
Abstract
Background
Genetic variation within GSTM2-5 genes may interfere with detoxification of environmental compounds, thereby having a detrimental effect on lung function following exposures such as tobacco smoke. We aim to investigate the influence of variants and associated methylation in the GSTM gene cluster with changes in lung function growth during adolescence.
Methods
Growth in forced expiratory volume (FEV1), forced vital capacity (FVC), and change in FEV1/FVC ratio measures were obtained from children in the Isle of Wight birth cohort at ages 10 and 18. Illumina GoldenGate assays were used to genotype 10 tagging polymorphisms from GSTM2 (rs574344 and rs12024479), GSTM3 (rs1537236, rs7483, and rs10735234), GSTM4 (rs668413, rs560018, and rs506008), and GSTM5 (rs929166 and rs11807) genes. Diplotypes were generated in the software Phase 3.0.2. DNA methylation was measured in over 450,000 CpG sites using the Infinium HumanMethylation450 BeadChip (Illumina 450K) in a subsample of 245 18-year olds from the Isle of Wight birth cohort. Gender, age, in utero smoke exposure, secondhand smoke exposure (SHS), and current smoking status were assessed via questionnaire; smoke exposures were validated with urine cotinine. We used linear mixed models to estimate the effect of GSTM diplotypes on lung function across time and examine interactions with tobacco smoke.
Results
1,121 (77%) out of 1,456 children had information on lung function at ages 10 or 18. After adjustment for false discovery rate, one diplotype in GSTM3 had a detrimental effect on changes in FEV1 (p=0.03), and another diplotype in GSTM3 reduced FVC (p=0.02) over time. No significant interactions with smoking were identified. SHS significantly modified the relationship between diplotypes and methylation levels in one GSTM2 CpG site; however, this site did not predict lung function outcomes at age 18. Joint effects of GSTM loci and CpG sites located within these loci on adolescent lung growth were detected.
Conclusions
Diplotypes within GSTM2-5 genes are associated with lung function growth across adolescence, but do not appear to modify the effect of tobacco smoke exposures on adolescent lung growth. Interactions between DNA methylation and diplotypes should be taken into account to gain further understanding on lung function in adolescence.
Similar content being viewed by others

Background
Pregnancy is a critical period of lung growth and development, making the fetus susceptible to environmental exposures [1]. Indeed, in utero exposure to tobacco smoke has shown detrimental effects on lung health [1–4]. In utero tobacco smoke exposure may affect morphogenesis and maturation of the lungs, leading to impaired lung health by adulthood [2]. The toxic effect of tobacco smoke exposure might be reduced through metabolizing xenobiotic products, thus minimizing oxidative stress, a physiological event caused by the imbalance between reactive oxygen species and the body’s ability to detoxify these products [5]. However, if detoxification is inefficient or absent within the mother or the child due to genetic polymorphisms, the exposed child may have a decreased ability to remove reactive oxygen species properly. Furthermore, effects of prenatal and early childhood exposure to tobacco smoke may persist through time, thus leading to reduced lung function growth even in adolescence. Additional exposures during adolescence, such as personal smoking, may further affect the growth trajectory in lung function in vulnerable individuals as well; therefore, it is important to control for these factors as well in order to determine the pure joint effects of tobacco smoke exposures and genetic polymorphisms responsible for detoxification.
The glutathione S-transferases (GST) are a superfamily of catalytic proteins responsible for detoxification of xenobiotic compounds, in conjugation with glutathione. There are seven classes of the GST superfamily: alpha, mu, pi, sigma, theta, omega, and zeta [5]. While the effects of glutathione S-transferase mu 1 (GSTM1) deletion have been extensively studied, results of studies examining these effects on various pulmonary outcomes are in conflict [6–11]. For instance, findings in a study among German schoolchildren aged 9 to 11 years only found a significant interaction between GSTM1 status and in utero smoke exposure on maximum mid expiratory flow (MMEF) levels [10], however Henderson et al. while confirming a detrimental effect of intrauterine tobacco smoke exposure on childhood lung function found no strong evidence of modification by maternal or child GSTM1 genotype [12]. Regarding growth, only one study found an association between GSTM1 gene function and lung function growth, where children with the GSTM1 deletion had slower lung function growth compared to those with the normal genotype [6]. In addition to copy number variation of the GSTM1gene, genetic variation in the form of single nucleotide polymorphisms (SNPs) within other members of the GST family also needs to be considered as it may lead to reduced expression of enzymes that detoxify harmful products that impact lung function growth.
One explanation for conflicting results of studies of the GSTM1 deletion is the failure to consider the impact of SNP variation in the adjacent GSTM2-5 loci. This is highlighted by the study of Breton et al. where GSTM2 was associated with growth in FEV1 and MMEF; GSTM4 was associated with FEV1, FVC, and MMEF; GSTM3 and GSTM5 were associated with MMEF; and in utero smoke exposure modified the effect of GSTM2 haplotypes on growth in FEV1 and FVC [13]. Because of these independent effects of variation within the GSTM2-5 loci on lung health, it is important to take this into account when examining the effect of GSTM1 variation on lung function growth in adolescence.
An alternative explanation for variation between studies in genetic epidemiology is variation in environmental exposure between cohorts [14]. Effects of genetic polymorphisms may be only seen in exposed populations, or vice versa, and in some circumstances, ‘flip-flop’ effects may be present where the effect of alleles on disease outcome is reversed depending on environmental exposure. Environmental exposure may also result in epigenetic effects such as altered DNA methylation that may either mask or synergize with the effects of germline genetic variation on phenotype [15]. For example in utero tobacco smoke exposure has been shown to result in changes in DNA methylation in cord blood DNA [16] that persist until adulthood (unpublished observations).
Thus far, only one study has reported the effects of genetic variation found in GSTM2-5 and their interaction effect with tobacco smoke exposure on lung function levels and growth in late adolescence, where in utero smoke exposure modified the effect of GSTM2 on lung function growth [13]. Since active smoking only had marginally significant effects on FEV1 and FEV1/FVC at age 18 in the Isle of Wight (IOW) cohort (unpublished observations), the effect may only be detected in those with a genetic susceptibility, acting through an epigenetic mechanism. To expand these findings, we analyzed data collected in the IOW birth cohort study. Specifically, we explore the interactive relationships between smoking status and individual GSTM diplotypes. Furthermore, we validated whether or not smoke exposures modified the effect of diplotypes on methylation levels of CpG sites within this gene cluster and determined whether or not methylation levels of these CpG sites affected lung function levels at age 18.
Methods
Study population
From January 1989 to February 1990 in the IOW, UK, 1,536 children were born; 1,456 mother-child pairs were enrolled into the cohort study. The local research ethics committee (National Research Ethics Service, NRES Committee South Central–Southampton B) approved the study and informed written parental consent was obtained for all participants at recruitment and subsequently at each follow-up. The IOW birth cohort has been described in detail elsewhere [17–19].
Lung function measurement
At ages 10 and 18 years, pulmonary function tests, which included forced expiratory volume in 1 second (FEV1), forced vital capacity (FVC), and FEV1/FVC ratio, were performed on all consenting children (n=1,121). Because mixed models can handle unbalanced data sets, characteristics of the analytic sample (children with pulmonary function test data available at age 10, age 18, or both) were compared with characteristics of the total cohort. Details of lung function measurement performed at ages 10 and 18 years were reported elsewhere [19]. Briefly, lung function measurements were conducted using a Koko Spirometer and software with a desktop portable device (PDS Instrumentation, Louisville, USA), according to American Thoracic Society guidelines [20]. Children were required to be free from respiratory infection for 14 days and were not taking oral steroids. In addition, they abstained from any β agonist medication for six hours and caffeine intake for at least four hours.
Laboratory analysis
Details on genomic DNA extraction from whole blood samples collected at age 18 and genotyping can be found in the online supplement (Additional file 1). Genomic DNA obtained from a subsample of female subjects (N=245) were bisulfite-converted and applied to the Infinium HumanMethylation450 BeadChip from Illumina (Illumina, San Diego, CA, USA), which is used to assess methylation in over 484,000 CpG sites found within approximately 24,000 genes. Arrays were processed using a standard protocol as described elsewhere [21], with multiple identical control samples assigned to each bisulphite conversion batch to assess assay variability and samples randomly distributed on microarrays to control against batch effects. The BeadChips were scanned using a BeadStation, and the methylation level (beta value) calculated for each queried CpG locus using the Methylation Module of BeadStudio software. All GSTM2-5 SNPs of interest were checked against a database containing SNPs that may artificially alter GSTM2-5 CpG site methylation produced from dbSNP build v130 [22]. Also, CpG sites were excluded from further analyses if nearby probe SNPs affected their methylation.
Covariates
Smoking during pregnancy was ascertained at enrollment, soon after the birth of the child. Information on tobacco smoking by mothers, by fathers or any other individual within the household was recorded at recruitment and updated at each follow-up at ages 1, 2, 4, 10, and 18 to assess secondhand smoke (SHS) exposure for ages 10 and 18 years. Personal smoking status of the offspring was collected via questionnaires and validated through urinary cotinine levels (ng/mmol) at age 18. Height in centimeters (cm) and weight in kilograms (kg) were assessed by study investigators at ages 1, 2, 4, 10, and 18 years; body mass index (BMI) was calculated. To ensure that effects of GSTM2-5 loci were independent of GSTM1, rs366631, a pseudo-SNP that acts as a GSTM1 deletion marker, was included as one of the covariates [23].
Statistical analysis
Haploview 4.2 [24] was used to assess each SNP for Hardy-Weinberg equilibrium (HWE) and create linkage disequilibrium (LD) blocks using the Gabriel et al. method.[25] SNPs with a HWE p-value ≥ 0.01 and with a minor allele frequency (MAF) ≥ 5% were considered for diplotype formation. Haplotypes were generated in Phase 2.1 [26, 27] and were linked as pairs if the probability of constituting a pair was >0.60. The advantage of using diplotypes is that each study subject has only one diplotype. Usage of haplotypes would double the sample size, thereby deflating variance and p-values [28]. Diplotypes with frequencies of 5% or less were combined into a “minor pairs” category. Diplotypes were characterized according to the sequence of the alleles involved (e.g. TC_TC or both haplotypes having thymidine and cytosine).
Statistical analyses were conducted using SAS 9.2 (Cary, NC) to assess effect of SNPs and diplotypes on lung function growth. Linear mixed models were applied to adjust for correlations between the repeated measurements. Models were built with an unstructured covariance matrix, which requires the least amount of constraints. Other covariance matrices were applied, but no other covariance matrix improved the fit (Akaike information criteria and Bayesian Schwarz information criterion).
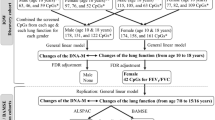
Separate models for changes of FEV1, FVC, and FEV1/FVC over time were created. Significance of interactions (α = 0.05) was assessed first, then confounders were removed if they did not change the estimates of diplotype categories by at least 10%. The most frequent diplotype served as the reference group, allowing us to assess the effects of less common pairs on lung function outcomes. Due to the number of hypothesis tests being conducted, diplotypes were declared significant at a false discovery rate (FDR) of 0.05 within each lung function outcome. The overall effect of the diplotype blocks were evaluated through the global F-test. To determine joint effects of diplotypes and smoke exposures, separate models were constructed for each type of diplotype-smoking interaction and were examined for significance through the global F-test. Part A of Figure 1 provides an overview of the analysis plan. To validate self-reported responses of maternal and active smoking, we compared cotinine levels of individuals belonging to exposed and unexposed groups using the Wilcoxon-Mann–Whitney test. Individuals who did not actively smoke were examined to assess the pure effect of SHS exposure.

Overview of data analysis plan. Part A describes analysis plan to assess main genetic effects of GSTM2-5 diplotypes and single nucleotide polymorphisms (SNPs) and their interactions with smoke exposures. Part B outlines the CpG site screening process and the plan to find methylation quantitative trait loci (methQTL), conditioned on smoking, and the joint effect of modifiable genetic variants (modGVs) and DNA methylation on lung function.
Methylation quantitative trait loci (methQTL) analyses were performed via Wilcoxon-Mann–Whitney tests to select CpG sites housed in the GSTM2-5 cluster that varied by diplotype among a subset of participants with methylation information (n=245). Because tobacco smoke exposure is postulated to interact with certain diplotypes and produce epigenetic changes, general linear models were used to assess the joint effect of diplotypes and tobacco smoke exposure on methylation levels of GSTM2-5 CpG sites. GSTM2-5 CpG sites that were the result of a significant interaction were then tested whether they modified the effect of diplotypes (modifiable genetic variants [modGV]) [15] on lung function levels at age 18 using general linear models, adjusting for confounders. Joint effects of these CpG sites and other GSTM2-5 modGVs were also examined. Part B of Figure 1 gives an overview of this analysis plan. Findings were verified through path analytical models. More details on path analysis are found in the supplement (Additional file 1).
Results
Data on lung function measures were available for 1,121 children (Table 1). No significant differences were found between this subsample and the total cohort (n = 1,456). All SNPs in the GSTM2-5 loci were in HWE and had a MAF ≥ 5% (Table 2). SNPs within each gene formed their own blocks based on linkage disequilibrium (LD) measures, where the r2 value was 0.8 or greater. A total of four blocks were generated (Additional file 1: Figure S2). The each block was comprised of the following SNPs: GSTM2 rs574344 and rs12024479; GSTM3 rs1537236, rs7483, and rs7537234; GSTM4 rs668413, rs560018, and rs506008; and GSTM5 rs929166 and rs11807; (Additional file 1: Table S1). All reported p-values were adjusted for multiple testing for an FDR of 0.05, when applicable. Only GSTM3 and GSTM5 show direct associations with gain in FEV1 or FVC (Table 3). Statistical analyses of the individual effects of SNPs within the GSTM2-5 cluster on gain in each lung function outcome yielded no significant findings (Additional file 2: Table S2). When looking at main effects of GSTM2-5 diplotypes by age, only GSTM5 diplotypes contributed to FEV1 levels at age 10 (Additional file 3: Table S3a). Several interactions between GSTM2-5 diplotypes and smoke exposures were detected, but only at age 18. SHS modified the relationship between GSTM2 and FEV1 and FVC levels (pinteraction = 0.004 and pinteraction = 0.0003, respectively) (Additional file 3: Table S4b); SHS modified the relationship between GSTM3 and FEV1 levels (pinteraction = 0.04) (Additional file 3: Additional file 3: Table S5b; and active smoking modified the relationship between GSTM5 and FEV1 and FVC levels (pinteraction = 0.004 and pinteraction = 0.05, respectively) (Additional file 3: Table S7c).
Multivariable analyses
GSTM2
Diplotypes within the GSTM2 locus did not significantly contribute to gain in FEV1, FVC, and change in FEV1/FVC ratio (Table 3). Additionally, no statistically significant interactions between diplotypes and tobacco smoke exposures were detected (Additional file 3: Tables S8a-c).
GSTM3
The overall effect of variations in diplotypes within GSTM3 neared significance for gain in both FEV1 and FVC (p = 0.12 and p = 0.08, respectively). After adjustment for false discovery rate, one diplotype was associated with significantly lower gain in FEV1 (GGG_GGG: -63.81 mL, p = 0.03) (Table 3). This diplotype also produced detrimental effects on gain in FVC, but it was not significant (GGG_GGG: -51.91, p = 0.09). An additional diplotype was associated with significantly lower gain in FVC (AGA_AAA: -83.12 mL, p = 0.02). In regard to tobacco smoke interactions, SHS exposure marginally modified the relationship between GSTM3 diplotypes and change in FEV1/FVC (pinteraction = 0.06) (Additional file 3: Table S9b). Statistically significant interactions between diplotypes within GSTM3 and in utero smoke exposure and personal smoking at age 18 were not observed (Additional file 3: Tables S9a and S9c).
GSTM4
Overall, diplotypes created in the GSTM4 locus had no significant effects on gain in FEV1, FVC, and change in FEV1/FVC ratio (Table 3). Additionally, no significant interactions with in utero smoke exposure, SHS exposure, and personal smoking at age 18 were observed (Additional file 3: Tables S10a-c).
GSTM5
Global F-tests indicated a significant contribution of GSTM5 diplotypes to gain in FEV1 (p = 0.02, Table 3). Only one pair showed a statistically significant positive relationship with gain in FEV1 (AA_CA: 53.26 mL, p = 0.04). Although the interaction between GSTM5 diplotypes and SHS exposure appeared to affect FEV1/FVC change, this relationship was not statistically significant (p = 0.08) (Additional file 3: Table S11b). No other statistically significant interactions were seen between this diplotype group and in utero smoke exposure and current smoking (Additional file 3: Tables S11a and S11c).
Methylation analyses
Although no effects of significant interactions on lung function growth were present in this study, the GSTM2×in utero smoke exposure interaction was found to be significant in Breton et al.[13], suggesting that this smoke exposure changed the function of this gene through epigenetic modifications. As a result, methylation levels in this gene cluster were investigated for their role in lung function. After removal of 25 CpG sites that may be affected by probe SNPs, methQTL analyses revealed that several CpG site methylation levels were dependent on genetic variants of the specific gene: diplotypes of the GSTM2 diplotypes predicted three CpG site methylation levels; GSTM3 diplotypes predicted five CpG site methylation levels; GSTM4 gene predicted five CpG site methylation levels; and GSTM5 diplotypes predicted one CpG site methylation levels (Additional file 3: Table S12).
When looking at the joint effect of GSTM2-5 diplotypes and tobacco smoke exposures (SHS and active smoking), only SHS exposure at the age of 18 modified the relationship between GSTM2 diplotypes and one CpG site found in GSTM2: cg06970744 (pinteraction = 0.01; Table 4). However, these sites did not predict lung function levels at age 18 (Additional file 3: Table S13). The joint effect of this CpG site and other modGVs had a significant relationship with FEV1 (cg06970744× GSTM5: pinteraction = 0.02) and FVC levels at age 18 (cg06970744× GSTM5: pinteraction = 0.02) (Table 5). Increasing levels of methylation at cg06970744 produced increased FEV1 and FVC levels for individuals with the CA_CA diplotype in GSTM5.
Results of the path analyses revealed no indirect effects of diplotypes on lung function through methylation at cg06970744 at age 18 (Additional file 3: Table S14).
Discussion
Results suggest that variation within the GSTM3 and GSTM5 loci had significant impact on lung function outcomes at age 18 after adjustment for confounding. Some diplotypes, but none of their interactions with smoke exposure (in utero, SHS, or active smoking exposure at 18), produced an independent, statistically significant relationship with FEV1 and FVC, but not with FEV1/FVC. Additional analyses suggest a multi-stage model. First, DNA methylation was affected by diplotypes conditionally on smoking exposure; second, an altered DNA methylation modified the effect of diplotypes on lung function (acting as modGVs), leading to significant effects of GSTM2 and GSTM5 on FEV1 and FVC. Path analyses show that methylation at cg06970744 did not lie on the pathway between GSTM5 diplotypes and lung function, further providing evidence that methylation at certain sites can modify the effect of genetic variation on an outcome. Also, methylation at this site appears to be unaffected by nearby SNPs (unpublished observations).
Several GSTM3 diplotypes had strong, negative effects on gain in lung function. SNPs in GSTM3 included a functional SNP (rs7483). Previous studies have found an association with this SNP and Alzheimer’s disease [29, 30]. Breton et al. have previously examined the association of rs7483 with lung function, and similar to the present study, no significant associations were found [13]. GSTM4 had a synonymous coding SNP (rs506008). This SNP produced positive, but insignificant effects on lung function across time and thus more than likely does not make contributions to lung function at the diplotype level. There are no reported associations between rs506008 and any disease outcomes. Although GSTM5 had no functional SNPs, there was a significant lung function improvement in those who possessed the AA_CA diplotype, demonstrating a need to investigate this particular region. Only one of the SNPs (rs11807) was previously reported in the literature, showing a strong association with hypertension [31]; however, no association with lung function had been reported.
Due to previous findings that showed lower lung function outcomes due to various tobacco smoke exposures [2–4, 32], we assessed the critical period where lung development may be severely impaired by tobacco smoke exposure. No two-way interaction effects of tobacco smoke with GSTM2-5 loci were seen, which is in contrast to findings by Breton et al., who reported interaction effects with GSTM2 and in utero tobacco smoke [13]. This discrepancy may be either due to misclassification of the exposure and/or insufficient sample size in our study or missing replication in the study by Breton et al.[13]. Because questions involving tobacco smoke exposure at age 18 were validated with urinary cotinine levels (Additional file 3: Table S9), there is no suggestion of major misclassification, as those who were active smokers or were exposed to SHS had significantly higher levels of cotinine compared to nonsmokers. The study by Breton et al. included at least one more repeated measurement [13], hence, it is possible that the present study does not have equal statistical power to detect interactions. However, there is a need of replication studies to examine the role of these genes on lung function in conjunction with tobacco smoke exposure, especially in different environments. It is also necessary to consider that other environmental and lifestyle exposures including air pollution and paracetamol (acetaminophen) use [33] may also alter oxidative stress [34] and mask or falsely indicate an effect of related exposures. Air pollution was not controlled in the study by Breton et al.[13], but was reported to modify the effect of a similarly functioning gene (GSS) on lung function growth in another study of this group [33]. Because it has been suggested in the literature that tobacco smoke exposure, especially in utero[35], may epigenetically modify these genes, hence changing the function of these genes, we tested the effect of CpG sites on lung function levels at age 18. Although there were no independent effects of these CpG site methylation levels on lung function outcomes, it is interesting to note that these effects were not seen until joint effects of GSTM2 and GSTM5 diplotypes were taken into account, indicating that DNA methylation may modify the effect of genetic variants on lung function, a mechanism that needs further investigation [15]. These findings may also help explain the lack of consistency between the present study’s findings and the findings of Breton et al.[13]. It may not be the genotype that produces inconsistent results, but rather different DNA methylation levels in different study groups that accounted for the discrepancy.
To further lend validity to this study, selection bias was not apparent regarding availability of lung function data. Approximately 95% (1456/1536) of mother-child pairs were enrolled into this study; and those who underwent pulmonary function tests were not significantly different from the total cohort (Table 1). Also, lung function measurements were obtained under standardized conditions in a prospective manner, decreasing the likelihood of information bias. With respect to genotyping data, all SNPs that were shared with Breton et al. were in HWE and had comparable frequencies with Caucasians in their sample [13]. These polymorphisms also agreed substantially in their association with FEV1 and FVC with our findings (Additional file 1: Figure S2 and Additional file 2: Figure S3). In regard to haplotypes, diplotypes (haplotype-pairs) were used, thus reducing uncertainty and subsequently misclassification of individuals, which is often encountered in haplotype association studies [36]. Also, our population was homogeneous, meaning controlling for population stratification is unnecessary and ensures HWE, which appeared to be an issue for Breton et al.[13], possibly resulting in different findings. While SNPs included in the haplotype analysis of the latter study were in HWE within ethnic groups, they were not in HWE when examining the entire population [13]. This is problematic because inclusion of SNPs that deviate from HWE may produce spurious associations between haplotypes and lung function [37, 38]. In addition, the findings by Breton et al.[13] may be due to population stratification. The authors identified ancestry indicators, controlled these as confounders but did not stratify their analysis by these markers. Nevertheless, population stratification addresses the possibility that haplotypes may reveal different associations in different ethnic/racial strata. This may also account for the lack of agreement of single SNP effects on lung function outcomes between the present study and the Breton et al. study [13].
Despite the strengths of our study, some limitations are present. First, because maternal smoking and active smoking during adolescence is obtained via self-reported questionnaires, misclassification of this exposure is possible. Our previous publications have shown that maternal smoking during pregnancy interacts with the IL13 and IL1RN genes and increases the risk of asthma and wheezing [39, 40]. Hence, ascertainment of maternal smoking seemed to provide valid information. However, maternal smoking × IL13 and IL1RN gene interaction assumes an effect of smoking on these genes. Against that, GSTM2-5 × maternal smoking interactions assume that the genes regulate the toxicity of tobacco smoke, possibly via epigenetic mechanisms. Also, based on results of the methylation analyses in the present study, the joint effect of maternal smoking and GSTM2-5 diplotypes did not influence methylation levels of CpG sites within the GSTM2-5 cluster; however, these methylation levels were captured at age 18 and therefore we cannot ascertain whether or not epigenetic changes had taken place due to this particular exposure nor can we determine that methylation levels at age 18 are representative of early-life methylation profiles. Regarding validation of other smoke exposures, passive smoke exposure and active smoking at age 18 was associated with increased cotinine levels. Also, active smoking at age 18 led to deficits in FEV1/FVC after adjustments for GSTM5 diplotype, GSTM1 genotype, sex, BMI, height, in utero smoke exposure, and SHS (Additional file 3: Table S16). Reduced FEV1/FVC has previously been observed in adolescent smokers [41].
Second, not all SNPs used in the Breton et al. study were genotyped in this work. Therefore combinations of SNPs that were not included in the haplotype construction may lead to different results. Variation in FEV1 in these results may be due to differences in population characteristics between the two studies, residual confounding, or as demonstrated by methylation levels, differences in expression levels of detoxification enzymes produced by this set of genes. Based on the results of this study, methylation levels at age 18 within this gene cluster can be ruled out because analyses revealed no significant effect of methylation on lung function levels at 18. The goal of this study was to expand upon findings by Breton et al. through accounting for methylation [13].
Conclusions
Based on results of this study, it is clear that certain genes within the GSTM2-5 loci explain some differences in lung function in late adolescence; however, joint effects with tobacco smoke exposure were not detected. This finding was validated through examination of joint effects of diplotypes and in utero smoke exposure, which did not significantly alter methylation levels within this gene cluster. Only one study has reported an interaction of maternal smoking with GSTM2-5 genes [13]. A past genetic association study has examined the interactive effects of maternal genotypes on other GST genes [12], but no studies have examined the joint effect of maternal genotype and GSTM2-5 genotypes in their offspring. Future studies are necessary to elucidate conflicting results, taking into account DNA methylation ×gene interactions, interaction with other xenobiotics affecting the glutathione levels, and interaction with the maternal genome.
References
Wu ZX, Hunter DD, Kish VL, Benders KM, Batchelor TP, Dey RD: Prenatal and early, but not late, postnatal exposure of mice to sidestream tobacco smoke increases airway hyperresponsiveness later in life. Environ Health Perspect. 2009, 117: 1434-1440.
Beyer D, Mitfessel H, Gillissen A: Maternal smoking promotes chronic obstructive lung disease in the offspring as adults. Eur J Med Res. 2009, 14 (Suppl 4): 27-31. 10.1186/2047-783X-14-S4-27.
Leung DY, Szefler SJ: In utero smoke (IUS) exposure has been associated with increased prevalence of asthma and reduced lung function in healthy children. J Allergy Clin Immunol. 2010, 126: 481-482. 10.1016/j.jaci.2010.08.004.
Gilliland FD, Berhane K, McConnell R, Gauderman WJ, Vora H, Rappaport EB, Avol E, Peters JM: Maternal smoking during pregnancy, environmental tobacco smoke exposure and childhood lung function. Thorax. 2000, 55: 271-276. 10.1136/thorax.55.4.271.
Hayes JD, Flanagan JU, Jowsey IR: Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005, 45: 51-88. 10.1146/annurev.pharmtox.45.120403.095857.
Gilliland FD, Gauderman WJ, Vora H, Rappaport E, Dubeau L: Effects of glutathione-S-transferase M1, T1, and P1 on childhood lung function growth. Am J Respir Crit Care Med. 2002, 166: 710-716. 10.1164/rccm.2112065.
Imboden M, Downs SH, Senn O, Matyas G, Brandli O, Russi EW, Schindler C, Ackermann-Liebrich U, Berger W, Probst-Hensch NM: Glutathione S-transferase genotypes modify lung function decline in the general population: SAPALDIA cohort study. Respir Res. 2007, 8: 2-10.1186/1465-9921-8-2.
Hersh CP, Demeo DL, Lange C, Litonjua AA, Reilly JJ, Kwiatkowski D, Laird N, Sylvia JS, Sparrow D, Speizer FE, et al: Attempted replication of reported chronic obstructive pulmonary disease candidate gene associations. Am J Respir Cell Mol Biol. 2005, 33: 71-78. 10.1165/rcmb.2005-0073OC.
Romieu I, Sienra-Monge JJ, Ramirez-Aguilar M, Moreno-Macias H, Reyes-Ruiz NI, del Rio-Navarro EB, Hernandez-Avila M, London SJ: Genetic polymorphism of GSTM1 and antioxidant supplementation influence lung function in relation to ozone exposure in asthmatic children in Mexico City. Thorax. 2004, 59: 8-10.
Kabesch M, Hoefler C, Carr D, Leupold W, Weiland SK, von Mutius E: Glutathione S transferase deficiency and passive smoking increase childhood asthma. Thorax. 2004, 59: 569-573. 10.1136/thx.2003.016667.
He JQ, Ruan J, Connett JE, Anthonisen NR, Pare PD, Sandford AJ: Antioxidant gene polymorphisms and susceptibility to a rapid decline in lung function in smokers. Am J Respir Crit Care Med. 2002, 166: 323-328. 10.1164/rccm.2111059.
Henderson AJ, Newson RB, Rose-Zerilli M, Ring SM, Holloway JW, Shaheen SO: Maternal Nrf2 and gluthathione-S-transferase polymorphisms do not modify associations of prenatal tobacco smoke exposure with asthma and lung function in school-aged children. Thorax. 2010, 65: 897-902. 10.1136/thx.2009.125856.
Breton CV, Vora H, Salam MT, Islam T, Wenten M, Gauderman WJ, Van den Berg D, Berhane K, Peters JM, Gilliland FD: Variation in the GST mu locus and tobacco smoke exposure as determinants of childhood lung function. Am J Respir Crit Care Med. 2009, 179: 601-607. 10.1164/rccm.200809-1384OC.
Holloway JW, Savarimuthu Francis S, Fong KM, Yang IA: Genomics and the respiratory effects of air pollution exposure. Respirology. 2012, 17: 590-600. 10.1111/j.1440-1843.2012.02164.x.
Karmaus W, Ziyab AH, Everson T, Holloway JW: Epigenetic mechanisms and models in the origins of asthma. Curr Opin Allergy Clin Immunol. 2012, In press
Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, Huang Z, Hoyo C, Midttun O, Cupul-Uicab LA, et al: 450K Epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012, 120: 1425-1431.
Arshad SH, Hide DW: Effect of environmental factors on the development of allergic disorders in infancy. J Allergy Clin Immunol. 1992, 90: 235-241. 10.1016/0091-6749(92)90077-F.
Arshad SH, Stevens M, Hide DW: The effect of genetic and environmental factors on the prevalence of allergic disorders at the age of two years. Clin Exp Allergy. 1993, 23: 504-511. 10.1111/j.1365-2222.1993.tb03238.x.
Scott M, Raza A, Karmaus W, Mitchell F, Grundy J, Kurukulaaratchy RJ, Arshad SH, Roberts G: Influence of atopy and asthma on exhaled nitric oxide in an unselected birth cohort study. Thorax. 2010, 65: 258-262. 10.1136/thx.2009.125443.
American Thoracic Society: Standardization of spirometry, 1994 update. Am J Respir Crit Care Med. 1995, 152: 1107-1136.
Bibikova M, Fan JB: GoldenGate assay for DNA methylation profiling. Methods Mol Biol. 2009, 507: 149-163. 10.1007/978-1-59745-522-0_12.
Supporting manifest file for HumanMethylation450 beadchip: http://supportres.illumina.com/documents/downloads/productfiles/humanmethylation450/humanmethylation450_15017482_v1-2.csv,
Huang RS, Chen P, Wisel S, Duan S, Zhang W, Cook EH, Das S, Cox NJ, Dolan ME: Population-specific GSTM1 copy number variation. Hum Mol Genet. 2009, 18: 366-372.
Barrett JC, Fry B, Maller J, Daly MJ: Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005, 21: 263-265. 10.1093/bioinformatics/bth457.
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, et al: The structure of haplotype blocks in the human genome. Science. 2002, 296: 2225-2229. 10.1126/science.1069424.
Stephens M, Scheet P: Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet. 2005, 76: 449-462. 10.1086/428594.
Stephens M, Smith NJ, Donnelly P: A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001, 68: 978-989. 10.1086/319501.
Sasieni PD: From genotypes to genes: doubling the sample size. Biometrics. 1997, 53: 1253-1261. 10.2307/2533494.
Maes OC, Schipper HM, Chong G, Chertkow HM, Wang E: A GSTM3 polymorphism associated with an etiopathogenetic mechanism in Alzheimer disease. Neurobiol Aging. 2010, 31: 34-45. 10.1016/j.neurobiolaging.2008.03.007.
Hong GS, Heun R, Jessen F, Popp J, Hentschel F, Kelemen P, Schulz A, Maier W, Kolsch H: Gene variations in GSTM3 are a risk factor for Alzheimer's disease. Neurobiol Aging. 2009, 30: 691-696. 10.1016/j.neurobiolaging.2007.08.012.
Delles C, Padmanabhan S, Lee WK, Miller WH, McBride MW, McClure JD, Brain NJ, Wallace C, Marcano AC, Schmieder RE, et al: Glutathione S-transferase variants and hypertension. J Hypertens. 2008, 26: 1343-1352. 10.1097/HJH.0b013e3282fe1d67.
Gold DR, Wang X, Wypij D, Speizer FE, Ware JH, Dockery DW: Effects of cigarette smoking on lung function in adolescent boys and girls. N Engl J Med. 1996, 335: 931-937. 10.1056/NEJM199609263351304.
Breton CV, Salam MT, Vora H, Gauderman WJ, Gilliland FD: Genetic variation in the glutathione synthesis pathway, air pollution, and children's lung function growth. Am J Respir Crit Care Med. 2011, 183: 243-248. 10.1164/rccm.201006-0849OC.
Shaheen S, Potts J, Gnatiuc L, Makowska J, Kowalski ML, Joos G, van Zele T, van Durme Y, De Rudder I, Wohrl S, et al: The relation between paracetamol use and asthma: a GA2LEN European case–control study. Eur Respir J. 2008, 32: 1231-1236. 10.1183/09031936.00039208.
Suter MA, Aagaard K: What changes in DNA methylation take place in individuals exposed to maternal smoking in utero?. Epigenomics. 2012, 4: 115-118. 10.2217/epi.12.7.
Lamina C, Kuchenhoff H, Chang-Claude J, Paulweber B, Wichmann HE, Illig T, Hoehe MR, Kronenberg F, Heid IM: Haplotype misclassification resulting from statistical reconstruction and genotype error, and its impact on association estimates. Ann Hum Genet. 2010, 74: 452-462. 10.1111/j.1469-1809.2010.00593.x.
Chakraborty R, Smouse PE: Recombination of haplotypes leads to biased estimates of admixture proportions in human populations. Proc Natl Acad Sci U S A. 1988, 85: 3071-3074. 10.1073/pnas.85.9.3071.
Li CC: Population subdivision with respect to multiple alleles. Ann Hum Genet. 1969, 33: 23-29. 10.1111/j.1469-1809.1969.tb01625.x.
Sadeghnejad A, Karmaus W, Arshad SH, Kurukulaaratchy R, Huebner M, Ewart S: IL13 gene polymorphisms modify the effect of exposure to tobacco smoke on persistent wheeze and asthma in childhood, a longitudinal study. Respir Res. 2008, 9: 2-10.1186/1465-9921-9-2.
Ramadas RA, Sadeghnejad A, Karmaus W, Arshad SH, Matthews S, Huebner M, Kim DY, Ewart SL: Interleukin-1R antagonist gene and pre-natal smoke exposure are associated with childhood asthma. Eur Respir J. 2007, 29: 502-508. 10.1183/09031936.00029506.
Yoo S, Kim HB, Lee SY, Kim BS, Kim JH, Yu J, Kim BJ, Lee DH, Seong MW, Hong SJ: Effect of active smoking on asthma symptoms, pulmonary function, and BHR in adolescents. Pediatr Pulmonol. 2009, 44: 954-961. 10.1002/ppul.21066.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2466/13/56/prepub
Funding
This study was funded in part by the National Institutes of Health R01 HL082925and AI061471. The 10-year follow-up of this study was funded by National Asthma Campaign, UK (Grant No 364) and the 18-year follow-up by NIH/NHLBI R01 HL082925 (PI: S. Hasan Arshad). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors gratefully acknowledge the cooperation of the children and parents who participated in this study and the Isle of Wight research team in collecting data and Nikki Graham for technical support. We thank the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant reference 090532/Z/09/Z and MRC Hub grant G0900747 91070) for the generation of the methylation data.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MA performed the research, statistical analyses, data interpretation and wrote the manuscript. WK, JWH, HZ, RJK, GR and SHA performed research, interpreted data, and critically read the manuscript. SE carried out the molecular genetic studies, interpreted data and critically read the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12890_2012_430_MOESM1_ESM.docx
Additional File 1: Sample collection, genotyping methods, and haplotype blocks within GSTM2-5. This additional file contains information on sample collection, genotyping, and haplotype block creation (Figure S2) and diplotype frequencies (Table S1).(DOCX 111 KB)
12890_2012_430_MOESM2_ESM.docx
Additional File 2: Statistical analyses and results of SNP effects. This additional file contains information on statistical methods used to analyze effects of SNPs on lung function growth (Table S2). Two plots were created to compare results from the present study with a study by Breton et al. (Figures S3 and S4).(DOCX 66 KB)
12890_2012_430_MOESM3_ESM.docx
Additional File 3: Effect of GSTM2-5 diplotypes x smoke interactions and CpG sites on lung function levels and lung function growth. This file contains supplementary tables on main effects of GSTM2-5 diplotypes on lung function levels at ages 10 and 18 (Tables S3a and S3b), interactions between GSTM2-5 diplotypes and smoke exposures on lung function levels at ages 10 and 18 (Tables S4a-S4c, Tables S5a-S5c, Tables S6a-S6c, and Tables S7a-S7c), interactions between GSTM2-5 diplotypes and smoke exposures on lung function growth (Tables S8a-S8c, Tables S5a-S5c, and Tables S6a-S6c), the influence of diplotypes on CpG site methylation (Table S12), the effect of CpG sites on lung function growth (Table S13), validation of smoke exposure categories via urinary cotinine (Table S15), and the effect of active smoking on lung function levels at age 18 (Table S16).(DOCX 128 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Alexander, M., Karmaus, W., Holloway, J.W. et al. Effect of GSTM2-5polymorphisms in relation to tobacco smoke exposures on lung function growth: a birth cohort study. BMC Pulm Med 13, 56 (2013). https://doi.org/10.1186/1471-2466-13-56
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2466-13-56