]. miR-125a and miR-125b have been identified and characterized as regulators of A20 expression and function and, consequently, NF-κB activity. The fine-tuning of A20 levels mediated by miR-125 expression had a striking impact on the K63-linked ubiquitination of TRAF2 and RIP1, IκBα (inhibitor of nuclear factor kappa B alpha) degradation, p65 nuclear accumulation, and transcription of NF-κB target genes[44].
A20 gene alterations in lymphocytic malignancy
Recent studies have shown that A20 is frequently inactivated by deletions and/or mutations in several lymphoma subtypes including marginal zone lymphoma (MZL), diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), extranodal marginal zone lymphoma (EMZL) of MALT, primary mediastinal B-cell lymphoma, and Hodgkin’s lymphoma (HL). A20 deficiency has also been found in Sezary syndrome, which is a cutaneous T cell lymphoma, particularly those characterized by constitutive NF-κB activation[1, 28, 33, 45–47].
A20 deletion and mutation
There are numerous reports demonstrating different A20 mutations and deletions in lymphocytic malignancy, and the most frequent A20 alteration was found in B cell lymphoma; however, the frequency of A20 abnormalities is relatively different in different reports, while the most frequency of A20 mutations was found in B cell lymphoma. The various A20 alterations in lymphocytic malignancies are summarized in Table1. The most frequent deletions or mutations involve exons 3, 6, and 7.
Promotor methylation
The methylation-mediated silencing of genes is an epigenetic mechanism implicated in cancer. The CpG island-associated A20 promoter was previously found in osteosarcoma cells[56]. Thus, promoter methylation is another A20 inactivation mechanism. A study by Chanudet E et al. demonstrated that A20 promoter methylation was observed in 26% (7/27) of MALT lymphoma cases including five ocular adnexal and two extra-ocular cases. Of these seven cases, six demonstrated a similar methylation pattern with prominent methylation at the 7th, 8th and 9th CpG sites. These findings are in line with the expected role of promoter methylation in the transcriptional silencing of the remaining A20 allele[50].
The role of A20 in the pathogenesis of lymphocytic malignancy
The association between A20 dysfunction and lymphocytic malignancy
Recent genome-wide association studies have demonstrated a strong link between A20 polymorphisms and a collection of chronic inflammatory disorders including autoimmune diseases[5]. The phenotype of mice with full or conditional A20 deletion illustrates that A20 expression is essential for preventing chronic inflammation and autoimmune pathology. In addition, polymorphisms within the A20 genomic locus have been associated with multiple inflammatory and autoimmune disorders including systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), Crohn’s disease and psoriasis[57]. Both SLE and RA are associated with a significantly increased risk of lymphoma, particularly MALT lymphoma. Moreover, MALT lymphomas of the thyroid and salivary glands also frequently arise in a background of an autoimmune disorder, and tumor cells may be directly involved in the autoimmune process[1]. These data imply that such a nonresolving inflammation, which lacks A20, is a critical component of cancer development.
The role of A20 inactivation in lymphocytic malignancy
The development of hematological malignancies is a multistep process that requires at least two genetic abnormalities for disease development. Increasing data regarding the role of gene alterations such as that for DNMT, TET2, IDH1/2, NPM1, ASXL1, FLT3, and EKLF for acute myeloid leukemia, STAT3 for non-GCB DLBCL, and PPP2R5C and BCL11B for T-ALL were described, and some of these genes were thought to be correlated with leukemogenesis, poor overall survival or as a prognostic or therapeutic target factor[58–62]. The novel data demonstrating the significant contribution of A20 inactivation resulting in the constitutive activation of the NF-κB pathway is considered to be associated with cancer pathogenesis; at a minimum, it plays a crucial role in the carcinogenesis of certain lymphoid malignancies. It is now clear that the genetic loss of A20 may predispose to certain lymphoid malignancies[34]. CLL with unmutated IGHV genes that demonstrate poorer survival mostly have an A20 deletion. In this case, A20 may be a biomarker and a potential target for the underlying mechanism and treatment of CLL[55].
Summary
A20 acts as a negative feedback regulator of NF-κB activation in response to multiple stimuli and is considered a tumor suppressor. A20 dysfunction may be related to lymphocytic malignancy, and the exact A20 roles and mechanisms in the pathogenesis of lymphocytic malignancy, particularly in those with autoimmune disorders, require further characterization. Moreover, evidence has been presented suggesting that A20 and A20-binding proteins may be used as biomarkers and potential novel therapeutic targets in lymphocytic malignancies.
References
Chanudet E, Huang Y, Zeng N, Streubel B, Chott A, Raderer M, Du M-Q: TNFAIP3 abnormalities in MALT lymphoma with autoimmunity. B J Haematol. 2011, 154 (4): 533-535. 10.1111/j.1365-2141.2011.08623.x.
Opipari AW, Boguski MS, Dixit VM: The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J B Chem. 1990, 265 (25): 14705-14708.
Dixit VM, Green S, Sarma V, Holzman LB, Wolf FW, O'Rourke K, Ward PA, Prochownik EV, Marks RM: Tumor necrosis factor-alpha induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. J B chem. 1990, 265 (5): 2973-2978.
Tewari M, Wolf FW, Seldin MF, O'Shea KS, Dixit VM, Turka LA: Lymphoid expression and regulation of A20, an inhibitor of programmed cell death. J Immunol. 1995, 154 (4): 1699-1706.
Vereecke L, Beyaert R, van Loo G: The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009, 30 (8): 383-391. 10.1016/j.it.2009.05.007.
Hymowitz SG, Wertz IE: A20: from ubiquitin editing to tumour suppression. Nat Rev Cancer. 2010, 10 (5): 332-341. 10.1038/nrc2775.
Evans PC, Ovaa H, Hamon M, Kilshaw PJ, Hamm S, Bauer S, Ploegh HL, Smith TS: Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem J. 2004, 378: 727-734. 10.1042/BJ20031377.
Wertz IE, O'Rourke KM, Zhou HL, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL: De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappa B signalling. Nature. 2004, 430 (7000): 694-699. 10.1038/nature02794.
Bosanac I, Wertz IE, Pan B, Yu C, Kusam S, Lam C, Phu L, Phung Q, Maurer B, Arnott D: Ubiquitin binding to A20 ZnF4 is required for modulation of NF-kappaB signaling. Mol Cell. 2010, 40 (4): 548-557. 10.1016/j.molcel.2010.10.009.
Beyaert R, Heyninck K, Van Huffel S: A20 and A20-binding proteins as cellular inhibitors of nuclear factor-kappa B-dependent gene expression and apoptosis. Biochem Pharmacol. 2000, 60 (8): 1143-1151. 10.1016/S0006-2952(00)00404-4.
Opipari AW, Hu HM, Yabkowitz R, Dixit VM: The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J Biol Chem. 1992, 267 (18): 12424-12427.
Jaattela M, Mouritzen H, Elling F, Bastholm L: A20 zinc finger protein inhibits TNF and IL-1 signaling. J Immunol. 1996, 156 (3): 1166-1173.
Song HY, Rothe M, Goeddel DV: The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proc Natl Acad Sci U S A. 1996, 93 (13): 6721-6725. 10.1073/pnas.93.13.6721.
Krikos A, Laherty CD, Dixit VM: Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J Biol Chem. 1992, 267 (25): 17971-17976.
Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, Ferran C: A20 blocks endothelial cell activation through a NF-kappaB-dependent mechanism. J Biol Chem. 1996, 271 (30): 18068-18073. 10.1074/jbc.271.30.18068.
Sarma V, Lin Z, Clark L, Rust BM, Tewari M, Noelle RJ, Dixit VM: Activation of the B-cell surface receptor CD40 induces A20, a novel zinc finger protein that inhibits apoptosis. J Biol Chem. 1995, 270 (21): 12343-12346. 10.1074/jbc.270.21.12343.
Laherty CD, Perkins ND, Dixit VM: Human T cell leukemia virus type I Tax and phorbol 12-myristate 13-acetate induce expression of the A20 zinc finger protein by distinct mechanisms involving nuclear factor kappa B. J Biol Chem. 1993, 268 (7): 5032-5039.
Miller WE, Mosialos G, Kieff E, Raab-Traub N: Epstein-Barr virus LMP1 induction of the epidermal growth factor receptor is mediated through a TRAF signaling pathway distinct from NF-kappaB activation. J Virol. 1997, 71 (1): 586-594.
Laherty CD, Hu HM, Opipari AW, Wang F, Dixit VM: The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating nuclear factor kappa B. J Biol Chem. 1992, 267 (34): 24157-24160.
Zhang SQ, Kovalenko A, Cantarella G, Wallach D: Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000, 12 (3): 301-311. 10.1016/S1074-7613(00)80183-1.
De Valck D, Heyninck K, Van Criekinge W, Vandenabeele P, Fiers W, Beyaert R: A20 inhibits NF-kappaB activation independently of binding to 14-3-3 proteins. Biochem Biophys Res Commun. 1997, 238 (2): 590-594. 10.1006/bbrc.1997.7343.
Ferran C, Stroka DM, Badrichani AZ, Cooper JT, Wrighton CJ, Soares M, Grey ST, Bach FH: A20 inhibits NF-kappaB activation in endothelial cells without sensitizing to tumor necrosis factor-mediated apoptosis. Blood. 1998, 91 (7): 2249-2258.
Heyninck K, De Valck D, Vanden Berghe W, Van Criekinge W, Contreras R, Fiers W, Haegeman G, Beyaert R: The zinc finger protein A20 inhibits TNF-induced NF-kappaB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-kappaB-inhibiting protein ABIN. J Cell Biol. 1999, 145 (7): 1471-1482. 10.1083/jcb.145.7.1471.
Liu YC, Penninger J, Karin M: Immunity by ubiquitylation: a reversible process of modification. Nat Rev Immunol. 2005, 5 (12): 941-952. 10.1038/nri1731.
Düwel M, Welteke V, Oeckinghaus A, Baens M, Kloo B, Ferch U, Darnay BG, Ruland J, Marynen P, Krappmann D: A20 negatively regulates T cell receptor signaling to NF-kappaB by cleaving Malt1 ubiquitin chains. J Immunol. 2009, 182 (12): 7718-7728. 10.4049/jimmunol.0803313.
Malinverni C, Unterreiner A, Staal J, Demeyer A, Galaup M, Luyten M, Beyaert R, Bornancin F: Cleavage by MALT1 induces cytosolic release of A20. Biochem Biophys Res Commun. 2010, 400 (4): 543-547. 10.1016/j.bbrc.2010.08.091.
Verstrepen L, Verhelst K, van Loo G, Carpentier I, Ley SC, Beyaert R: Expression, biological activities and mechanisms of action of A20 (TNFAIP3). Biochem Pharmacol. 2010, 80 (12): 2009-2020. 10.1016/j.bcp.2010.06.044.
Kato M, Sanada M, Kato I, Sato Y, Takita J, Takeuchi K, Niwa A, Chen Y, Nakazaki K, Nomoto J: Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009, 459 (7247): 712-716. 10.1038/nature07969.
Coornaert B, Baens M, Heyninck K, Bekaert T, Haegman M, Staal J, Sun L, Chen ZJ, Marynen P, Beyaert R: T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat Immunol. 2008, 9 (3): 263-271. 10.1038/ni1561.
Chu Y, Vahl JC, Kumar D, Heger K, Bertossi A, Wojtowicz E, Soberon V, Schenten D, Mack B, Reutelshofer M: B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood. 2011, 117 (7): 2227-2236. 10.1182/blood-2010-09-306019.
Song XT, Evel-Kabler K, Shen L, Rollins L, Huang XF, Chen SY: A20 is an antigen presentation attenuator, and its inhibition overcomes regulatory T cell-mediated suppression. Nat Med. 2008, 14 (3): 258-265. 10.1038/nm1721.
Wang X, Friesen J, Nishanth G, Thi XN, Matuschewski K, Schluter D: Hepatocyte-specific knockout of A20 augments defense against malaria liver stage infections. Int J Med Microbiol. 2011, 301: 42-43.
Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A: Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009, 459 (7247): 717-721. 10.1038/nature07968.
Shembade N, Harhaj EW: Regulation of NF-kappaB signaling by the A20 deubiquitinase. Cell Mol Immunol. 2012, 9 (2): 123-130. 10.1038/cmi.2011.59.
Komander D, Reyes-Turcu F, Licchesi JD, Odenwaelder P, Wilkinson KD, Barford D: Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 2009, 10 (5): 466-473. 10.1038/embor.2009.55.
Inomata M, Niida S, Shibata K, Into T: Regulation of Toll-like receptor signaling by NDP52-mediated selective autophagy is normally inactivated by A20. Cell Mol Life Sci. 2012, 69 (6): 963-979. 10.1007/s00018-011-0819-y.
Shembade N, Harhaj E: A20 inhibition of NF-κB and inflammation: Targeting E2:E3 ubiquitin enzyme complexes. Cell Cycle. 2010, 9 (13): 10-11. 10.4161/cc.9.13.12269.
Shembade N, Harhaj NS, Parvatiyar K, Copeland NG, Jenkins NA, Matesic LE, Harhaj EW: The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat Immunol. 2008, 9 (3): 254-262. 10.1038/ni1563.
Bousquet M, Harris MH, Zhou BY, Lodish HF: MicroRNA miR-125b causes leukemia. Proc Natl Acad Sci U S A. 2010, 107 (50): 21558-21563. 10.1073/pnas.1016611107.
Guo SQ, Lu J, Schlanger R, Zhang H, Wang JY, Fox MC, Purton LE, Fleming HH, Cobb B, Merkenschlager M: MicroRNA miR-125a controls hematopoietic stem cell number. Proc Natl Acad Sci U S A. 2010, 107 (32): 14229-14234. 10.1073/pnas.0913574107.
O'Connell RM, Chaudhuri AA, Rao DS, Gibson WS, Balazs AB, Baltimore D: MicroRNAs enriched in hematopoietic stem cells differentially regulate long-term hematopoietic output. Proc Natl Acad Sci U S A. 2010, 107 (32): 14235-14240. 10.1073/pnas.1009798107.
Ooi AG, Sahoo D, Adorno M, Wang Y, Weissman IL, Park CY: MicroRNA-125b expands hematopoietic stem cells and enriches for the lymphoid-balanced and lymphoid-biased subsets. Proc Natl Acad Sci U S A. 2010, 107 (50): 21505-21510. 10.1073/pnas.1016218107.
Chaudhuri AA, So AY, Sinha N, Gibson WS, Taganov KD, O'Connell RM, Baltimore D: MicroRNA-125b potentiates macrophage activation. J Immunol. 2011, 187 (10): 5062-5068. 10.4049/jimmunol.1102001.
Kim SW, Ramasamy K, Bouamar H, Lin AP, Jiang DF, Aguiar RCT: MicroRNAs miR-125a and miR-125b constitutively activate the NF-kappa B pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20). Proc Natl Acad Sci U S A. 2012, 109 (20): 7865-7870. 10.1073/pnas.1200081109.
Honma K, Tsuzuki S, Nakagawa M, Tagawa H, Nakamura S, Morishima Y, Seto M: TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood. 2009, 114 (12): 2467-2475. 10.1182/blood-2008-12-194852.
Novak U, Rinaldi A, Kwee I, Nandula SV, Rancoita PM, Compagno M, Cerri M, Rossi D, Murty VV, Zucca E: The NF-κB negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood. 2009, 113 (20): 4918-4921. 10.1182/blood-2008-08-174110.
Schmitz R, Hansmann ML, Bohle V, Martin-Subero JI, Hartmann S, Mechtersheimer G, Klapper W, Vater I, Giefing M, Gesk S: TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med. 2009, 206 (5): 981-989. 10.1084/jem.20090528.
Nakajima S, Saito Y, Takahashi S, Hiramatsu N, Kato H, Johno H, Yao J, Paton AW, Paton JC, Kitamura M: Anti-inflammatory subtilase cytotoxin up-regulates A20 through the unfolded protein response. Biochem Biophys Res Commun. 2010, 397 (2): 176-180. 10.1016/j.bbrc.2010.05.069.
Yan QG, Huang YX, Watkins AJ, Kocialkowski S, Zeng NY, Hamoudi RA, Isaacson PG, de Leval L, Wotherspoon A, Du MQ: BCR and TLR signaling pathways are recurrently targeted by genetic changes in splenic marginal zone lymphomas. Haematologica. 2012, 97 (4): 595-598. 10.3324/haematol.2011.054080.
Chanudet E, Huang Y, Ichimura K, Dong G, Hamoudi RA, Radford J, Wotherspoon A, Isaacson PG, Ferry J, Du MQ: A20 is targeted by promoter methylation, deletion and inactivating mutation in MALT lymphoma. Leukemia. 2010, 24 (2): 488-489. 10.1038/leu.2009.278.
Braun FC, Grabarczyk P, Mobs M, Braun FK, Eberle J, Beyer M, Sterry W, Busse F, Schroder J, Delin M: Tumor suppressor TNFAIP3 (A20) is frequently deleted in Sezary syndrome. Leukemia. 2011, 25 (9): 1494-1501. 10.1038/leu.2011.101.
Giulino L, Mathew S, Ballon G, Chadburn A, Barouk S, Antonicelli G, Leoncini L, Liu YF, Gogineni S, Tam W: A20 (TNFAIP3) genetic alterations in EBV-associated AIDS-related lymphoma. Blood. 2011, 117 (18): 4852-4854. 10.1182/blood-2010-10-310995.
Montesinos-Rongen M, Schmitz R, Brunn A, Gesk S, Richter J, Hong K, Wiestler OD, Siebert R, Kuppers R, Deckert M: Mutations of CARD11 but not TNFAIP3 may activate the NF-kappaB pathway in primary CNS lymphoma. Acta Neuropathol. 2010, 120 (4): 529-535. 10.1007/s00401-010-0709-7.
Philipp C, Edelmann J, Buhler A, Winkler D, Stilgenbauer S, Kuppers R: Mutation analysis of the TNFAIP3 (A20) tumor suppressor gene in CLL. Int J Cancer. 2011, 128 (7): 1747-1750. 10.1002/ijc.25497.
Frenzel LP, Claus R, Plume N, Schwamb J, Konermann C, Pallasch CP, Claasen J, Brinker R, Wollnik B, Plass C: Sustained NF-kappaB activity in chronic lymphocytic leukemia is independent of genetic and epigenetic alterations in the TNFAIP3 (A20) locus. Int J Cancer. 2011, 128 (10): 2495-2500. 10.1002/ijc.25579.
Al-Romaih K, Somers GR, Bayani J, Hughes S, Prasad M, Cutz JC, Xue H, Zielenska M, Wang Y, Squire JA: Modulation by decitabine of gene expression and growth of osteosarcoma U2OS cells in vitro and in xenografts: identification of apoptotic genes as targets for demethylation. Cancer Cell Int. 2007, 7: 14-10.1186/1475-2867-7-14.
Vereecke L, Beyaert R, van Loo G: Genetic relationships between A20/TNFAIP3, chronic inflammation and autoimmune disease. Biochem Soc Trans. 2011, 39 (4): 1086-1091. 10.1042/BST0391086.
Takahashi S: Current findings for recurring mutations in acute myeloid leukemia. J Hematol Oncol. 2011, 4: 36-10.1186/1756-8722-4-36.
Ayala R, Martinez-Lopez J, Gilsanz F: Acute myeloid leukemia and transcription factors: role of erythroid Kruppel-like factor (EKLF). Cancer Cell Int. 2012, 12 (1): 25-10.1186/1475-2867-12-25.
Wu ZL, Song YQ, Shi YF, Zhu J: High nuclear expression of STAT3 is associated with unfavorable prognosis in diffuse large B-cell lymphoma. J Hematol Oncol. 2011, 4 (1): 31-10.1186/1756-8722-4-31.
Zheng H, Chen Y, Chen S, Niu Y, Yang L, Li B, Lu Y, Geng S, Du X, Li Y: Expression and distribution of PPP2R5C gene in leukemia. J Hematol Oncol. 2011, 4: 21-10.1186/1756-8722-4-21.
Huang X, Chen S, Shen Q, Yang L, Li B, Zhong L, Geng S, Du X, Li Y: Analysis of the expression pattern of the BCL11B gene and its relatives in patients with T-cell acute lymphoblastic leukemia. J Hematol Oncol. 2010, 3 (1): 44-10.1186/1756-8722-3-44.
Acknowledgements
The study was supported by grants from the National Natural Science Foundation of China (No. 91129720) and the Guangdong Science & Technology Project (2012B050600023).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
The concept of this paper was devised by YQL. FZ, LJY and YQL contributed to the intellectual input of the paper. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhang, F., Yang, L. & Li, Y. The role of A20 in the pathogenesis of lymphocytic malignancy. Cancer Cell Int 12, 44 (2012). https://doi.org/10.1186/1475-2867-12-44
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2867-12-44