
Abstract
Long standing diabetes leads to structural and functional alterations in both the micro- and the macro-vasculature. Vascular endothelial cells (ECs) are the primary target of the hyperglycemia-induced adverse effects. Vascular stem cells that give rise to endothelial progenitor cells (EPCs) and mesenchymal progenitor cells (MPCs) represent an attractive target for cell therapy for diabetic patients. A number of studies have reported EPC dysfunction as a novel participant in the culmination of the diabetic complications. The controversy behind the identity of EPCs and the similarity between these progenitor cells to hematopoietic cells has led to conflicting results. MPCs, on the other hand, have not been examined for a potential role in the pathogenesis of the complications. These multipotent cells, however, do show a therapeutic role. In this article, we summarize the vascular changes that occur in diabetic complications highlighting some of the common features, the key findings that illustrate an important role of vascular stem cells (VSCs) in the pathogenesis of chronic diabetic complications, and provide mechanisms by which these cells can be used for therapy.
Similar content being viewed by others

Chronic diabetic complications
Diabetes is a chronic and debilitating metabolic disease that presently has no cure. Currently, the total number of people with diabetes is upwards of 221 million and in North America alone, more than 10% of the population is affected [1]. This amounts to a staggering economic burden, estimated to reach $17 billion a year by 2020 in Canada [2], and almost $116 billion in the United States [3]. Although the incidence in North America is quite alarming, close to 80% of diabetes-related deaths occur in low- and middle- income countries due to poor management of complications and lower standards of healthcare [4]. Despite great efforts to combat this disease, the World Health Organization projects that diabetes-related deaths will more than double by the year 2030 [4].
The most important discovery in the diabetes field was that of insulin in 1921. Exogenous insulin significantly alleviated diabetic coma and ketoacidosis, and saved millions. However, diabetic patients are still not morbidity-free due to the chronic secondary complications that arise in every diabetic patient. These long-term complications manifest as micro- (retinopathy, neuropathy, nephropathy, and cardiomyopathy) and macro- (atherosclerosis) vascular dysfunctions [5]. Although the clinical features of the complications are quite varied, the underlying cause is an aberration in the vasculature of the target organs. Two major clinical trials paved the way to better understanding the cause of the diabetic complications: the Diabetes Control and Complications Trial (DCCT) and the United Kingdom Perspective Diabetes Study (UKPDS), completed in 1993 and 1997 respectively. In both trials, type 1 and type 2 diabetic patients were put under intensive glycemic control, and in both cases, there was delayed progression and/or inhibition of the onset of diabetic complications [6, 7]. It is true that other factors, such as hyperlipidemia and hyperinsulinemia, may contribute to the pathogenesis of diabetic complications. The results of the clinical trials and years of research in animal models of diabetes and cultured cells confirm the notion that hyperglycemia is the primary cause of the micro- and macro-angiopathy we see in long-term diabetes.
Molecular basis of the vascular dysfunction in diabetic complications
Endothelial cells (ECs) are a critical component of the vascular unit. These specialized cells form the inner lining of blood vessels, sit on a basement membrane, and are surrounded by supportive perivascular cells (pericytes or smooth muscle cells) (Figure 1). ECs not only function as a barrier- producing an interface between circulating blood and the perfused tissue- but also play a prominent role in tissue functioning as well as organogenesis. These cells are involved in various important vascular processes such as regulating blood flow and pressure, permeability, blood fluidity, the thrombotic/fibrinolytic balance, and leukocyte traffic [8, 9].

Schematic of large and small blood vessels. Large vessels contain a prominent elastic tissue and multiple layers of the contractile cells (smooth muscle cells). Small capillaries may or may not have contractile cell (pericyte) coverage. Endothelial cells in capillaries sit on a thin basement membrane.
Due to their anatomical location in the blood vessel, ECs are not surprisingly the first to encounter circulating glucose. Glucose transporters (Gluts) facilitate the uptake of glucose [10–12]. The predominant Glut in the vascular ECs is Glut1 [13, 14]. Although Gluts are typically expressed in a tissue-specific manner, Glut1 is ubiquitously expressed under normal growth conditions [15]. Unlike many other glucose transporters, Glut1 activity and expression level does not change with increased or decreased plasma glucose levels. This indicates that hyperglycemia may have profound detrimental effects on vascular ECs specifically, as glucose uptake may not be actively regulated [13, 14]. There are certain conditions, however, that may alter Glut1 expression. For example, hypoxia increases Glut1 levels in ECs [16]. This may be one of the mechanisms behind uncontrollable dysfunction of the ECs in diabetic complications.
In vitro studies have shown that exposure to high levels of glucose lead to biochemical alterations in mature vascular ECs [17]. These alterations manifest as increased production of extracellular matrix proteins, such as collagen and fibronectin, increased production of the procoagulant protein von Willebrand Factor (vWF), and altered cellular activities [18–20]. In addition to a reduction in proliferation and migration [21], a number of studies have provided evidence that hyperglycemia can directly promote EC apoptosis [22–24]. This apoptotic pathway is believed to be activated by increased oxidative stress, increased intracellular Ca2+, mitochondrial dysfunction, changes in intracellular fatty acid metabolism, activation of mitogen activated protein kinase (MAPK) signaling pathways, and impaired phosphorylation/activation of protein kinase B (also known as Akt) [25, 26].
One of the earliest functional changes, which precedes any structural change in the vasculature of the target organs, is the impairment of endothelial-dependent vasodilation [18]. This impairment arises because of two inter-regulated mechanisms: decreased production of vasodilators and increased production of vasoconstrictors. Diminished levels of nitric oxide (NO) and increases in endothelin-1 (ET-1), the most potent endogenous vasoconstrictor, have been demonstrated in vascular ECs cultured in high glucose [18]. We and others have shown that the enzymes involved in NO production are upregulated in the ECs upon glucose challenge [27]. However, uncoupling of the enzymatic reaction and possible sequestration of NO by oxidative stress leads to significantly reduced NO [18, 28]. Another well-established pathway leading to increased EC damage in diabetes is the oxidative stress pathway. Hyperglycemic ECs produce reactive oxygen species (ROS) such as hydroxyl radicals, superoxide anions, and hydrogen peroxide [19]. The overproduction of ROS may also be attributed to the activation of alternate metabolic/signaling pathways such as the polyol pathway and hexoseamine pathway, and signaling through protein kinase C, AGE formation, and PARP activation [17, 29, 30] (Figure 2). Each of these pathways may potentiate each other, culminating in increased ET-1 activity, reduced NO bioavailability, oxidative stress, and EC dysfunction. Remarkably, the biochemical changes that we see in high glucose-treated ECs are reminiscent of the chronic complications that present in the diabetic patients.

Mechanisms of glucose-induced oxidative stress in ECs. Hyperglycemia leads to cell death by the overproduction of ROS and impairment in the ROS neutralizing enzymes. Multiple pathways may lead to ROS production. A consequence of activating these oxidant pathways is the depletion of co-factors required by the anti-oxidant enzyme systems. The net effect is an imbalance in ROS production and ROS clearance [AGE=advanced glycation end product; ATP=adenosine-5'-triphosphate; iNOS=inducible nitric oxide synthase; LOX-1=receptor for oxidized-low density lipoprotein; NAD=nicotinamide adenine dinucleotide; NADPH=reduced NAD phosphate; ox-LDL=oxidized-low density lipoprotein; PARP=poly (ADP-ribose) polymerase; RAGE=receptor for AGE].
We now know that altered ECs provide a backbone for the long-term vascular dysfunctions that arise in the diabetic patients (Figure 3). Changes in the structure and function of ECs leads to subsequent aberration of entire vascular networks, and tissues will begin to shows signs of poor blood flow and ischemia [31]. Normally, an adaptive response would be expected under these conditions. There is a vascular response in diabetic patients, although it varies depending on the organ system involved. For example, the retina and kidneys typically exhibit enhanced blood vessel formation, while this process is impaired in the heart and lower limbs [31, 32]. The selectivity in the target organ system in diabetes suggests the importance of both the tissue microenvironment and the intrinsic properties of the ECs [18].

Mechanisms leading to vascular disruption in diabetes. High glucose causes various biochemical and molecular changes in the vascular ECs, resulting in functional and structural alterations of the target organ vascular bed. Impaired vasoregulation and loss of vessel integrity leads to reduced blood flow and ischemia. In response, the target organ exhibits neovascularization (diabetic retinopathy/nephropathy) or fibrosis (diabetic cardiomyopathy/neuropathy) [AGE=advanced glycation end product; BM=basement membrane; EC=endothelial cell; ECM=extracellular matrix; EDV=endothelial-dependent vasodilation; ET-1=endothelin-1; MAPK=mitogen-activated protein kinase; PKB=protein kinase B; PKC=protein kinase C].
Growth factors and extracellular matrix (ECM) proteins are two major regulators of the balance that exists between neovascularization and scar formation/fibrosis in diabetic complications. Vascular endothelial growth factor (VEGF) is an EC-specific mitogen that promotes angiogenesis in a number of disease models. In parallel with a lack of angiogenesis that occurs in the heart in diabetes, a reduced expression of VEGF and its receptors is reported in the myocardium [33]. This is in direct contrast to elevated VEGF levels in the retina [34], correlating with uncontrolled retinal neovascularization. In addition to growth factors, the ECM regulates the vascular cells and may contribute to the differential effects of high glucose levels in diabetic complications. Binding of EC surface integrins to the ECM proteins regulates cell survival/apoptosis, growth, and cytoskeletal changes [35]. Therefore, angiogenesis is highly dependent on the interactions between the cellular components of the vascular unit and the surrounding scaffolding proteins. In fact, ECM changes that are believed to promote neovascularization during tumorigenesis are mimicked in retinal vascular development, and include increases in fibronectin and laminin [36]. Retinal basement membranes of diabetic animals show a similar protein profile- with elevated collagen IV, laminin and fibronectin as early as 8weeks following the onset of diabetes [37]. Along with ECM protein heterogeneity, increased ECM deposition in the heart may contribute to the impaired angiogenic response. Cardiac fibroblasts, which are present in significant numbers, may be responsible for this unregulated deposition of ECM proteins through the action ET-1 which has been shown in vitro to increase production of ECM components by fibroblasts [38, 39].
Regardless of the organ system, vascular ECs are the primary mediators of hyperglycemic damage, and they undergo functional and structural changes. Subsequent impaired vasoregulation, increased permeability, ECM expansion, and dysfunction of entire vascular networks causes reduced blood flow to the target organ, setting the stage for uncontrolled progression of the complications. To stop the progression of these complications and to repair the damage, we would need to either replace the damaged ECs or create brand new vascular networks. Considering mounting evidence of stem/progenitor cells in various tissues including the blood vessel wall [40], we can speculate that the reason diabetic patients exhibit impaired repair mechanisms is because these stem/progenitor cells are also affected.
Vascular stem cells (VSCs): current evidence and promise
Stem cells are defined by their ability to both self-renew and differentiate into functionally mature cells [41]. The potential of the cells is determined by the hierarchy and specialization level. Stem cells have been identified in a variety of post-embryonic tissues, including bone marrow, blood, fat, and skin [42, 43]. Finding these stem cell populations presents the opportunity for non-invasive tissue repair and tissue regeneration including the vascular tissue. In terms of regenerating brand new vascular networks (de novo formation), we must first find a suitable cell source. Ideally, it would be one cell type or a subpopulation that can produce both endothelial cells and the supportive perivascular cells. The notion of a specific ascular tem ell (VSC) able to produce mature/functional cells of the blood vessels is slowly gaining momentum. Several groups have demonstrated the existence of a common vascular precursor cell in both mouse and human studies. Kattman et al. used cell tracing studies in mice and showed that cardiomyocytes arise from a cell population expressing the VEGF receptor-2 (VEGFR2/Flk1) [44], indicating that they develop from a progenitor that also has vascular potential. They followed up these studies with an embryonic stem cell differentiation model, in which they isolated cardiovascular progenitors (brachyury+; VEGFR2+) from human embryoid bodies (EBs) and successfully demonstrated the potential for generating cardiomyocytes, endothelial cells, and vascular smooth muscle cells [44]. Yamashita and colleagues showed that VEGFR2+ cells, derived from embryonic stem cells, could differentiate into both endothelial and mural cells through differing culture conditions [45]. These cells were also able to reproduce the vascular organization process when placed in three-dimensional culture systems [45]. Similarly, Ferreira et al. demonstrated that vascular progenitors (CD34+) isolated from EBs will give rise to endothelial and smooth muscle-like cells, and have the ability to form vascular networks when implanted in vivo[46].
The exact identity of the VSCs is still not clear. There is ample evidence that these VSCs are found in the bone marrow and circulation and are quite distinct from hematopoietic stem cells. Selection of CD133+ cells from the circulation purifies a population(s) of cells that under different culture conditions, will produce lineage-restricted endothelial progenitor cells (EPCs) and mesenchymal/mesodermal progenitor cells (MPCs) [47–51]. However, cells expressing pan hematopoietic marker CD45 fail to yield endothelial cells [52–54]. It is unknown thus far whether one or more stem cell subtypes reside within this CD133+ population that are limited in their capacity to produce endothelial and mesenchymal cell types. More importantly, stem cell-derived EPCs and MPCs form functional vascular networks [45, 46, 52]. Whether this is a feasible avenue for diabetic patients is just recently being probed.
Endothelial progenitor cells (EPCs) and diabetic complications
Progenitor, or precursor, cells are committed (lineage-restricted) and highly proliferative derivatives of stem cells. These cells may be capable of doubling their population every 1015 hours [55]. However, unlike fully mature cells, progenitors may express markers of full maturity in addition to select stem cell markers [55]. For example, EPCs share markers of both stem and mature endothelial lineages [56]. With the potential use of EPCs, either clinically or as a biomarker, accurate identification and reproducible classification is of great importance. Despite advances in research on this subject, a lack of consensus remains on how EPCs should be defined. Traditionally, EPCs have been identified as the spindle-shaped or polymorphic cells that appear within 24days in culture after isolation of the mononuclear cell (MNC) fraction from blood or bone marrow [57] (Figure 4). The cells are characterized by Ulex europaeus agglutinin binding and DiI-labeled acetylated-low density lipoprotein (LDL) uptake [58–60]. These two properties are considered functional characteristics of ECs. However, along with expressing some EC markers, these short-term EPC colonies also express monocyte-specific marker CD14 and/or hematopoietic cell marker CD45. Further, acetylated-LDL uptake is a known feature of monocytes which was identified in 1979 [61]. Ulex europaeus agglutinin is a lectin which binds to the EC surface via fucose resides. Also, these fucose residues are not specific to the ECs [62].

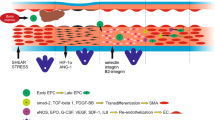
Early and Late EPCs. Schematic illustrating the two major types of cell colonies arising from blood and bone marrow mononuclear cells. Short term colonies appear within 7days of culture and are comprised of spindle shape or polymorphic cells. These early EPCs (also called angiogenic EPCs) express a number of endothelial and hematopoietic markers but fail to proliferate in culture. Late colonies, appearing from 721days, are comprised of epitheloid cells with high proliferative capacity. These late EPCs (also called vasculogenic EPCs) express all markers of mature endothelial cells but lack hematopoietic marker expression.
So what are EPCs? The single most important property (or functional attribute) of EPCs is the ability to incorporate into blood vessels. In other words, EPCs are vasculogenic. In comparison, early EPCs may be considered angiogenic as they may facilitate angiogenesis through elaboration of growth factors. We and others have characterized marker expression and cellular activities of vasculogenic EPCs extensively. EPCs display properties of both ECs and unipotent progenitor cells. EPCs differ from mature ECs in CD133 expression (positive on EPCs but readily lost upon culture) [48, 49], proliferation/growth kinetics (EPCs show lower population doubling time and higher cumulative population doublings) [53, 63], and response to endostatin (EPCs are stimulated whereas mature ECs are inhibited) [48]. Over time, EPCs resemble mature ECs in terms of marker expression and all cellular activities [48, 52, 53]. Much of the controversy behind angiogenic and vasculogenic EPCs could be negated by performing functional cellular activity tests (summarized in Figure 5). These include assessing the expression of endothelial-specific markers [48, 52, 53], activation by cytokine challenge [48], and most importantly, the ability of the cells to create blood vessels [52, 53].

Characterization scheme for EPCs and MPCs. (a) EPCs are defined by a set of morphological and phenotypic characteristics. These cells show properties of bone fide endothelial cells including expression and localization of CD31 and ve-cadherin on the cell membrane and von willebrand factor in the Wieble Palade bodies. Vascular endothelial growth factor is a mitogen for endothelial cells and EPCs. Also, EPCs induce adhesion molecules when challenged with cytokines, similar to mature endothelial cells. The progenitor properties include cloncal growth potential and in vivo vasculogenesis. (b) MPCs are also defined by morphological and phenotypic characteristics of mature mesenchymal cells (such as perivascular cells) and mesodermal progenitors. Similar to mature smooth muscle cells, MPCs express CD90, -smooth muscle actin, and calponin. Upon treatment with specific growth factors, such as plateled-derived growth factor and epidermal growth factor, MPCs proliferate and also exhibit chemotaxis. The progenitor phenotype involves the ability of the cells to give rise to mesenchymal lineage-specific cells such as adipocytes, osteocytes, and chondrocytes.
EPCs may be involved in vascular dysfunction in chronic diabetic complications (reviewed in [64]). It has been demonstrated that type 1 and type 2 diabetics maintain a lower circulating number of EPCs when compared with healthy subjects [56, 65–67]. In two similar studies, flow cytometric analysis was used to quantify EPCs (CD34 +/VEGFR2+/CD31+) in diabetic patients. These studies showed that EPCs were reduced by 44% and 40%, respectively [65, 68]. More recently, the number of circulating CD34+/VEGFR2+ cells were shown to correlate with glycemic control in type 2 diabetic patients [69]. This study also highlighted the negative relationship between circulating CD34+/VEGFR2+ cells and arterial stiffness in diabetic patients. Since these surface markers are not exclusive to EPCs, the reduced number may be inclusive of altered levels of hematopoietic stem/progenitor cells. In fact, a fairly large study with 120 patients with ischemic heart disease showed reduced levels of bone marrow-derived CD34+/CD45+ cells which also correlated with glycated hemoglobin HbA1c levels [70]. In vitro experimental studies using early EPCs have also shown a lower angiogenic ability [65], and impaired adherence to the mature EC monolayer in diabetes [71, 72]. Though less work has been done on the late vasculogenic EPCs, we have shown that high levels of glucose do not alter the cell growth, proliferation, or migration of late EPCs [73, 74]. The identical condition, however, increased ET receptor expression in the mature ECs and enhanced glucose-induced mature EC death. These findings show vasculogenic EPCs may be resistant to the adverse effects of high glucose.
Though EPC number may be reduced in long-term diabetes, there is still promise for their therapeutic potential. If the cellular activity of the EPCs remains intact in a diabetic setting, administration of ex vivo expanded EPCs should essentially work to improve vascular dysfunction. Not only has successful expansion of adult blood-derived EPCs been shown in vitro, but their ability to form fully functional vascular networks has also been demonstrated in vivo[47, 53]. It is important to note that in order to form stable and durable networks, EPCs require co-implantation with a source of perivascular cell. MPCs, being derived from the same CD133+ fraction as EPCs, may be a suitable candidate for this task.
Mesenchymal progenitor cells (MPCs) and diabetic complications
MPCs are multipotent cells that are derived, along with EPCs, from the CD133+ population of circulating cells [51]. MPCs can be isolated in large quantities from adult human bone marrow [75]. In addition, MPCs have been identified in liver [76], spleen [76], and adipose tissue [77]. Like EPCs and other progenitor cell types, MPCs share properties of both stem cells and mature cells. MPCs can be characterized by a combination of phenotypic and functional properties, including expression of cell surface markers, cell adhesion molecules, and differentiation potential (Figure 5). Because there is not one marker that is specific to MPCs, all parameters must be taken into consideration when properly identifying this cell population. Mesenchymal cells in culture typically exude a spindle-like morphology [75], however, some heterogeneity has been noted depending on the tissue source and especially when arising from differing species. MPCs are negative for both the endothelial marker CD31 and the hematopoietic marker CD45 [52]. Analysis of mRNA and/or protein can be used to demonstrate expression of -smooth muscle actin (SMA), calponin, CD90, PDGFR, and NG2[47, 52]. Functionally, MPCs differentiate into the mesenchymal lineage cells including adipocytes, osteocytes, and chondrocytes [47, 52, 75].
Not much is known about a possible pathogenic role of MPCs in diabetic complications. In terms of therapeutic benefit, however, recent studies show improvement and amelioration of complications, including cardiomyopathy, nephropathy, neuropathy, and wound healing by MPCs. Using a rat model of diabetic cardiomyopathy, MPCs were administered intravenously and shown to attenuate cardiac remodeling and improve myocardial function through a marked increase in the activity of matrix metalloproteinase (MMP)-2 and decrease in MMP-9[78]. In addition, reduced levels of VEGF, insulin-like growth factor-1 (IGF-1), adrenomedullin (AM), and hepatocyte growth factor (HGF) were found [78]. The MPCs differentiated into both cardiomyocytes and vascular ECs, improving myocardial perfusion and regeneration in the diabetic heart [78]. MPCs have also successfully improved diabetic nephropathy in mice. After systemic injection, the precursor cells were shown to engraft in the damaged kidneys and differentiate into renal cells, improving renal function and the regeneration of glomerular structures [79, 80]. Furthermore, MPCs improve diabetic polyneuropathy through increased secretion of angiogenic cytokines such as bFGF and VEGF when injected intramuscularly [81]. In a model of skin wound healing, administration of MPCs in streptozotocin-induced diabetic rats completely normalized the delayed wound closure time [82]. This effect was mediated, in part, through reduced number of infiltrating CD45+ cells into the wounds. This study involved normal MPCs (i.e. cells isolated from non-diabetic rats) and the question remains whether diabetes causes alteration of the functional properties of MPCs. This is a new field of research with not much known. However, a recent study showed that AGEs (Figure 2) may increase the generation of reactive oxygen species and reduce the proliferation and migration of MPCs [83]. Whether this plays a role in human diabetes or in animal models of diabetic complications, requires further studies.
Given the advantages that MPCs have over other cell types (differentiation potential and capability for regulation of the immune response), they are likely to be good therapeutic candidates in diabetic complications. The recent studies in animal models do show promise. Several studies have also reported that treatment with MPCs can enhance angiogenesis through paracrine effects [84–86]. The paracrine role may involve the release of angiogenic factors to facilitate EPC homing and the restructuring of vascular networks [52].
Concluding remarks
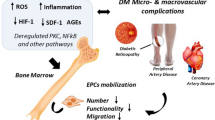
Examining the long-term effects of diabetes has led to implication of vascular ECs as the primary target of hyperglycemia-induced damage. Subsequently, entire vascular networks in target organs become dysfunctional and provide the foundation for the complications we see in the patients. Experimental evidence shows that stem/progenitor cells isolated from diabetic mice are able to restore vascular homeostasis [87, 88]. This suggests that the main stem cell deficit in diabetes is reduced number. This reduction may take place somewhere between the bone marrow and circulation. If we can find a way utilize these two cells types to repair vascular damage and restore blood vessel functioning, there is hope that the chronic complications can be attenuated (Figure 6).

The potential of VSCs for therapeutic use in diabetics. A schematic of our working hypothesis showing that CD133+ VSCs are non-invasively isolated from diabetic patients and differentiated into endothelial and mesenchymal lineages by defined media. EPCs and MPCs are then expanded ex vivo and re-implanted in the patients to repair the damage and restore vascular homeostasis [middle box shows the immunophenotype of VSCs; VEGF=vascular endothelial growth factor (obligatory factor for endothelial lineage)].
The success of therapeutic vascularization will rely on many factors, one of which being the ability of engineered blood vessels to form stable and functional anastomoses with the host vasculature. Neovascularization has been successfully shown thus far using human umbilical vein endothelial cells (HUVECs), as well as human microvascular endothelial cells (HDMECs) [89, 90]. However, there are limitations to the clinical use of these particular EC types because of the lower yield. Considering the ease with which EPCs can be isolated from adult peripheral blood, an opportunity is presented to obtain these cells non-invasively and in large enough quantities for expansion ex vivo. Following expansion, the cells can be implanted into the diabetic patient to restore vascular homeostasis. It has previously been shown that adult and cord blood-derived EPCs have the ability to form functional vascular networks in vivo[53, 91]. Importantly, this requires co-implantation of perivascular cells in order to maintain stable, functional networks. MPCs are an ideal candidate for a viable source of perivascular cell because, like EPCs, they can be isolated with minimal complications from sites such as bone marrow [75] and even adult blood [92]. We have previously shown the success of subcutaneous co-implantation of EPCs and MPCs into the backs of athymic nu/nu mice, resulting in the creation of human microvessels that formed functional anastomoses with the host vasculature [52].
Abbreviations
- AGE:
-
Advanced glycation end products
- AM:
-
Adrenomedullin
- DCCT:
-
Diabetes Control and Complications Trial
- EB:
-
Embryoid body
- EC:
-
Endothelial cell
- ECM:
-
Extracellular matrix
- EPC:
-
Endothelial progenitor cell
- ET:
-
Endothelin
- Glut:
-
Glucose transporter
- HDMEC:
-
Human dermal microvascular endothelial cell
- HGF:
-
Hepatocyte growth factor
- HUVEC:
-
Human umbilical vein endothelial cell
- IGF:
-
Insulin-like growth factor
- LDL:
-
Low-density lipoprotein
- MAPK:
-
Mitogen-activated protein kinase
- MMP:
-
Matrix metalloproteinase
- MPC:
-
Mesenchymal progenitor cell
- NO:
-
Nitric oxide
- PARP:
-
Poly (ADP-ribose) polymerase
- PDGFR:
-
Platelet-derived growth factor receptor
- ROS:
-
Reactive oxygen species
- UKPDS:
-
United Kingdom Perspective Diabetes Study
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
VEGF receptor
- VSC:
-
Vascular stem cell
- vWF:
-
von Willeband Factor.
References
Amos AF, McCarty DJ, Zimmet P: The rising global burden of diabetes and its complications: estimates and projections to the year 2010. Diabet Med. 1997, 14 (Suppl 5): S1-S85.
An economic tsunami, the cost of diabetes in Canada. Book An economic tsunami, the cost of diabetes in Canada. 2009, (Editor ed.^eds.). City: Canadian Diabetes Association
2011 National Diabetes Fact Sheet, National Center for Chronic Disease Prevention and Health Promotion. Book 2011 National Diabetes Fact Sheet, National Center for Chronic Disease Prevention and Health Promotion. 2011, (Editor ed.^eds.), vol. 2011. City: American Diabetes Association
King H, Rewers M: Global estimates for prevalence of diabetes mellitus and impaired glucose tolerance in adults. WHO Ad Hoc Diabetes Reporting Group. Diabetes Care. 1993, 16: 157-177. 10.2337/diacare.16.1.157.
Engerman R, Bloodworth JM, Nelson S: Relationship of microvascular disease in diabetes to metabolic control. Diabetes. 1977, 26: 760-769. 10.2337/diabetes.26.8.760.
Albers JW, Herman WH, Pop-Busui R, Feldman EL, Martin CL, Cleary PA, Waberski BH, Lachin JM: Effect of prior intensive insulin treatment during the Diabetes Control and Complications Trial (DCCT) on peripheral neuropathy in type 1 diabetes during the Epidemiology of Diabetes Interventions and Complications (EDIC) Study. Diabetes Care. 33: 1090-1096.
Ratner RE: Glycemic control in the prevention of diabetic complications. Clin Cornerstone. 2001, 4: 24-37. 10.1016/S1098-3597(01)90027-4.
Florey : The endothelial cell. Br Med J. 1966, 2: 487-490. 10.1136/bmj.2.5512.487.
Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, et al: Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998, 91: 3527-3561.
Joost HG, Thorens B: The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (review). Mol Membr Biol. 2001, 18: 247-256. 10.1080/09687680110090456.
Augustin R: The protein family of glucose transport facilitators: Its not only about glucose after all. IUBMB Life. , 62: 315-333.
Thorens B, Mueckler M: Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab. , 298: E141-E145.
Mandarino LJ, Finlayson J, Hassell JR: High glucose downregulates glucose transport activity in retinal capillary pericytes but not endothelial cells. Invest Ophthalmol Vis Sci. 1994, 35: 964-972.
Kaiser N, Sasson S, Feener EP, Boukobza-Vardi N, Higashi S, Moller DE, Davidheiser S, Przybylski RJ, King GL: Differential regulation of glucose transport and transporters by glucose in vascular endothelial and smooth muscle cells. Diabetes. 1993, 42: 80-89. 10.2337/diabetes.42.1.80.
Wertheimer E, Sasson S, Cerasi E, Ben-Neriah Y: The ubiquitous glucose transporter GLUT-1 belongs to the glucose-regulated protein family of stress-inducible proteins. Proc Natl Acad Sci U S A. 1991, 88: 2525-2529. 10.1073/pnas.88.6.2525.
Takagi H, King GL, Aiello LP: Hypoxia upregulates glucose transport activity through an adenosine-mediated increase of GLUT1 expression in retinal capillary endothelial cells. Diabetes. 1998, 47: 1480-1488. 10.2337/diabetes.47.9.1480.
Brownlee M: Biochemistry and molecular cell biology of diabetic complications. Nature. 2001, 414: 813-820. 10.1038/414813a.
Khan ZA, Chakrabarti S: Therapeutic targeting of endothelial dysfunction in chronic diabetic complications. Recent Pat Cardiovasc Drug Discov. 2006, 1: 167-175. 10.2174/157489006777442531.
Khan ZA, Farhangkhoee H, Mahon JL, Bere L, Gonder JR, Chan BM, Uniyal S, Chakrabarti S: Endothelins: regulators of extracellular matrix protein production in diabetes. Exp Biol Med (Maywood). 2006, 231: 1022-1029.
Cagliero E, Maiello M, Boeri D, Roy S, Lorenzi M: Increased expression of basement membrane components in human endothelial cells cultured in high glucose. J Clin Invest. 1988, 82: 735-738. 10.1172/JCI113655.
Graier WF, Grubenthal I, Dittrich P, Wascher TC, Kostner GM: Intracellular mechanism of high D-glucose-induced modulation of vascular cell proliferation. Eur J Pharmacol. 1995, 294: 221-229. 10.1016/0014-2999(95)00534-X.
Du XL, Sui GZ, Stockklauser-Farber K, Weiss J, Zink S, Schwippert B, Wu QX, Tschope D, Rosen P: Introduction of apoptosis by high proinsulin and glucose in cultured human umbilical vein endothelial cells is mediated by reactive oxygen species. Diabetologia. 1998, 41: 249-256. 10.1007/s001250050900.
Lorenzi M, Cagliero E, Toledo S: Glucose toxicity for human endothelial cells in culture. Delayed replication, disturbed cell cycle, and accelerated death. Diabetes. 1985, 34: 621-627. 10.2337/diabetes.34.7.621.
Baumgartner-Parzer SM, Wagner L, Pettermann M, Grillari J, Gessl A, Waldhausl W: High-glucosetriggered apoptosis in cultured endothelial cells. Diabetes. 1995, 44: 1323-1327. 10.2337/diabetes.44.11.1323.
Yang Z, Mo X, Gong Q, Pan Q, Yang X, Cai W, Li C, Ma JX, He Y, Gao G: Critical effect of VEGF in the process of endothelial cell apoptosis induced by high glucose. Apoptosis. 2008, 13: 1331-1343. 10.1007/s10495-008-0257-y.
Favaro E, Miceli I, Bussolati B, Schmitt-Ney M, Cavallo Perin P, Camussi G, Zanone MM: Hyperglycemia induces apoptosis of human pancreatic islet endothelial cells: effects of pravastatin on the Akt survival pathway. Am J Pathol. 2008, 173: 442-450. 10.2353/ajpath.2008.080238.
Chen S, Khan ZA, Barbin Y, Chakrabarti S: Pro-oxidant role of heme oxygenase in mediating glucose-induced endothelial cell damage. Free Radic Res. 2004, 38: 1301-1310. 10.1080/10715760400017228.
El-Remessy AB, Abou-Mohamed G, Caldwell RW, Caldwell RB: High glucose-induced tyrosine nitration in endothelial cells: role of eNOS uncoupling and aldose reductase activation. Invest Ophthalmol Vis Sci. 2003, 44: 3135-3143. 10.1167/iovs.02-1022.
Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, et al: Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000, 404: 787-790. 10.1038/35008121.
van den Oever IA, Raterman HG, Nurmohamed MT, Simsek S: Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus. Mediators Inflamm 2010, 2010: 792393.
Aronson D: Hyperglycemia and the pathobiology of diabetic complications. Adv Cardiol. 2008, 45: 1-16.
Alberti KG, Zimmet PZ: New diagnostic criteria and classification of diabetesagain?. Diabet Med. 1998, 15: 535-536. 10.1002/(SICI)1096-9136(199807)15:7<535::AID-DIA670>3.0.CO;2-Q.
Chou E, Suzuma I, Way KJ, Opland D, Clermont AC, Naruse K, Suzuma K, Bowling NL, Vlahos CJ, Aiello LP, King GL: Decreased cardiac expression of vascular endothelial growth factor and its receptors in insulin-resistant and diabetic States: a possible explanation for impaired collateral formation in cardiac tissue. Circulation. 2002, 105: 373-379. 10.1161/hc0302.102143.
Khan ZA, Chakrabarti S: Growth factors in proliferative diabetic retinopathy. Exp Diabesity Res. 2003, 4: 287-301. 10.1155/EDR.2003.287.
Boudreau NJ, Jones PL: Extracellular matrix and integrin signalling: the shape of things to come. Biochem J. 1999, 339 (Pt 3): 481-488.
Jiang B, Liou GI, Behzadian MA, Caldwell RB: Astrocytes modulate retinal vasculogenesis: effects on fibronectin expression. J Cell Sci. 1994, 107 (Pt 9): 2499-2508.
Nishikawa T, Giardino I, Edelstein D, Brownlee M: Changes in diabetic retinal matrix protein mRNA levels in a common transgenic mouse strain. Curr Eye Res. 2000, 21: 581-587. 10.1076/0271-3683(200007)2111-ZFT581.
Katwa LC, Guarda E, Weber KT: Endothelin receptors in cultured adult rat cardiac fibroblasts. Cardiovasc Res. 1993, 27: 2125-2129. 10.1093/cvr/27.12.2125.
Guarda E, Katwa LC, Myers PR, Tyagi SC, Weber KT: Effects of endothelins on collagen turnover in cardiac fibroblasts. Cardiovasc Res. 1993, 27: 2130-2134. 10.1093/cvr/27.12.2130.
Chandrasekhar KS, Zhou H, Zeng P, Alge D, Li W, Finney BA, Yoder MC, Li J: Blood vessel wall-derived endothelial colony-forming cells enhance fracture repair and bone regeneration. Calcif Tissue Int. 2011, 89: 347-357. 10.1007/s00223-011-9524-y.
Wu Y, Wang J, Scott PG, Tredget EE: Bone marrow-derived stem cells in wound healing: a review. Wound Repair Regen. 2007, 15 (Suppl 1): S18-S26.
Tepper OM, Galiano RD, Kalka C, Gurtner GC: Endothelial progenitor cells: the promise of vascular stem cells for plastic surgery. Plast Reconstr Surg. 2003, 111: 846-854. 10.1097/01.PRS.0000039576.63523.08.
Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, Alfonso ZC, Fraser JK, Benhaim P, Hedrick MH: Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002, 13: 4279-4295. 10.1091/mbc.E02-02-0105.
Kattman SJ, Huber TL, Keller GM: Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev Cell. 2006, 11: 723-732. 10.1016/j.devcel.2006.10.002.
Yamashita J, Itoh H, Hirashima M, Ogawa M, Nishikawa S, Yurugi T, Naito M, Nakao K: Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature. 2000, 408: 92-96. 10.1038/35040568.
Ferreira LS, Gerecht S, Shieh HF, Watson N, Rupnick MA, Dallabrida SM, Vunjak-Novakovic G, Langer R: Vascular progenitor cells isolated from human embryonic stem cells give rise to endothelial and smooth muscle like cells and form vascular networks in vivo. Circ Res. 2007, 101: 286-294. 10.1161/CIRCRESAHA.107.150201.
Khan ZA, Boscolo E, Picard A, Psutka S, Melero-Martin JM, Bartch TC, Mulliken JB, Bischoff J: Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest. 2008, 118: 2592-2599.
Khan ZA, Melero-Martin JM, Wu X, Paruchuri S, Boscolo E, Mulliken JB, Bischoff J: Endothelial progenitor cells from infantile hemangioma and umbilical cord blood display unique cellular responses to endostatin. Blood. 2006, 108: 915-921. 10.1182/blood-2006-03-006478.
Wu X, Rabkin-Aikawa E, Guleserian KJ, Perry TE, Masuda Y, Sutherland FW, Schoen FJ, Mayer JE, Bischoff J: Tissue-engineered microvessels on three-dimensional biodegradable scaffolds using human endothelial progenitor cells. Am J Physiol Heart Circ Physiol. 2004, 287: H480-H487. 10.1152/ajpheart.01232.2003.
Peichev M, Naiyer AJ, Pereira D, Zhu Z, Lane WJ, Williams M, Oz MC, Hicklin DJ, Witte L, Moore MA, Rafii S: Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood. 2000, 95: 952-958.
Tondreau T, Meuleman N, Delforge A, Dejeneffe M, Leroy R, Massy M, Mortier C, Bron D, Lagneaux L: Mesenchymal stem cells derived from CD133-positive cells in mobilized peripheral blood and cord blood: proliferation, Oct4 expression, and plasticity. Stem Cells. 2005, 23: 1105-1112. 10.1634/stemcells.2004-0330.
Melero-Martin JM, De Obaldia ME, Kang SY, Khan ZA, Yuan L, Oettgen P, Bischoff J: Engineering robust and functional vascular networks in vivo with human adult and cord blood-derived progenitor cells. Circ Res. 2008, 103: 194-202. 10.1161/CIRCRESAHA.108.178590.
Melero-Martin JM, Khan ZA, Picard A, Wu X, Paruchuri S, Bischoff J: In vivo vasculogenic potential of human blood-derived endothelial progenitor cells. Blood. 2007, 109: 4761-4768. 10.1182/blood-2006-12-062471.
Case J, Mead LE, Bessler WK, Prater D, White HA, Saadatzadeh MR, Bhavsar JR, Yoder MC, Haneline LS, Ingram DA: Human CD34+AC133+VEGFR-2+ cells are not endothelial progenitor cells but distinct, primitive hematopoietic progenitors. Exp Hematol. 2007, 35: 1109-1118. 10.1016/j.exphem.2007.04.002.
Zampetaki A, Kirton JP, Xu Q: Vascular repair by endothelial progenitor cells. Cardiovasc Res. 2008, 78: 413-421. 10.1093/cvr/cvn081.
Fadini GP, Agostini C, Avogaro A: Characterization of endothelial progenitor cells. Biochem Biophys Res Commun. 2005, 336: 1-2. 10.1016/j.bbrc.2005.07.119.
Lopez-Holgado N, Alberca M, Sanchez-Guijo F, Villaron E, Almeida J, Martin A, Armellini A, Garcia C, Blanco B, Sanchez-Abarca I, et al: Short-term endothelial progenitor cell colonies are composed of monocytes and do not acquire endothelial markers. Cytotherapy. 2007, 9: 14-22. 10.1080/14653240601047726.
Capla JM, Grogan RH, Callaghan MJ, Galiano RD, Tepper OM, Ceradini DJ, Gurtner GC: Diabetes impairs endothelial progenitor cell-mediated blood vessel formation in response to hypoxia. Plast Reconstr Surg. 2007, 119: 59-70. 10.1097/01.prs.0000244830.16906.3f.
Kusuyama T, Omura T, Nishiya D, Enomoto S, Matsumoto R, Takeuchi K, Yoshikawa J, Yoshiyama M: Effects of treatment for diabetes mellitus on circulating vascular progenitor cells. J Pharmacol Sci. 2006, 102: 96-102. 10.1254/jphs.FP0060256.
Wang CH, Ting MK, Verma S, Kuo LT, Yang NI, Hsieh IC, Wang SY, Hung A, Cherng WJ: Pioglitazone increases the numbers and improves the functional capacity of endothelial progenitor cells in patients with diabetes mellitus. Am Heart J. 2006, 152: e1051-e1058.
Goldstein JL, Ho YK, Basu SK, Brown MS: Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci U S A. 1979, 76: 333-337. 10.1073/pnas.76.1.333.
Jackson CJ, Garbett PK, Nissen B, Schrieber L: Binding of human endothelium to Ulex europaeus I-coated Dynabeads: application to the isolation of microvascular endothelium. J Cell Sci. 1990, 96 (Pt 2): 257-262.
Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, Krasich R, Temm CJ, Prchal JT, Ingram DA: Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007, 109: 1801-1809. 10.1182/blood-2006-08-043471.
Bernardi S, Severini GM, Zauli G, Secchiero P: Cell-based therapies for diabetic complications. Exp Diabetes Res. 2012, 2012: 872504.
Loomans CJ, de Koning EJ, Staal FJ, Rookmaaker MB, Verseyden C, de Boer HC, Verhaar MC, Braam B, Rabelink TJ, van Zonneveld AJ: Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004, 53: 195-199.
Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S: Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001, 89: E1-E7. 10.1161/hh1301.093953.
Fadini GP, Sartore S, Albiero M, Baesso I, Murphy E, Menegolo M, Grego F, Vigili de Kreutzenberg S, Tiengo A, Agostini C, Avogaro A: Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler Thromb Vasc Biol. 2006, 26: 2140-2146. 10.1161/01.ATV.0000237750.44469.88.
Fadini GP, Miorin M, Facco M, Bonamico S, Baesso I, Grego F, Menegolo M, de Kreutzenberg SV, Tiengo A, Agostini C, Avogaro A: Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. 2005, 45: 1449-1457. 10.1016/j.jacc.2004.11.067.
Yue WS, Lau KK, Siu CW, Wang M, Yan GH, Yiu KH, Tse HF: Impact of glycemic control on circulating endothelial progenitor cells and arterial stiffness in patients with type 2 diabetes mellitus. Cardiovasc Diabetol. 2011, 10: 113-10.1186/1475-2840-10-113.
Bozdag-Turan I, Turan RG, Turan CH, Ludovicy S, Akin I, Kische S, Arsoy NS, Schneider H, Ortak J, Rehders T, et al: Relation between the frequency of CD34 bone marrow derived circulating progenitor cells and the number of diseased coronary arteries in patients with myocardial ischemia and diabetes. Cardiovasc Diabetol. 2011, 10: 107-10.1186/1475-2840-10-107.
Tamarat R, Silvestre JS, Le Ricousse-Roussanne S, Barateau V, Lecomte-Raclet L, Clergue M, Duriez M, Tobelem G, Levy BI: Impairment in ischemia-induced neovascularization in diabetes: bone marrow mononuclear cell dysfunction and therapeutic potential of placenta growth factor treatment. Am J Pathol. 2004, 164: 457-466. 10.1016/S0002-9440(10)63136-7.
Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, Levine JP, Gurtner GC: Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002, 106: 2781-2786. 10.1161/01.CIR.0000039526.42991.93.
Keats E, Khan ZA: High glucose causes impairment of mature endothelial cells but not adult endothelial progenitor cells. Can J Diabetes. 2011, 35: A126.
Keats E, Khan ZA: Unique cellular responses of adult blood-derived endothelial progenitor cells and mature endothelial cells to high glucose. Diabetes. 2011, 60: 1624-10.2337/db10-1656.
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR: Multilineage potential of adult human mesenchymal stem cells. Science. 1999, 284: 143-147. 10.1126/science.284.5411.143.
Campagnoli C, Roberts IA, Kumar S, Bennett PR, Bellantuono I, Fisk NM: Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood. 2001, 98: 2396-2402. 10.1182/blood.V98.8.2396.
Traktuev DO, Merfeld-Clauss S, Li J, Kolonin M, Arap W, Pasqualini R, Johnstone BH, March KL: A population of multipotent CD34-positive adipose stromal cells share pericyte and mesenchymal surface markers, reside in a periendothelial location, and stabilize endothelial networks. Circ Res. 2008, 102: 77-85. 10.1161/CIRCRESAHA.107.159475.
Zhang N, Li J, Luo R, Jiang J, Wang JA: Bone marrow mesenchymal stem cells induce angiogenesis and attenuate the remodeling of diabetic cardiomyopathy. Exp Clin Endocrinol Diabetes. 2008, 116: 104-111. 10.1055/s-2007-985154.
Ezquer FE, Ezquer ME, Parrau DB, Carpio D, Yanez AJ, Conget PA: Systemic administration of multipotent mesenchymal stromal cells reverts hyperglycemia and prevents nephropathy in type 1 diabetic mice. Biol Blood Marrow Transplant. 2008, 14: 631-640. 10.1016/j.bbmt.2008.01.006.
Lee RH, Seo MJ, Reger RL, Spees JL, Pulin AA, Olson SD, Prockop DJ: Multipotent stromal cells from human marrow home to and promote repair of pancreatic islets and renal glomeruli in diabetic NOD/scid mice. Proc Natl Acad Sci U S A. 2006, 103: 17438-17443. 10.1073/pnas.0608249103.
Shibata T, Naruse K, Kamiya H, Kozakae M, Kondo M, Yasuda Y, Nakamura N, Ota K, Tosaki T, Matsuki T, et al: Transplantation of bone marrow-derived mesenchymal stem cells improves diabetic polyneuropathy in rats. Diabetes. 2008, 57: 3099-3107. 10.2337/db08-0031.
Kuo YR, Wang CT, Cheng JT, Wang FS, Chiang YC, Wang CJ: Bone marrow-derived mesenchymal stem cells enhanced diabetic wound healing through recruitment of tissue regeneration in a rat model of streptozotocin-induced diabetes. Plast Reconstr Surg. 2011, 128: 872-880. 10.1097/PRS.0b013e3182174329.
Yang K, Wang XQ, He YS, Lu L, Chen QJ, Liu J, Shen WF: Advanced glycation end products induce chemokine/cytokine production via activation of p38 pathway and inhibit proliferation and migration of bone marrow mesenchymal stem cells. Cardiovasc Diabetol. 2010, 9: 66-10.1186/1475-2840-9-66.
Falanga V, Iwamoto S, Chartier M, Yufit T, Butmarc J, Kouttab N, Shrayer D, Carson P: Autologous bone marrow-derived cultured mesenchymal stem cells delivered in a fibrin spray accelerate healing in murine and human cutaneous wounds. Tissue Eng. 2007, 13: 1299-1312. 10.1089/ten.2006.0278.
Wu Y, Chen L, Scott PG, Tredget EE: Mesenchymal stem cells enhance wound healing through differentiation and angiogenesis. Stem Cells. 2007, 25: 2648-2659. 10.1634/stemcells.2007-0226.
Javazon EH, Keswani SG, Badillo AT, Crombleholme TM, Zoltick PW, Radu AP, Kozin ED, Beggs K, Malik AA, Flake AW: Enhanced epithelial gap closure and increased angiogenesis in wounds of diabetic mice treated with adult murine bone marrow stromal progenitor cells. Wound Repair Regen. 2007, 15: 350-359. 10.1111/j.1524-475X.2007.00237.x.
Naruse K, Hamada Y, Nakashima E, Kato K, Mizubayashi R, Kamiya H, Yuzawa Y, Matsuo S, Murohara T, Matsubara T, et al: Therapeutic neovascularization using cord blood-derived endothelial progenitor cells for diabetic neuropathy. Diabetes. 2005, 54: 1823-1828. 10.2337/diabetes.54.6.1823.
Huang P, Li S, Han M, Xiao Z, Yang R, Han ZC: Autologous transplantation of granulocyte colony-stimulating factor-mobilized peripheral blood mononuclear cells improves critical limb ischemia in diabetes. Diabetes Care. 2005, 28: 2155-2160. 10.2337/diacare.28.9.2155.
Koike N, Fukumura D, Gralla O, Au P, Schechner JS, Jain RK: Tissue engineering: creation of long-lasting blood vessels. Nature. 2004, 428: 138-139. 10.1038/428138a.
Nor JE, Peters MC, Christensen JB, Sutorik MM, Linn S, Khan MK, Addison CL, Mooney DJ, Polverini PJ: Engineering and characterization of functional human microvessels in immunodeficient mice. Lab Invest. 2001, 81: 453-463. 10.1038/labinvest.3780253.
Au P, Daheron LM, Duda DG, Cohen KS, Tyrrell JA, Lanning RM, Fukumura D, Scadden DT, Jain RK: Differential in vivo potential of endothelial progenitor cells from human umbilical cord blood and adult peripheral blood to form functional long-lasting vessels. Blood. 2008, 111: 1302-1305.
Simper D, Stalboerger PG, Panetta CJ, Wang S, Caplice NM: Smooth muscle progenitor cells in human blood. Circulation. 2002, 106: 1199-1204. 10.1161/01.CIR.0000031525.61826.A8.
Acknowledgements
The authors would like to acknowledge support from the Canadian Diabetes Association (Z.K.) and the Lawson Health Research Institute (Z.K. and E.K.). Z.K. is a recipient of a New Investigator Award from the Heart and Stroke Foundation of Canada. All authors read and approved the final manuscript.
Authors contributions
EK and ZK both wrote and finalized the paper. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors confirm that there are no competing financial interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Keats, E.C., Khan, Z.A. Vascular stem cells in diabetic complications: evidence for a role in the pathogenesis and the therapeutic promise. Cardiovasc Diabetol 11, 37 (2012). https://doi.org/10.1186/1475-2840-11-37
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2840-11-37