
Abstract
Background
Lipase from Rhizopus chinensis is a versatile biocatalyst for various bioconversions and has been expressed at high-level in Pichia pastoris. However, the use of R. chinensis lipase in industrial applications is restricted by its low thermostability. Directed evolution has been proven to be a powerful and efficient protein engineering tool for improvement of biocatalysts. The present work describes improvement of the thermostability of R. chinensis lipase by directed evolution using P. pastoris as the host.
Results
An efficient, fast and highly simplified method was developed to create a mutant gene library in P. pastoris based on in vivo recombination, whose recombination efficiency could reach 2.3 × 105 /μg DNA. The thermostability of r27RCL was improved significantly by two rounds of error-prone PCR and two rounds of DNA shuffling in P. pastoris. The S4-3 variant was found to be the most thermostable lipase, under the conditions tested. Compared with the parent, the optimum temperature of S4-3 was two degrees higher, Tm was 22 degrees higher and half-lives at 60°C and 65°C were 46- and 23- times longer. Moreover, the catalytic efficiency kcat/Km of S4-3 was comparable to the parent. Stabilizing mutations probably increased thermostability by increasing the hydrophilicity and polarity of the protein surface and creating hydrophobic contacts inside the protein.
Conclusions
P. pastoris was shown to be a valuable cell factory to improve thermostability of enzymes by directed evolution and it also could be used for improving other properties of enzymes. In this study, by using P. pastoris as a host to build mutant pool, we succeeded in obtaining a thermostable variant S4-3 without compromising enzyme activity and making it a highly promising candidate for future applications at high temperatures.
Similar content being viewed by others


Background
Lipases (triacylglycerol ester hydrolases EC 3.1.1.3) are enzymes that hydrolyze the ester bonds of water-insoluble substrates at the interface between substrate and water. They are well known because of their remarkable levels of activity and stability in non-aqueous environments, which makes them as attractive enzyme for use in industrial applications, such as in food, pharmaceutical, chemical and detergent industries [1–3].
Rhizopus lipases have been widely used in food industries [4, 5]. Nevertheless, most Rhizopus lipases are mesozymes as they are produced by mesophilic organisms and exhibit unfavorable thermostability [6]. For example, lipase RAL from R. arrhizus showed poor thermostability when incubated at 30°C for 30 min [7]. Lipase ROL from R. oryzae showed highest activity at 35°C and about 35% of the lipase activity was lost after incubation at 45°C for 30 min [8]. Another lipase proROL derived from R. oryzae DSM853 exhibited better thermostability with a Tm value of 57.5°C [9]. In our previous study, a lipase gene proRCL from R. chinensis CCTCC M201021 was cloned and expressed at high-level in Pichia pastoris as r27RCL, whose activity was about 580 times higher than that of wild-type R. chinensis lipase [10]. This recombinant enzyme showed a high potential for industrial usage, as an additive for bread baking, synthesis of eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), sorbitan oleate, and ethyl esters [11–15]. However, the use of lipase r27RCL from R. chinensis is restricted by its low thermostability.
Directed evolution has been proven to be a powerful and efficient protein engineering tool for improvement of biocatalysts [16]. Several lipases from Rhizopus sp. were subjected to directed evolution in an effort to produce thermostable variants [7, 17]. The half-life of RAL expressed in P. pastoris was prolonged by 12-fold at 50°C [7]. The Tm value of RNL from R. niveus expressed in Saccharomyces cerevisiae was improved from 47°C to 57°C [17].
The production of an active Rhizopus lipase has been performed in Escherichia coli[18], S. cerevisiae[19] and P. pastoris[20–22], among which the expression of Rhizopus lipase in P. pastoris was the best. The P. pastoris expression system offers several advantages, as a desirable host in directed evolution, for easy library screening and further industrial application, such as the ability to perform eukaryotic protein modifications, high levels of protein expression at the intra- or extracellular level and an alcohol oxidase 1 (AOX1) gene promoter tightly regulated by methanol [23, 24]. However, mutant library construction in P. pastoris usually requires time-consuming multistep procedures which include an intermediate bacterial host (E. coli) and restriction digestion procedures [25].
In this work, we developed a faster and simplified mutant library production method, based on which, the thermostability of the lipase r27RCL from R. chinensis CCTCC M201021 was improved by directed evolution.
Results and discussion
Library construction
A novel approach for lipase mutant library construction in P. pastoris was established based on the formation of a recombination cassette in vivo (Figure 1). Three pairs of overlapping primers were used to generate the mutant gene (fragment B/C) and two different vector fragments (A and D). Primer BC-F had 66 bp overlapped pairs with the 5'-end of the gene sequence (“a” in Figure 1) of fragment D and primer BC-R had 60 bp overlapped pairs with the 3'-end of the gene sequence (“b” in Figure 1) of fragment A. Therefore, fragment B/C generated by primers BC-F and BC-R had 66 bp and 60 bp overlapped sequences with vector fragments D and A. The lipase gene fragment B (or C) and vector fragments A and D were mixed and transformed into P. pastoris competent cells by electroporation. In vivo, the fragments A, B/C and D self-reassembled to form a recombination cassette relying on the 60 bp and 66 bp homologous arms between them (Steps 1 and 2 in Figure 1). The self-reassembled expression cassette was integrated into the genome of P. pastoris by homologous recombination of the flanking AOX1 gene sequences (Steps 3 and 4 in Figure 1).

The novel approach of construction of the lipase mutant library in P. pastoris based on the formation of a recombination cassette in vivo . The hatched areas (a) and (b) were overlapped sequences between amplified fragments.
Recombination efficiency is one of the most important factors in directed evolution. In order to improve the recombination efficiency, the effects of the fragment molar ratio and the length of the overlapping sequences for recombination were investigated. As shown in Table 1, the recombination efficiency was improved with the increased molar ratio of fragment B/C:A:D in the DNA mixture by electroporation. About 2.3 × 105 /μg DNA recombinants were obtained when the molar ratio reached 10:1:1 and the values dropped to 1.8 × 105 /μg DNA, 1.2 × 105 /μg DNA or 1.0 × 105 /μg DNA when the molar ratio was 5:1:1, 3:1:1 or 1:1:1. In order to obtain high recombination efficiency, the fragment molar ratio B/C:A:D of 10:1:1 was chosen. The recombination efficiency was low with shorter overlapping sequences (<55 bp) due to the difficulty in formation of a recombination cassette in vivo, while too long overlapping sequences (>70 bp) resulted in PCR technical difficulties with blurred and weak target bands when amplified with such long primers. Medium length of overlapping sequences (55-70 bp) generated satisfied recombination efficiency without PCR technical difficulties (Table 1). It is well known that homologous recombination is a common characteristic of yeast. [Baudin et al. 26] demonstrated that the amount of homology necessary to promote efficient recombination-mediated gene disruption in S. cerevisiae is in the order of 30–50 bp. [Oldenburg et al. 27] reported that 20 bp of homology is sufficient to generate the PCR-directed recombination in S. cerevisiae, at least under certain conditions (vector:insert 1:6).
In this work, in order to make directed evolution practical in the host organism P. pastoris an efficient, fast and highly simplified method was developed to create a gene mutant library in P. pastoris. This method went a crucial step beyond previous approaches [25, 28–30] because it completely discarded time-consuming multi-step procedures including construction of recombinant plasmid, transformation of an intermediate bacterial host (E. coli), plasmid isolation and linearization. In our approach, only three PCR amplification processes and an electroporation step were required. [Fernandez et al. 31] also reported a method to produce a mutant library in P. pastoris, which reassembled a recombination cassette in vitro and produced about 6000 colonies per 1 μg of linear DNA. The method developed in our research was more convenient and efficient since self-reassembly of the linear recombinant cassette occurred in vivo.
Thermostable variants
The thermostability of r27RCL was improved by combining two rounds of error-prone PCR and two rounds of DNA shuffling (Figure 2). Error-prone PCR was used to introduce an average of 1 to 2 amino acid substitutions per proRCL gene and DNA shuffling could combine beneficial mutations as well as create new mutations. After creating the first-generation mutant library using error-prone PCR, approximately 5000 clones were screened for increased thermostability at 45°C. Two variants (ep1-1 and ep1-26) were identified and confirmed to have longer half-lives at 60°C (Table 2). ep1-1 (P168L, V329A) and ep1-26 (S234F) with a total of three substitutions were chosen as templates for generating the second-generation mutant library using error-prone PCR. Only 0.1% of the second generation mutant pool (5200 clones) screened at 50°C exhibited better performance than the first-generation parents. The identified variants ep2-3, ep2-4, ep2-8 and ep2-12 were selected for DNA shuffling. It was interesting to note that ep2-8 (S234F, P168L, K219D) and ep2-12 (S234F, V329A, K161R) both acquired mutations that were derived from ep1-1 and ep1-26 via in vivo DNA shuffling. From each generation of DNA shuffling, approximately 10000 clones were screened for increased thermostability. The third-generation evolution produced three variants (S3-1, S3-4, S3-18) whose thermostability slightly increased, whereas the last generation of evolution yielded three variants (S4-3, S4-13, S4-20) with significantly improved thermostability (Figure 2). S4-3 was the most thermostable variant. It contained five mutations (A129S, A230T, S234F, L180H, H317P) observed in previous generations plus one new mutation (T218S) generated by the fourth round of DNA shuffling.

Lineage of variants and amino acid substitutions by directed evolution. Newly- introduced mutations in each generation are marked with asterisks.
Thermostabilities of the evolved lipases
Table 2 lists the t1/2 values of the parent and the evolved thermostable variants during four generations of directed evolution. At 60°C the t1/2 of the parent was 4 min. The variants from the first and the second generations had half-lives of thermal inactivation two- to five-fold longer than the parent. The half-lives of the three variants discovered from the third round of directed evolution (DNA shuffling) were 10 to 14 times longer than that of the parent at 60°C. The last round of mutagenesis resulted in three variants that had the longest half-lives of thermal inactivation, 40 to 46 times longer than the parent at 60°C, among which, S4-3 was the most thermostable enzyme. The enzymatic properties of the purified S4-3 were then studied and its thermo-mechanism was explored.
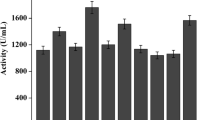
Initial activity vs thermostability of variants
In order to determine the relationship between thermostability and enzyme activity, the initial activities of all 12 variants were measured. As shown in Figure 3, five variants exhibited improved Tm as well as higher activity. The activities of the other 7 variants were not affected much by increased thermostability, among which S3-1, with the lowest activity, still retained about 90% activity compared to the parent enzyme. These results provided strong evidence that improvement of thermostability and maintenance of enzyme activity could be simultaneously achieved, and improved thermostability did not occur at the expense of activity at low temperatures [32–34].

Initial activity vs T m of the parent and variants. The cross point of dashed lines and solid lines represent parent enzyme r27RCL. Variants exhibited higher activity and improved Tm were squared with solid lines. The relative activity of the parent taking p NPP as substrate was set as 100%.
Enzymatic properties of the thermostable variant S4-3
The most thermostable variant in the last generation, S4-3, was purified and its enzymatic properties were investigated. As shown in Figure 4, the residual activities of S4-3 and the parent were determined after incubation at different temperatures for 30 min. S4-3 displayed greatly- increased thermostability in the range of 20°C -60°C and retained 50% residual activity at 67°C, whereas the parent started to denature at 40°C and was completely inactivated at 60°C after 30 min heat-treatment.

The effect of temperature on the stability of the parent and S4-3.
The thermal inactivation of S4-3 and the parent were characterized by incubation at 60°C and 65°C (Figure 5). The thermal inactivation curves of the parent at 60°C and 65°C were much steeper than those of S4-3, and the half-lives of S4-3 at 60°C and 65°C were 23- and 46-fold higher than those of the parent. In addition, the optimum temperature of S4-3 was also increased by 2°C compared with that of the parent (data not shown).

Thermostability of the parent and S4-3 at 60° C (a) and 65°C (b).
Kinetic parameters of the parent and S4-3 towards p NPP were calculated and listed in Table 3. S4-3 exhibited a 32% decrease in Km whereas a 33% increase in kcat, resulted in almost identical catalytic efficiency kcat/Km compared to the parent.
Thermo-mechanism of S4-3
The three-dimensional homology model of r27RCL shows that the five mutations A129S, A230T, S234F, L180H, T218S were all present on the surface of the enzyme and far from the catalytic domain (S265-D324-H377) except for the mutation H317P (Figure 6). It is consistent with the notion that the initial steps in protein unfolding during the irreversible thermal denaturation primarily involve surface-located parts of the protein [35, 36].

Three-dimensional structure of r27RCL. Locations of six amino acid substitutions (A129S, A230T, S234F, L180H, T218S, H317P) in S4-3 were shown in red sticks and the catalytic residues were shown in blue sticks (in the oval).
The contributions of mutations A230T and A129S toward thermostability could be interpreted based on the structure model which located them at the end of α-helix and in a loop on the protein surface. The substitution of A230 for T230 and A129 for S129 may have increased the hydrophilicity and polarity of the protein, which enhanced the thermostability of the enzyme [37]. Mutation L180H located in a loop on the protein surface introduced a positively-charged residue which may increase protein stability by creating a more favorable surface charge distribution [38, 39], as found in engineered thermostable cold shock proteins [40]. The mutation S234F was located in an α-helix on the protein surface. It was surprising that replacement of an uncharged hydrophilic residue for a nonpolar hydrophobic residue (S234F) at the protein surface increased protein stability but this effect has been observed previously [40, 41]. The H317P mutation is buried inside the protein in a β-strand. There are two assumptions that could be made to explain the improved thermostability. The first assumption was the addition of a hydrophobic residue. In the stabilized variant S4-3, hydrophilic histidine was replaced by hydrophobic proline, whose side-chain engaged in hydrophobic contacts with the side chain of Y293. This interaction was further improved by surrounding hydrophobic amino acids such as L137 and V341 and therefore enhanced protein stability. The alternative explanation was a reduction of the entropy of unfolding. The proline residue, with its pyrrolidine ring, had the lowest conformational entropy, thus the mutation histidine to proline should decrease the entropy of protein in the unfolded state and stabilize the protein [37].
It must be noted that the above-assumed thermo-mechanisms of S4-3 have been observed in many evolved thermostable enzymes and naturally-occuring thermophilic and hyperthermophilic proteins [35, 42]. Determination of the crystal structure of S4-3 should further confirm its thermo-mechanism. But, the crystal structure of r27RCL has not been reported before. Even if it is available, it is still difficult to predict the specific thermostabilizing mutations based on its structure because there are so many structural features that stabilize proteins [37] and the contribution of each feature to the total free energy of thermostability is quite small.
Conclusion
P. pastoris proved to be a valuable cell factory for the directed evolution of the lipase from R. chinensis for thermostability improvement, especially when combined with the novel method of library construction described here. In the present study, the thermostability of r27RCL was improved significantly by two rounds of error-prone PCR and two rounds of DNA shuffling in P. pastoris. The S4-3 variant is the most thermostable lipase from Rhizopus sp., under the conditions tested. Compared with the parent r27RCL, the optimum temperature of S4-3 was two degrees higher, Tm was 22 degrees higher and half-lives at 60°C and 65°C were 46- and 23- times longer, respectively. Moreover, the catalytic efficiency kcat/Km of S4-3 was comparable to the parent. In summary, S4-3 maintained good catalytic properties and exhibited much greater tolerability and stability at high temperatures, which made this variant a promising candidate for industrial applications at high temperatures.
Methods
Strains, plasmid, reagents and cultures
Restriction endonucleases, Taq DNA polymerase were purchased from Takara. All chemicals were of analytical grade or higher quality and purchased from Sigma. P. pastoris GS115 (His-Mut+) and the pPIC9K expression vector are from Invitrogen BV. Recombinant plasmid pPIC9K-proRCL and strain GS115/ pPIC9K-proRCL were constructed by [Yu et al.10]. The media used in this study include YPD (10g/L yeast extract, 20g/L peptone, 20g/L dextrose), MD (13.4g/L YNB, 0.016 μM biotin, 20g/L dextrose), BMGY (10g/L yeast extract, 20g/L peptone, 10g/L glycerol, pH 6.0 100 mM potassium phosphate, 13.4g/L YNB, 0.016 μM biotin), BMMY (10g/L yeast extract, 20g/L peptone, 10g/L methanol, pH 6.0 100 mM potassium phosphate, 13.4g/L YNB, 0.016μM biotin), BMMYA (10g/L yeast extract, 20g/L peptone, 5g/L methanol, pH 6.0 100 mM potassium phosphate, 13.4g/L YNB, 0.016 μM biotin, 6g/L agar).
Production of proRCL variants by error-prone PCR
The first two rounds of random mutagenesis of proRCL gene were carried out by error-prone PCR as described elsewhere [43]. A 50 μL reaction mixture contained 5 μL PCR Gold buffer, 0.7 mM MnCl2, 3 mM MgCl2, 0.5 mM dATP/dGTP, 2.5 mM dCTP/dTTP, 40 pmol of each primer (BC-F and BC-R), 5 ng of plasmid pPIC9K-proRCL, and 2.5 U of Taq polymerase. PCR cycle conditions were: initial denaturation for 3 min at 94°C; followed by 30 cycles of 30 s at 94°C, 30 s of annealing at 55°C and 1 min of extension at 72°C; and a 10 min final extension at 72°C. The template for the second round of error-prone PCR used an equimolar mixture of positive variants from the first generation of random mutagenesis.
Production of proRCL variants by DNA shuffling
The last two rounds of random mutagenesis of proRCL gene were carried out by DNA shuffling based on the previously methodology reported by [Zhao and Arnold 44]. The DNA template was an equimolar mixture of positive variants from the former round. The template was digested with 0.1 U DNase I in 200 μL of 50 mM Tris-HCl pH 7.4, 1 mM MgCl2 for 10 min at 37°C. Fragments of 50-100 bp were purified by gel extraction (QIAGEN). Then purified fragments were resuspended in PCR mixture (without primers) containing 5 μL PCR Gold buffer, 200 μM of each dNTP and 1.25 U of Taq polymerase. PCR cycle conditions were: initial denaturation for 3 min at 94°C; followed by 10 cycles of 30 s at 94°C, 30 s of annealing at 45°C and 1 min of extension at 72°C; 10 cycles with annealing temperatures of 48°C and 51°C, respectively; and a 10 min final extension at 72°C. Then, 1-3 μL of primerless PCR product was added to a 50 μL PCR mixture with 2 μM primers (BC-F/BC-R). Other PCR conditions were similar to those described above, and a single product comprising full-length proRCL was obtained.
Production of his4 3'-end containing fragment (vector fragment A and D)
Expression vector fragments were prepared by PCR amplification. The plasmid pPIC9K-proRCL was first amplified using flanking primers A-F/A-R or D-F/D-R to produce vector fragment A or D (Figure 1). The PCR was carried out in a final volume of 50 μL containing a mixture of 50 ng plasmid pPIC9K-proRCL, 1 μM of each primers, 5 μL PrimeSTAR DNA polymerase buffer, 100 μM of each dNTP and 1.25 U of PrimeSTAR DNA polymerase. PCR cycle conditions were: initial denaturation for 3 min at 94°C; followed by 30 cycles of 30 s at 94°C, 30 s of annealing at 50°C and 2.5 min or 7.5 min extension at 72°C for fragment D or A, respectively; and a 10 min final extension at 72°C.
Library construction
P. pastoris was transformed with 2 μg of fragments A, D and B/C in a 10:1:1 molar ratio by electroporation according to the protocol reported by [Wu and Letchworth 45]. Briefly, the competent cells, pretreated with 0.1 M lithium acetate and 10 mM dithiothreitol, were transformed with 2 μg DNA fragments by electroporation with instrument settings of 1.5 kV, 25 μF and 186 Ω. Transformed yeast colonies appeared in 4-6 days at 30°C on MD plates. The variants were confirmed by DNA sequencing (Sangon Biotech Company).
Overlapping sequence length
In order to optimise the recombination efficiency, the length of the overlapping sequences between vector and gene fragments was optimized. It was noteworthy that the lipase gene fragment was be produced by normal PCR (called L) instead of ep-PCR. We generated fragment L using 10 bp, 25 bp, 40 bp, 60 bp, 70 bp and 85 bp of overlapping primers listed in Table 4. These fragments were mixed with fragment A and D in a 10:1:1 molar ratio and electroporated into P. pastoris.
Pre-screening
Colonies from libraries of each generation were replicated with sterile toothpicks from MD plates onto YPD plates (for the storage of variants) and BMMYA plates (for gene expression and screening), simultaneously. The parent strain was used as a positive control. YPD and BMMYA plates were then incubated at 28-30°C for 2 d and 5 d, respectively. Then, BMMYA plates from each ep-PCR and DNA shuffling generation were subjected to heat-treatment at 45°C, 50°C, 55°C and 65°C, respectively, for 60 min. Screening was performed with 15 mL of top agar (5g/L) containing 160 μL of Fast blue RR (89g/L in dimethyl sulfoxide) and 80 μL α-naphthyl acetate (40g/L in dimethyl formamide) [46]. Positive colonies exhibited dark brown coloration within two minutes after pouring the top agar and were further screened using the 96 MTPs screening method.
Screening for evolved variants
All positive candidates screened from top-agar staining were picked and cultivated in deep-well microtiter plates containing 900 μL of BMGY medium, at 28°C, 300 rpm for 16 h to reach an OD600 of 1. Then, cultures were harvested by centrifugation at 3000 g at 4°C for 10 min and the cells were suspended in 300 μL of BMMY medium for gene expression. The cultures were kept at 28°C, 300 rpm for 84 h with supply of 100 μL fresh BMMY medium and 10g/L methanol every 24 h.
Each culture was centrifuged at 5000g at 4°C for 10 min. The supernatant was then transferred into 96-well microtiter plates for heat-treatment at 60°C or 65°C for 60 min. Initial and residual lipase activities were measured. All the assays were done in triplicate and significant differences (p < 0.05) were measured. Only variants that showed both higher ratio of residual activity and similar initial activity compared to the parent enzyme were selected.
Enzyme purification
A single colony from each variant was cultivated in 100 mL of BMGY medium shaken at 28°C and 250 rpm in 500 mL glass flasks. When cultures reached an OD600 of 1, the cells were centrifuged and resuspended in 25 mL of BMMY medium to obtain an OD600 of 4 and shaken at 28°C and 250 rpm in 250 mL glass flasks for 84 h. The cultures were supplemented with methanol (5g/L) to induce the expression of lipase every 12 h. The culture was then centrifuged and the supernatant was collected for protein purification.
Cell free medium was concentrated and interchanged with 10 mM Tris-HCl buffer (pH 7.5) by ultra filtration through a 10-kDa membrane (Millipore). The concentrated solution was loaded onto a SP-Sepharose column (AKTA, 1 cm × 20 cm) equilibrated with 20 mM Tris-HCl buffer (pH 7.5) and eluted with 0-0.5 M NaCl in the same buffer. Fractions containing lipase activity were pooled, concentrated and loaded on a Phenyl-sepharose 6 FF column (AKTA, 1.6 cm × 20 cm) equilibrated in 50 mM Tris-HCl buffer (pH 7.5) containing 1.6 M ammonium sulfate. Lipase was then eluted in an ammonium sulfate concentration gradient decreasing from 1.6 to 0 M in 50 mM Tris-HCl buffer (pH 7.5) and 4 mL fractions were collected at a flow rate of 0.8 mL/min.
Lipase properties
Lipase activity was measured on emulsified p- nitrophenyl palmitate (p NPP) according to [Kordel et al.47]. Unless stated otherwise, the typical enzymatic reaction was carried out at 40°C, pH 8.5. One enzyme unit was defined as the amount of enzyme releasing 1 μmol of p-nitrophenol per minute under the assay conditions. Optimal temperature (Topt) was determined by measuring the enzyme activity at pH 8.5 under various temperatures (20-60°C). Tm values were determined as described previously [43]. 10 U purified enzyme solutions were incubated for 30 min in the temperature range of 20-80°C, cooled in the ice bath for 20 min and equilibrated at room temperature for 5 min. Then, residual activity was determined at 40°C and expressed as a percentage of the initial activity. The temperature at which 50% of lipase activity lost was the Tm value. Purified enzymes were incubated at specific temperatures (60°C and 65°C) for half-life (t1/2) determination. 10 U purified enzyme solutions were taken at various time intervals, cooled in the ice bath for 20 min and equilibrated at room temperature for 5 min. Then, residual activity was determined and the incubation time at which 50% of lipase activity lost was the t1/2 value. Inactivation was followed until > 80% of the activity was lost. The Michaelis-Menten kinetic parameters kcat and Km were calculated using p NPP as substrate. Lineweaver-Burk plots were used to determine kcat and Km parameters, assuming that the reactions followed a simple Michaelis-Menten kinetics.
Thermo-mechanism analysis
A three-dimensional model of R. chinensis lipase was built by SWISS-MODEL protein automated modelling program [48] on the basis of crystal structure of lLGY (crystal structure of lipase II from Rhizopus niveus solved with a resolution of 2.20 Å) [49], which showed the highest homology of 80.38% to R. chinensis lipase.
Abbreviations
- 96 MTPs:
-
96 micro test plates
- p NPP p:
-
-nitrophenyl palmitate.
References
Sharma R, Chisti Y, Banerjee UC: Production, purification, characterization, and applications of lipases. Biotechnol Adv. 2001, 19: 627-662.
Hasan F, Shah AA, Hameed A: Industrial applications of microbial lipases. Enzyme Microb Technol. 2006, 39: 235-251.
Jaeger KE, Eggert T: Lipases for biotechnology. Curr Opin Biotech. 2002, 13: 390-397.
Mukherjee KD: Lipase-catalyzed reactions for modification of fats and other lipids. Biocatal Biotransfor. 1990, 3: 277-293.
Gandhi NN: Applications of lipase. J Am Oil Chem Soc. 1997, 74: 621-634.
Kamiya N, Ogawa T, Nagamune T: Enhancement of apparent thermostability of lipase from Rhizopus sp. by the treatment with a microbial transglutaminase. Biotechnol Lett. 2001, 23: 1629-1632.
Niu W, Li Z, Tan T: Secretion of pro-and mature Rhizopus arrhizus lipases by Pichia pastoris and properties of the proteins. Mol Biotechnol. 2006, 32: 73-81.
Hiol A, Jonzo MD, Rugani N, Druet D, Sarda L, Comeau LC: Purification and characterization of an extracellular lipase from a thermophilic Rhizopus oryzae strain isolated from palm fruit. Enzyme Microb Technol. 2000, 26: 421-430.
Beer HD, McCarthy JEG, Bornscheuer UT, Schmid RD: Cloning, expression, characterization and role of the leader sequence of a lipase from Rhizopus oryzae. Biochimica et Biophysica Acta. 1998, 1399: 173-180.
Yu XW, Wang LL, Xu Y: Rhizopus chinensis lipase: gene cloning, expression in Pichia pastoris and properties. J Mol Catal B: Enzym. 2009, 57: 304-311.
Xu Y, Wang D, Mu XQ, Zhao GA, Zhang KC: Biosynthesis of ethyl esters of short-chain fatty acids using whole-cell lipase from Rhizopus chinensis CCTCC M201021 in non-aqueous phase. J Mol Catal B: Enzym. 2002, 18: 29-37.
Xu Y, Wang D, Mu XQ, Ni YQ: Efficient esterification of sorbitan oleate by lipase in a solvent-free system. J Am Oil Chem Soc. 2003, 80: 647-651.
Sun SY, Xu Y, Wang D: Purification and biochemical characterization of an intracellular lipase by Rhizopus chinensis under solid-state fermentation and its potential application in the production of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). J Chem Technol Biotechnol. 2009, 84: 435-441.
Sun SY, Xu Y, Wang D: Novel minor lipase from Rhizopus chinensis during solid-state fermentation: Biochemical characterization and its esterification potential for ester synthesis. Bioresource Technol. 2009, 100: 2607-2612.
Li Z, Tang X, Huang W, Liu JG, Tilley M, Yao Y: Rheology, microstructure, and baking characteristics of frozen dough containing Rhizopus chinensis lipase and transglutaminase. Cereal Chem. 2011, 88: 596-601.
Hibbert EG, Dalby PA: Directed evolution strategies for improved enzymatic performance. Microb Cell Fact. 2005, 4: 29-34.
Kohno M, Enatsu M, Funatsu J, Yoshiizumi M, Kugimiya W: Improvement of the optimum temperature of lipase activity for Rhizopus niveus by random mutagenesis and its structural interpretation. J Biotechnol. 2001, 87: 203-210.
Di Lorenzo M, Hidalgo A, Haas M, Bornscheuer UT: Heterologous production of functional forms of Rhizopus oryzae lipase in Escherichia coli. Appl Environ Microbiol. 2005, 71: 8974-8977.
Ueda M, Takahashi S, Washida M, Shiraga S, Tanaka A: Expression of Rhizopus oryzae lipase gene in Saccharomyces cerevisiae. J Mol Catal B: Enzym. 2002, 17: 113-124.
Minning S, Serrano A, Ferrer P, Sola C, Schmid RD, Valero F: Optimization of the high-level production of Rhizopus oryzae lipase in Pichia pastoris. J Biotechnol. 2001, 86: 59-70.
Resina D, Serrano A, Valero F, Ferrer P: Expression of a Rhizopus oryzae lipase in Pichia pastoris under control of the nitrogen source-regulated formaldehyde dehydrogenase promoter. J Biotechnol. 2004, 109: 103-113.
Resina D, Bollok M, Khatri N, Valero F, Neubauer P, Ferrer P: Transcriptional response of P. pastoris in fed-batch cultivations to Rhizopus oryzae lipase production reveals UPR induction. Microb Cell Fact. 2007, 6: 21-31.
Macauley-Patrick S, Fazenda ML, McNeil B, Harvey LM: Heterologous protein production using the Pichia pastoris expression system. Yeast. 2005, 22: 249-270.
Krainer FW, Dietzsch C, Hajek T, Herwig C, Spadiut O, Glieder A: Recombinant protein expression in Pichia pastoris strains with an engineered methanol utilization pathway. Microb Cell Fact. 2012, 11: 22-35.
Niu WN, Li ZP, Zhang DW, Yu MR, Tan TW: Improved thermostability and the optimum temperature of Rhizopus arrhizus lipase by directed evolution. J Mol Catal B: Enzym. 2006, 43: 33-39.
Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C: A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic acids Res. 1993, 21: 3329-3330.
Oldenburg KR, Vo KT, Michaelis S, Paddon C: Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic acids Res. 1997, 25: 451-452.
Musidlowska-Persson A, Bornscheuer UT: Recombinant porcine intestinal carboxylesterase: cloning from the pig liver esterase gene by site-directed mutagenesis, functional expression and characterization. Protein Eng. 2003, 16: 1139-1145.
Cristea M, Engström Å, Su C, Hörnsten L, Oliw EH: Expression of manganese lipoxygenase in Pichia pastoris and site-directed mutagenesis of putative metal ligands. Arch Biochem Biophys. 2005, 434: 201-211.
Liu Z, Pscheidt B, Avi M, Gaisberger R, Hartner FS, Schuster C, Skranc W, Gruber K, Glieder A: Laboratory evolved biocatalysts for stereoselective syntheses of substituted benzaldehyde cyanohydrins. Chem Bio Chem. 2008, 9: 58-61.
Fernández L, Jiao N, Soni P, Gumulya Y, de Oliveira LG, Reetz MT: An efficient method for mutant library creation in Pichia pastoris useful in directed evolution. Biocatal Biotransfor. 2010, 28: 122-129.
Bloom JD, Labthavikul ST, Otey CR, Arnold FH: Protein stability promotes evolvability. Proc Natl Acad Sci. 2006, 103: 5869-5874.
Zumárraga M, Bulter T, Shleev S, Polaina J, Martínez-Arias A, Plou FJ, Ballesteros A, Alcalde M: In vitro evolution of a fungal laccase in high concentrations of organic cosolvents. Chem Biol. 2007, 14: 1052-1064.
García-Ruiz E, Maté D, Ballesteros A, Martinez AT, Alcalde M: Evolving thermostability in mutant libraries of ligninolytic oxidoreductases expressed in yeast. Microb Cell Fact. 2010, 9: 17-
Eijsink VGH, Bjork A, Gaseidnes S, Sirevag R, Synstad B, Burg B, Vriend G: Rational engineering of enzyme stability. J Biotechnol. 2004, 113: 105-120.
Johannes TW, Woodyer RD, Zhao H: Directed evolution of a thermostable phosphite dehydrogenase for NAD (P) H regeneration. Appl Environ Microbiol. 2005, 71: 5728-5734.
Li W, Zhou X, Lu P: Structural features of thermozymes. Biotechnol Adv. 2005, 23: 271-281.
Törrönen A, Rouvinen J: Structural and functional properties of low molecular weight endo-1, 4-β-xylanases. J Biotechnol. 1997, 57: 137-149.
Turunen O, Vuorio M, Fenel F, Leisola M: Engineering of multiple arginines into the Ser/Thr surface of Trichoderma reesei endo-1, 4-β-xylanase II increases the thermotolerance and shifts the pH optimum towards alkaline pH. Protein Eng. 2002, 15: 141-145.
Martin A, Sieber V, Schmid FX: In-vitro selection of highly stabilized protein variants with optimized surface. J Mol Biol. 2001, 309: 717-726.
Martin A, Kather I, Schmid FX: Origins of the high stability of an in vitro-selected cold-shock protein. J Mol Biol. 2002, 318: 1341-1349.
Vieille C, Zeikus GJ: Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol Mol Biol Rev. 2001, 65: 1-43.
Zhao H, Arnold FH: Directed evolution converts subtilisin E into a functional equivalent of thermitase. Protein Eng. 1999, 12: 47-53.
Zhao H, Arnold FH: Optimization of DNA shuffling for high fidelity recombination. Nucleic Acids Res. 1997, 25: 1307-1308.
Wu SX, Letchworth GJ: High efficiency transformation by electroporation of Pichia pastoris pretreated with lithium acetate and dithiothreitol. Biotechniques. 2004, 36: 152-154.
Khalameyzer V, Fischer I, Bornscheuer U, Altenbuchner J: Screening, nucleotide sequence, and biochemical characterization of an esterase from Pseudomonas fluorescens with high activity towards lactones. Appl Environ Microbiol. 1999, 65: 477-482.
Kordel M, Hofmann B, Schomburg D, Schmid RD: Extracellular lipase of Pseudomonas sp. strain ATCC 21808: purification, characterization, crystallization, and preliminary X-Ray diffraction data. J Bacteriol. 1991, 173: 4836-4841.
Bordoli L, Schwede T: Automated protein structure modeling with SWISS-MODEL workspace and the protein model portal. Methods Mol Biol (Clifton, NJ). 2012, 857: 107-136.
Kohno M, Funatsu J, Mikami B, Kugimiya W, Matsuo T, Morita Y: The crystal structure of lipase II from Rhizopus niveus at 2.2 Å resolution. J Biochem. 1996, 120: 505-510.
Acknowledgments
Financial support from the National Key Basic Research and Development Program of China (973 Program) (No. 2011CB710800), the National High Technology Research and Development Program of China (863 Program) (No. 2012AA022207, 2011AA02A209 and 2011AA02A210), the Fundamental Research Funds for the Central Universities (JUSRP11014), the Programme of Introducing Talents of Discipline to Universities (111 Project) ( 111-2-06 ), and the Ministry of Education, R.P. China, and from NSFC (20802027) are greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Yu made substantial contributions to design the experiments and draft the manuscript. Wang carried out this research work, interpreted the data and drafted the manuscript. Zhang carried out the purification experiment. Xu and Xiao revised the manuscript. All authors read and approved the final manuscript.
Xiao-Wei Yu, Rui Wang contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Yu, XW., Wang, R., Zhang, M. et al. Enhanced thermostability of a Rhizopus chinensis lipase by in vivo recombination in Pichia pastoris. Microb Cell Fact 11, 102 (2012). https://doi.org/10.1186/1475-2859-11-102
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2859-11-102