
Abstract
Background
The pattern of lung function development from pre-adolescence to adulthood plays a significant role in the pathogenesis of respiratory diseases. Inconsistent findings in genetic studies on lung function trajectories, the importance of DNA methylation (DNA-M), and the critical role of adolescence in lung function development motivated the present study of pre-adolescent DNA-M with lung function trajectories. This study investigated epigenome-wide associations of DNA-M at cytosine-phosphate-guanine dinucleotide sites (CpGs) at childhood with lung function trajectories from childhood to young adulthood.
Methods
DNA-M was measured in peripheral blood at age 10 years in the Isle of Wight (IOW) birth cohort. Spirometry was conducted at ages 10, 18, and 26 years. A training/testing-based method was used to screen CpGs. Multivariable logistic regressions were applied to assess the association of DNA-M with lung function trajectories from pre-adolescence to adulthood. To detect differentially methylated regions (DMRs) among CpGs, DMR enrichment analysis was conducted. Findings were further tested in the Avon Longitudinal Study of Parents and Children (ALSPAC) cohort. Pathway analyses were performed on the mapped genes of the identified CpGs and DMRs. Biological relevance of the identified CpGs was assessed with gene expression. All analyses were stratified by sex.
Results
High and low trajectories of FVC, FEV1, and FEV1/FVC in each sex were identified. At PBonferroni < 0.05, DNA-M at 96 distinct CpGs (41 in males) showed associations with FVC, FEV1, and FEV1/FVC trajectories in IOW cohort. These 95 CpGs (cg24000797 was disqualified) were further tested in ALSPAC; 44 CpGs (19 in males) of these 95 showed the same directions of association as in the IOW cohort; and three CpGs (two in males) were replicated. DNA-M at two and four CpGs showed significant associations with the corresponding gene expression in males and females, respectively. At PFDR < 0.05, 23 and 10 DMRs were identified in males and females, respectively. Pathways were identified; some of those were linked to lung function and chronic obstructive lung diseases.
Conclusion
The identified CpGs at pre-adolescence have the potential to serve as candidate markers for lung function trajectory prediction and chronic lung diseases.
Similar content being viewed by others
Introduction
The patterns of lung function development, from pre-adolescence to adulthood, play a major role in the pathogenesis of respiratory diseases. Recent studies have highlighted that reduced lung function development in young adulthood predisposes to respiratory and other chronic diseases in later life and is also associated with early mortality [1, 2]. Lung function grows dramatically throughout childhood and reaches its peak in adolescence or early adulthood. After a brief period of stable lung function in early adulthood, a gradual decline ensues with aging [3,4,5]. Previous studies have demonstrated that early decline of lung function, and/or failure to reach maximal level lung function (even with a normal rate of decline), is associated with the development of chronic obstructive pulmonary disease (COPD) in later life [3,4,5,6], suggesting that the origins of COPD lie, in part, in early life [6, 7]. COPD is projected to become the third leading cause of death worldwide by 2030 [8, 9], highlighting how insights into the trajectories of lung function development from childhood-to-young adulthood would be beneficial for COPD prediction, prevention, and management.
Encouraged by the significance and advantage of longitudinal designs, we and others examined the temporal trend of lung function growth and decline through multiple important stages of life: childhood, adolescence, and adulthood [4, 10,11,12]. These studies demonstrated that there are distinct groups of individuals with a persistently low lung function trajectory from childhood-to-adulthood, suggested a potential connection with COPD in later life. One study showed weak evidence that persistently low FEV1 trajectory is associated with genetic factors in addition to multiple childhood exposures [10]. A recent study based on repeated measurement of lung function in adults reported that genetic variants associated with cross-sectional lung function measurements were not associated with a longitudinal decline of lung function [13]. These inconsistent findings in genetic studies and the clear impact of environmental factors on lung function motivated the investigation of the role of epigenetic factors such as DNA methylation (DNA-M) in determining variation in lung function between people and over time.
DNA-M represents an epigenetic mechanism that regulates gene expression, which consequently influences disease risk [14, 15]. Growing evidence indicates that DNA-M in whole blood is associated with lung function and its related diseases such as asthma and COPD [15,16,17,18]. Pre-adolescence adverse exposure is shown to be associated with adulthood chronic lung diseases [19]. As an epigenetic memory of past exposures, the role of pre-adolescence DNA-M on lung function trajectories from pre-adolescence to young adulthood is unknown [19]. We hypothesized that differential methylation at certain cytosine-phosphate-guanine dinucleotide sites (CpGs) in childhood is associated with the trajectories of lung function. Given that lung function growth and decline is sex-dependent and such dependence is attributable to multiple biological determinants, including dimensional/anatomical, immunological, and hormonal determinants [20,21,22,23], we examined the hypothesis in male and female participants, separately [12, 24]. The study was carried out in the birth cohort located on the Isle of Wight (IOW) in the UK. To assess the potential of generalizability, an independent UK birth cohort, the Avon Longitudinal Study of Parents and Children (ALSPAC) cohort, was used for replication.
Results
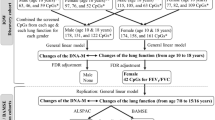
In the complete IOW cohort (n = 1456), lung function measurements at ages 10, 18, and 26 years were available for 980 (67.3%), 838 (57.6%), and 547 (37.6%) participants, respectively. A total of 377 male and 432 female participants were included for trajectory analyses, and each of the participants had spirometry tests at two or more of the three ages (Fig. 1). The analysed sub-sample (n = 809) was not statistically different from the complete cohort (n = 1456) regarding FVC, FEV1, and FEV1/FVC at the corresponding ages (Table 1).

Flow chart of study participants of the IOW cohort for each step of the analysis
Lung function trajectories
In trajectory analyses, two distinct lung function trajectories from pre-adolescence to adulthood (10 to 26 years of age) were identified in the IOW cohort, labelled as ‘low trajectory’ and ‘high trajectory’ for FVC, FEV1, and FEV1/FVC in both male and female participants (Fig. 2). Among the 377 male participants, 199 (52.8%), 204 (54.1%), and 96 (25.5%) were assigned to low FVC, FEV1, and FEV1/FVC trajectories (Fig. 2: gray “dashed lines”), respectively, using probability > 0.5 to define class membership. Among these male participants, at least 82% and 92% of them had a trajectory assignment probability ≥ 0.7 for low and high trajectories, respectively.

Distinct lung function trajectories from childhood-to-adulthood following comparable patterns in the IOW and ALSPAC cohorts. Note: Among the subjects assigned to each trajectory with a probability > 0.5, most assignments were with a probability ≥ 0.7, much higher than 0.5. In the following, we provide, for each sex and lung function parameter, the percentages of assignments with an assignment probability ≥ 0.7: (1) Among the male participants who were assigned to persistently low lung function trajectories with a probability > 0.5, 173 of 199 (86.9%) for FVC; 178 of 204 (87.3%) for FEV1, and 79 of 96 (82.3%) for FEV1/FVC had a probability ≥ 0.7 of belonging to their trajectory class. (2)The males assigned to high lung function trajectories with a probability > 0.5, 165 of 178, (92.7%) for FVC, 156 of 173 (90.2%) for FEV1, and 267 of 281 (95.0%) for FEV1/FVC had a probability ≥ 0.7 of belonging to the high lung function trajectory group. (3) Among the female participants assigned to each trajectory with a probability > 0.5, 190 of 215 (88.4%) for FVC, 188 of 205 (91.7%) for FEV1, and 78 of 95 (82.1%) for FEV1/FVC in persistently low lung function trajectory group had a probability ≥ 0.7 of belonging to their trajectory class. (4) The females assigned to persistently high lung function trajectories with a probability > 0.5, 195 of 217, (89.9%) for FVC, 191 of 227 (84.1%) for FEV1, and 311 of 337 (92.3%) for FEV1/FVC had a probability ≥ 0.7 of belonging to the normal lung function trajectory group
Similarly, among the 432 female participants, 215 (49.8%), 205 (47.5%), and 95 (22.0%) were assigned to low FVC, FEV1, and FEV1/FVC trajectories, respectively (Fig. 2: gray “two-dot-dashed lines”) using probability > 0.5 to define the group. More than 82% and 84% of the female participants had a probability ≥ 0.7 of being assigned to the low and high trajectories, respectively, for all three lung function parameters.
Pre-adolescence DNA-M and lung function trajectories
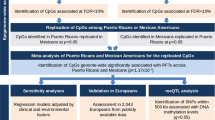
In total, 176 of the 377 male and 136 of the 432 female participants included in the analyses had DNA-M data available at age 10 years (Fig. 1). In screening, 119 distinct CpGs for males (33 CpGs for FVC, 37 for FEV1, and 51 for FEV1/FVC) and 56 distinct CpGs for females (22 CpGs for FVC, 21 for FEV1, and 16 for FEV1/FVC, Fig. 3) passed and were included in final analyses for their associations with lung function trajectories, with the effects of confounders adjustment. There was no overlap between the 119 and 56 CpGs identified in males and females, respectively.

Flow chart of statistical analyses with the number of identified CpGs at each step. Note: *Number of significant CpGs were mentioned in an order for FVC, FEV1, and FEV1/ FVC changes, respectively
Using multivariable logistic regression models, DNA-M levels at age 10 years of 11, 13, and 17 CpGs in males, and 21, 21, and 16 CpGs in females were statistically significantly associated with FVC, FEV1, and FEV1/FVC trajectories from pre-adolescence to adulthood, respectively, after correcting for multiple testing using the Bonferroni approach. Among the 96 distinct CpGs identified in the IOW cohort, 95 were further examined in the ALSPAC (cg24000797 was excluded during quality control in ALSPAC).
Testing IOW cohort findings in the ALSPAC
In total, 4,861 participants (males = 2216) in ALSPAC had FVC, FEV1, and FEV1/FVC measurements for more than a single time point at ages 8-, 15-, and 24-year follow-up and were included in the trajectory analyses. Of these participants, 691 had DNA-M data at age 7 years.
We identified two trajectories, low and high, for FVC, FEV1, and FEV1/FVC across 8, 15, and 24 years, comparable to those from the IOW cohort (Fig. 2). Next, for the 95 CpGs identified in the IOW cohort, we tested the association of DNA-M at age 7 years with lung function trajectories. Among the 95 CpGs, DNA-M at 44 distinct CpGs (2 CpGs overlapped between FEV1 and FVC in females) showed the consistent association with lung function trajectories with those in the IOW cohort in terms of the direction of association. These include 19 CpGs (6 CpGs for FVC, 7 for FEV1, and 6 for FEV1/FVC) in males, and 25 CpGs (11 CpGs for FVC, 9 for FEV1, and 7 for FEV1/FVC) in females (Table 2, Fig. 4, Additional file 1: Table S1). These 44 CpGs were noted as IOW-ALSPAC consistent CpGs. Among these CpGs, cg14669749 and cg21131402 showed statistically significant associations with FEV1/FVC trajectories in males and cg23987789 with FEV1 trajectories in females. DNA-M at three CpGs showed marginal statistical significance in females, two for FEV1/FVC trajectories (cg23190164, p = 0.08 and cg24479027, p = 0.09) and one for FVC (cg05597624, p = 0.088). At 74% of the 19 identified CpGs in males, a higher DNA-M was associated with a higher odds of being in the low lung function trajectories, while in females, the percentage was 44%.

Bar plots of log odds ratios (*ORs) of IOW-ASPAC consistent CpGs and their mapped genes. Note: Each box plot showed CpGs which showed consistent associations between the IOW and ALSPAC cohorts for the association of DNA-M at childhood with lung function trajectories from childhood to adulthood in males and females, separately. *In ALSPAC the log odds ratios were smaller than the IOW cohort. For better visualization of bars in the ALSPAC figure, the log ORs were multiplied by 10
Association with gene expression
In a time-lagged assessment of DNA-M at age 10 years with gene expression at 26 years (n = 35 males and 41 females), 15 of the 19 identified CpGs in males and another 15 CpGs of the 25 identified in females had expression data on the CpGs mapped genes. In males, of the 15 CpGs, DNA-M at cg16709691 (LMF1) and cg12655437 (SMAD2) was associated with the expression of genes (Table 3; both p values < 0.03). Among the 15 CpGs in females, DNA-M at cg16049690 (BTNL9), cg07562175 (FBRSL1), cg13168117 (KLHL30), and cg23987789 (VAMP3) was associated with gene expression (Table 3; all p values < 0.05). At these six CpGs, higher DNA-M at cg16709691 (LMF1) and cg16049690 (BTNL9) was associated with lower gene expression, while higher DNA-M at cg12655437 (SMAD2), cg07562175 (FBRSL1), cg13168117 (KLHL30), and cg23987789 (VAMP3) was associated with higher gene expression (Table 3).
Detection of differentially methylated regions
For DMR enrichment analysis, a frequency of 20 and above was focused in screening to secure a sufficient number of CpGs. In females, 486, 518, and 461 CpGs and in males, 419, 559, and 842 CpGs for FVC, FEV1, and FEV1/FVC trajectory, respectively, were selected after screening and included in DMR analyses for each trajectory. After controlling FDR of 0.05, 23 statistically significant DMRs in males and 10 in females were identified (Table 4 with breakdown for each lung function trajectory). In total, 78 CpGs were in the 33 (23 + 10) identified DMRs (Additional file 1: Table S2), of which two CpGs (cg09707262 and cg02304879) were also among the 44 CpGs individually identified CpGs. The CpGs in the identified DMRs with the mapped genes and chromosomes, and the corresponding p values of DMRs for each sex are presented in Additional file 1: Figure S1.
Biological pathways of mapped genes
The 44 IOW-ALSPAC consistent CpGs (19 CpGs for males and 25 for females) and the 78 CpGs in the 33 identified DMRs (23 DMRs in males and 10 in females) were mapped to 42 (Table 2) and 33 genes (Table 4), respectively. The distinct 73 genes were included in pathway enrichment analysis (focusing on pathways with at most 2000 genes) via bioinformatics tool ToppFun. After controlling the FDR of 0.05, six and 12 pathways were identified (Table 5) in males and females, respectively.
Discussion
Two major distinct lung function trajectories from pre-adolescence to adulthood in each sex were identified using latent class trajectory analyses in two population-based birth cohort. We showed that pre-adolescence DNA-M at 44 CpGs was associated with the trajectories. These CpGs mapped to 42 genes, which were enriched in 18 KEGG and REACTOME pathways. We identified 23 and 10 DMRs associated with the lung function trajectories in males and females, respectively. We further evaluated the functional effects of the identified CpGs by integrating gene expression. DNA-M at two CpGs in males and four in females at age 10 years was longitudinally associated with gene expression at age 26 year among the distinct set of 15 CpGs in each sex. Since in our study lung function trajectories cover from pre-adolescence transition till adulthood, and DNA-M was measured at an age close to the transition rather than a number of years before adolescence, the identified 44 CpG sites have a strong potential of high sensitivity to predict an individual’s lung function development from pre-adolescence to young adulthood.
The high and low trajectories of lung function identified in this study were the same as in our previous study [12] except that in this study subjects who had lung function measurement only at one-time point were excluded to improve the accuracy of trajectory assignment. The identified trajectories were also consistent with the main two trajectories of the previous reports from population-based studies including ours [4, 10, 12]. In a large-scale study, Belgrave et al. [10] (age 5–24 years) identified 4 distinct trajectories of FEV1; persistently high; normal; below average; and persistently low. In our study, on the other hand, two (high and low) trajectories of FVC, FEV1, and FEV1/FVC represented the data the best, which was likely due to the relatively smaller sample size. Moreover, like Belgrave et al. study, in this study, participants in the low FVC and FEV1 trajectory group did not achieve maximally attainable FVC and FEV1 and in the low FEV1/FVC trajectory group showed an accelerated decline from age 10 to 26 years (15% and 11% decline in males and females, respectively), compared to the declines in the high trajectory (9% and 10%, respectively) (Fig. 2), suggesting a risk of future COPD. The observations in this study also support the findings in the previous longitudinal studies by Lange et al. and Bui et al. [6, 11] that the persistently low lung function trajectory is associated with the risk of COPD in adults.
To our knowledge, this is the first study examining the association of pre-adolescence DNA-M with lung function trajectories ranging from pre-adolescence to post-adolescence. The identified CpGs and DMRs at childhood may provide insight into the pathogenesis of variations in lung function growth in adolescence. In addition, the associations of methylation at some identified CpGs with gene expression, such as cg16709691 (LMF1), cg12655437 (SMAD2), cg16049690 (BTNL9), cg07562175 (FBRSL1), cg13168117 (KLHL30), and cg23987789 (VAMP3) manifest the functional importance of the CpGs as biomarkers. Among these genes, SMAD2, FBRSL1, and VAMP3 were associated with lung function, its related pathway, and COPD in previous studies [25,26,27,28]. Although most individually identified CpGs through logistic regressions were different from those in the DMRs due to different assumptions and statistical approaches between these two analyses, their mapped genes jointly involved at the biological pathways (Table 5).
Among the listed biological pathways linked to the mapped genes (Table 4), several pathways play a significant role in lung function and/or COPD, including downregulation of SMAD2/3: SMAD4 transcriptional activity, circadian entrainment, GABA B receptor activation, and activation of G protein-gated potassium channels [25, 29,30,31,32]. For example, downregulation of SMAD2/3: SMAD4 transcriptional activity plays a role in the regulation of TGF-β1-induced collagen expression in lung. Excessive collagen deposition is one of the characteristics of idiopathic pulmonary fibrosis that lead to impaired lung function later in life. The association of cg12655437 with SMAD2 expression in this study revealed the pathway as functionally meaningful. Another pathway, the circadian rhythm regulates physiological diurnal variation of lung function through the autonomous peripheral circadian clock mechanisms. Clara cells in the bronchioles play a major role in such variations of lung function. These physiological oscillations are driven by transcriptional factors and genes such as PER3.
The CpGs showing consistent direction of associations with statistical significance at 0.05 or < 0.1 in both cohorts included cg14669749 (SKI), cg21131402 (C12orf50), cg23987789 (VAMP3), cg23190164 (LGR5), cg24479027 (ABR), and cg05597624 (RNF220). Some of these genes, such as VAMP3, LGR5, PER3, and SDC1, were found to be involved in the different physiological functions of lung and chronic lung disease [27, 28, 33,34,35,36]. Among these genes, VAMP3 is found as one of the soluble N-ethylmaleimide-sensitive factor attachment protein receptors regulating mucin granule exocytosis. Mucin secretion is an innate immunity mechanism, which is harmfully upregulated in obstructive lung diseases including COPD [27, 28]. In addition, being in the intergenic region, the significant positive association of methylation at cg23987789 with the expression of VAMP3 revealed a potential of this CpGs’ functionally regulatory role. LGR5 is related to the WNT signalling cascades, which are the critical regulators of different developmental and pathophysiological processes in lung. Dysregulated LGR5 expression influences to reduced WNT-β catenine signalling cascades, which is further linked to chronic lung disease including COPD [33, 34]. Among the annotated genes of the DMRs and also identified in circadian entrainment pathway, in females, PER3 has been previously associated with childhood and adolescence lung function (FEV1) [35]; in males, SDC1 was found as a differentially expressed genes in COPD development by robust rank aggregation method and in KEGG pathway in the previous study [36].
Although overall patterns of lung function trajectories in males and females were similar, for each identified trajectory, there existed large differences in volumes and flows between the two sex (Fig. 2). Such differences were expected and acknowledged in the literature [20,21,22,23] and were the major driving factors for the stratified study design. The uniqueness of identified CpGs for each sex led us to postulate the possibility of either different underlying epigenetic mechanisms in males and females in the regulation of gene activity and may act as the biomarkers of physiology and/or exposures that influence lung function trajectory. Another strength of this study was the time-lagged study design. Because of this, CpGs identified in this study have the potential to serve as candidate predictors for future lung function trajectories and will be beneficial to the detection of early lung diseases and subjects with a higher risk of developing those diseases.
Some issues related to the study designs and data analyses are worth discussing. In this study, participants with lung function measurements available only at one-time point were excluded from the analyses. The exclusion of subjects with missing data plus stratification by sex made the sample size smaller for each group, especially for females (n = 136). However, including participants with lung function measurement available at two or more time points ensured a high probability of trajectory group assignment rather random group assignment with a probability around 0.50. Also, based on the comparison with the whole study cohort, the study samples represented the whole cohort indicating that such restriction (≥ 2 repeated lung function measurements) did not bring statistically significant selection bias into the study samples. The use of a screening process, together with stringent control of multiple testing via the Bonferroni approach instead of controlling FDR, and the utilization of the replication cohort ensured that the findings from our study are robust with the potential of being generalized. Besides, in the IOW cohort, age 10 was treated as the pre-adolescence age, since almost all children (males 98% and females 92%) included in this study had not entered any phase of puberty. The children with minimal pubertal events were not excluded from the present study, since excluding them was not expected to alter the findings and conclusions but might have decreased testing power. Another perspective related to age is that, in the IOW cohort, the analyses were based on data collected at ages 10, 18, and 26 years representing pre- and post-adolescence. In the ALSPAC, the corresponding ages were 7/8, 15, and 24 years. The decline phase of lung function might have not started yet at age 24 years in the ALSPAC for some subjects (Fig. 3). This inconsistency between the two cohorts might be the cause of a lack of replication for some CpGs. Finally, the identified CpGs had minimal overlapping among FVC, FEV1, and FEV1/FVC trajectories, although in females, some mapped genes of identified CpGs associated with FVC, FEV1, and/or FEV1/FVC trajectories were involved in common pathways across these three different lung function parameters. Since DNA-M was measured in whole blood rather than in airways, although several studies support this non-invasive sampling approach of assessing DNA-M, the relevance of epigenetic changes measured in leukocytes in whole blood to gene expression in the lung remains unanswered and deserves further investigations of their biological evidence.
Conclusion
Our study identified 44 CpGs with pre-adolescence DNA-M shown to be associated with lung function trajectories from pre-adolescence to young adulthood. These CpGs have a strong potential as candidate markers in future studies focusing on predicting an individual’s lung function trajectory. A well-designed study plan is warranted to comprehensively assess these CpGs’ joint contributions on lung function patterns.
Methods
Study subjects and design
The IOW cohort—Discovery cohort
The Isle of Wight (IOW) birth cohort is a population-based birth cohort established in 1989, UK. The study was originally approved by the IOW Local Research Ethics Committee at recruitment, and further assessments of this cohort are approved by the National Research Ethics Service, Committee South Central—Southampton B (06/Q1701/34). Informed written consent was obtained from participants or their parents before participating. The study enrolled 1456 eligible children of 1536 born between January 1989 and February 1990 (after exclusion of adoptions, infant deaths, and denial). Details of the IOW birth cohort of 1989 have been described elsewhere [37]. Longitudinal monitoring of allergic diseases, phenotypic measures, genetic, and assessments of environmental exposures were conducted at birth, ages 1, (94.4%), 2 (84.5%), 4 (83.6%), 10 (94.3%), 18 (90.2%), and 26 (70.9%) years.
Lung function
Pre-bronchodilator spirometric measurements, including forced vital capacity (FVC), forced expiratory volume in one second (FEV1), and the ratio of FEV1 over FVC (FEV1/FVC), were conducted at ages 10 (n = 980), 18 (n = 838), and 26 (n = 546) years and included in the study. FVC and FEV1 were measured using a Koko spirometer and software with a portable desktop device (both PDS Instrumentation, Louisville, KY, USA), and the FEV1/FVC ratio was calculated. Spirometry was conducted and evaluated according to the American Thoracic Society (ATS) guidelines [38, 39]. To conduct spirometry, participants were required to be free of respiratory infection for two weeks, not taking oral steroids, not taking any β-agonist for six hours and caffeine for at least 4 h.
Measurement of DNA methylation (DNA-M)
Peripheral blood samples collected at ages 10 years from n = 330 randomly selected subjects were used for DNA extraction via a standard salting out procedure [40]. DNA concentration was estimated by Qubit quantitation. For each sample, one-microgram DNA was bisulphite-treated for cytosine to thymine conversion using the EZ 96-DNA methylation kit (Zymo Research, Irvine, CA, USA), following the manufacturer’s protocol. DNA samples were randomly distributed on microarrays to control against batch effects. DNA-M was measured using HumanMethylation450K and HumanMethylationEPIC BeadChips (Illumina, Inc., SanDiego, CA, USA). Arrays were processed using a standard protocol as described elsewhere [41], with multiple identical control samples assigned to each bisulphite conversion batch to assess assay variability.
Preprocessing of DNA-M
Probes not reaching a detection p value of 10–16 in at least 95% of samples were excluded. The same criterion was applied to exclude samples, i.e. a sample with detection p value of > 10–16 in more than 5% of CpGs was excluded. CpGs on sex chromosomes were also excluded to avoid bias. DNA-M was then pre-processed using the “CPACOR” pipeline for data from both platforms [42]. DNA-M intensities were quantile normalized using the R computing package, minfi [43]. DNA-M β values for each CpG were then calculated as a ratio of methylated (M) over the sum of methylated and unmethylated (U) probes (β = M/[c + M + U]) interpreted as the percentage of methylation [44], where c was used as a constant to prevent zero in the denominator. Principal components (PCs) inferred based on control probes were used to represent latent variables explaining chip-to-chip and technical (batch) effects on DNA-M variations. Since DNA-M data were from two different platforms (450 k and EPIC), we determined the PCs based on DNA-M at shared control probes between the two platforms. In total, 195 control probes were overlapped between 220 control probes from the 450 K BeadChips and 204 from the EPIC array. These 195 shared probes were then used to calculate the control probe PCs, top 15 of which were used to represent latent batch factors [42].
After pre-processing, a total of 473,864 and 847,155 CpGs were available in the 450 K and EPIC methylation array data, respectively, and 439,635 overlapping CpGs were identified between the two platforms. CpGs with a single nucleotide polymorphisms (SNP) overlapping the detection probe with minor allele frequency ≥ 0.7% (corresponding to at least 10 subjects in the IOW cohort with n = 1456) within 10 base pairs of the targeted CpG were excluded due to potential bias that those SNPs brought to the measurement of DNA-M. After excluding probe SNPs, 402,714 CpGs were included in the statistical analyses.
Gene expression data
RNA-seq gene expression data for subjects at age 26 years were available in IOW cohort, which was used to evaluate biological relevance of CpGs shown to have time-lagged associations with lung function. We used paired-end (2 × 75 bp) RNA sequencing with the Illumina Tru-Seq Stranded mRNA Library Preparation Kit with IDT for Illumina Unique Dual Index (UDI) barcode primers following manufacturer’s recommendations. RNA samples were extracted from whole blood of the IOW cohort participants at age 26 years. All samples were sequenced second time using the identical protocol, and for each sample, the output from both runs was combined. FASTQC were run to assess the quality of the FASTQ files [45]. Reads were mapped against Human Genome (GRch37 version 75) using HISAT2 (v2.1.0) aligner [46]. The alignment files, produced in the Sequence Alignment Map (SAM) format, were converted into the Binary Alignment Map (BAM) format using SAMtools (v1.3.1) [47]. HTseq (v0.11.1) was used to count the number of reads mapped to each gene in the same reference genome used for alignment [48]. Normalized read count FPKM (Fragments Per Kilobase of transcript per Million mapped reads) were calculated using the countToFPKM package (https://github.com/AAlhendi1707/countToFPKM) and were included for subsequent data for analysis.
Confounders
Based on prior knowledge in the published literature, variables potentially associated with lung function trajectories in addition to DNA-M were included in the model as confounders. The potential confounders were birth weight, gestational age, duration of breastfeeding, maternal smoking exposure during pregnancy, recurrent chest infection collected at ages 1/2 years, second-hand smoking exposure at age 10 years (childhood), height and body mass index (BMI) at age 10 years, exposure to pets at age 10 years, age of puberty onset, and socioeconomic status (SES) [4, 11, 12].
Gestational age information was recorded during delivery in the hospital. Birth weight was measured immediately after birth. Heights and weights at age 10 were measured before spirometric measurements, and BMI was calculated from height and weight accordingly. The age of puberty onset was estimated based on self-reports about age of initiation of five pubertal markers for each sex, a growth spurt, body hair growth, skin changes, deepening voice of male, facial hair of male, breast development of female, and initiation of menstruation of female. The National Institute of Child and Human Development questionnaire from the Study of Early Child Care and Youth Development was used to identify pubertal stages. Information on second-hand smoke exposure in childhood was collected from parents. SES was classified using the composite “SES-cluster” method based on the following three variables: (a) the British socioeconomic classes [1,2,3,4,5,6] derived from parental occupation reported at birth; (b) the number of children in the index child’s bedroom (collected at age 4 years); and (c) family income at age 10 years [49]. This composite variable captures the family social class across the entire study period. Pet exposure information was collected at age 10 years through questionnaires.
The ALSPAC cohort—Replication cohort
ALSPAC is a population-based birth cohort study established in 1991 in Avon, UK, approximately 75 miles from the IOW [50, 51]. All pregnant women who were expecting to deliver between 1 April 1991 and 31 December 1992, and residing in the Avon region of the South West of England were eligible to be recruited. In total, 14,541 pregnant women were recruited for the study, of those 13,761 were eligible with 10,321 having DNA sampled. Information on environment, lifestyle, and health of the child and family was collected through annual questionnaires since the child’s birth. At age 7 an additional 913 children were enrolled. The total sample size for analyses using any data collected after the age of 7 is therefore 15,454 pregnancies, resulting in 15,589 foetuses. Of these 14,901 were alive at 1 year of age. From age 7 years, the participants were invited to an annual research clinic to obtain the exposure and other demographic data annually. Spirometry (Vitalograph 2120; Vitalograph, Maids Moreton, UK) was performed at 8, 15, and 24 years of age according to ATS standards [39, 52, 53], the same method as that applied in the IOW cohort. The study website contains details of all the data that are available through a fully searchable data dictionary and variable search tool (http://www.bristol.ac.uk/alspac/researchers/our-data/).
DNA-M data of children at ages 7 (n = 968) years were included in the study. DNA-M in peripheral blood was assessed using the Infinium HumanMethylation450K BeadChip [54]. The procedure for DNA sample preparation was comparable to that applied in the IOW cohort. The pre-processing of DNA-M was performed by adjusting batch effect, excluding CpGs with detection p value ≥ 0.01, and excluding samples that were flagged a sex-mismatch based on X-chromosome methylation. Details of pre-processing, quality control, and quantile normalization of DNA-M data have been described elsewhere [54, 55].
Statistical analyses
Descriptive analyses
To evaluate whether subjects included in the study reasonably represented those in the complete study cohort, we compared lung function tests at ages 10, 18, and 26 years in the study samples with those of the complete cohort using one-sample t tests.
Determining distinctive lung function trajectories
Our previous publication [12] of lung function trajectory was based on at least single spirometry test to attain a maximum sample size. In this study, subjects with at least two-time point tests were included for trajectory analyses to improve the average posterior probability and to avoid the random assignment of the subjects into a trajectory. An unsupervised semi-parametric mixture modelling implemented in the SAS procedure PROC TRAJ [56] was applied to identify developmental lung function trajectories of FVC, FEV1, and FEV1/FVC over time (10, 18, and 26 years) for males and females separately [57], the same approach applied in our previous study [12]. This method combines the latent growth curve and mixture modelling approaches to detect distinct groups of trajectories [56]. All possible models were evaluated each with different numbers of groups (i.e. 2, 3, and 4) and different shapes of the trajectories (linear, quadratic, and cubic) for each group. Trajectory parameters were estimated using the maximum likelihood approach [58, 59]. The best model was selected based on two criteria, being as parsimonious as possible to summarize the distinctive features and with high Bayesian information criterion (BIC) [57, 60, 61]. To improve the quality of identified trajectories, in addition to BIC, probability of trajectory assignment as well as sample sizes in each group was further incorporated; the average posterior probabilities of assignment to a group were set at least 0.7, and the sample size of each group was required to be at least 5% of the total sample size [60]. Individuals were assigned to one of the trajectories/groups based on their highest estimated group-membership probabilities. The assigned group (categorical variable) of distinct lung function trajectories was used in subsequent analyses.
Association analyses
To assess the association of DNA-M at an earlier age with lung function trajectories at later ages in the IOW cohort, we followed a two-step analytical approach. In the first step, CpGs were screened to exclude CpGs potentially not associated with lung function trajectories using ttScreening (R package 3.5.1 version) [62, 63]. This method utilizes training and testing data in robust linear regressions with surrogate variables included in the regressions to adjust for unknown effects. The training and testing steps were repeated 100 times. A CpG that was statistically significant in both training and testing steps at least 50 times was included in the final set for subsequent regression analyses. The screening was performed for each lung function parameter, stratified by sex.
In second step, CpGs that passed screening were further assessed in logistic regression models in SAS 9.4 for the trajectories of each lung function parameter stratified by sex and adjusting for the above-mentioned confounders. Lung function trajectory was treated as the outcome variable, and the DNA-M at each CpG that passed screening was used as an independent variable. Multiple testing was corrected using the Bonferroni method with an experiment-wise significance level of 0.05. In all analyses, DNA-M adjusted for cell types, principle components, and batch effects at each CpG was used.
Replication analysis—in the ALSPAC cohort
The identified CpGs from the IOW cohort were further examined in the ALSPAC. Comparable analytical methods as those implemented in the IOW cohort were applied except for the exclusion of two covariates in regression analyses, recurrent chest infection, and pet exposure at childhood, which were unavailable in the ALSPAC.
Gene expression analysis
To assess the potential biological relevance of the identified CpGs, we examined the time-lagged association of DNA-M at those CpGs with the expression of their mapped genes. Linear regressions were applied to DNA-M at age 10 years with gene expression at age 26 years to each CpG which showed the consistent direction of association between the IOW cohort and ALSPAC.
Analyses of differentially methylated regions (DMRs)
To detect regional differential methylation signals among the CpGs that passed screening, an R package DMRcate was used [64]. The default settings in DMRcate include having at least two CpGs in a region and a minimum length of 1000 nucleotides. In our study, a DMR was considered to be statistically significant if the false discovery rate (FDR)-adjusted p value was < 0.05 [64]. Since DMR analyses focus on contribution of a region as a whole unit, a significant DMR can be detected even if there is no genome-wide significant individual CpGs in the region.
Pathway analyses
Genes annotated to the CpGs explored in ALSPAC with respect to the direction of odds ratios (ORs in the log scale) and DMRs were identified based on Illumina's manifestation file and SNIPPER (https://csg.sph.umich.edu/boehnke/snipper/) version 1.2. Bioinformatic assessment of the genes was conducted using the online bioinformatics tool ToppFun, available in the ToppGene Suite [65]. Multiple testing was adjusted by controlling FDR of 0.05.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ATS:
-
American Thoracic Society
- ALSPAC:
-
Avon Longitudinal Study of Parents and Children
- BMI:
-
Body mass index
- CpGs:
-
Cytosine-phosphate-guanine dinucleotide site or sites
- COPD:
-
Chronic obstructive pulmonary disease
- DMR:
-
Differentially methylated regions
- DNA-M:
-
DNA methylation
- FVC:
-
Forced vital capacity
- FEV1 :
-
Forced expiratory volume in one second
- FDR:
-
False discovery rate
- IOW:
-
Isle of Wight
- ORs:
-
Odds ratios
- PCs:
-
Principal components
- ttScreening :
-
Training and testing screening
References
Vasquez MM, Zhou M, Hu C, Martinez FD, Guerra S. Low lung function in young adult life is associated with early mortality. Am J Respir Crit Care Med. 2017;195(10):1399–401.
Agustí A, Noell G, Brugada J, Faner R. Lung function in early adulthood and health in later life: a transgenerational cohort analysis. Lancet Respir Med. 2017;5(12):935–45.
Belgrave DC, Buchan I, Bishop C, Lowe L, Simpson A, Custovic A. Trajectories of lung function during childhood. Am J Respir Crit Care Med. 2014;189(9):1101–9.
Berry CE, Billheimer D, Jenkins IC, Lu ZJ, Stern DA, Gerald LB, et al. A distinct low lung function trajectory from childhood to the fourth decade of life. Am J Respir Crit Care Med. 2016;194:607–12.
McGeachie MJ, Yates KP, Zhou X, Guo F, Sternberg AL, Van Natta ML, et al. Patterns of growth and decline in lung function in persistent childhood asthma. N Engl J Med. 2016;374(19):1842–52.
Lange P, Celli B, Agustí A, Boje Jensen G, Divo M, Faner R, et al. Lung-function trajectories leading to chronic obstructive pulmonary disease. N Engl J Med. 2015;373(2):111–22.
Martinez FD. Early-life origins of chronic obstructive pulmonary disease. N Engl J Med. 2016;375(9):871–8.
Global surveillance, prevention and control of chronic respiratory diseases: a comprehensive approach. World Health Organization; 2007.
Quaderi SA, Hurst JR. The unmet global burden of COPD. Glob Health Epidemiol Genomics. 2018;3:e4–e4:1–3.
Belgrave DCM, Granell R, Turner SW, Curtin JA, Buchan IE, Le Souëf PN, et al. Lung function trajectories from pre-school age to adulthood and their associations with early life factors: a retrospective analysis of three population-based birth cohort studies. Lancet Respir Med. 2018;6(7):526–34.
Bui DS, Lodge CJ, Burgess JA, Lowe AJ, Perret J, Bui MQ, et al. Childhood predictors of lung function trajectories and future COPD risk: a prospective cohort study from the first to the sixth decade of life. Lancet Respir Med. 2018;6:535–44.
Karmaus W, Mukherjee N, Janjanam VD, Chen S, Zhang H, Roberts G, et al. Distinctive lung function trajectories from age 10 to 26 years in men and women and associated early life risk factors—a birth cohort study. Respir Res. 2019;20(1):98.
John C, Soler Artigas M, Hui J, Nielsen SF, Rafaels N, Paré PD, et al. Genetic variants affecting cross-sectional lung function in adults show little or no effect on longitudinal lung function decline. Thorax. 2017;72:400–8.
Everson TM, Lyons G, Zhang H, Soto-Ramirez N, Lockett GA, Patil VK, et al. DNA methylation loci associated with atopy and high serum IgE: a genome-wide application of recursive Random Forest feature selection. Genome Med. 2015;7:89.
Zhang H, Tong X, Holloway JW, Rezwan FI, Lockett GA, Patil V, et al. The interplay of DNA methylation over time with Th2 pathway genetic variants on asthma risk and temporal asthma transition. Clin Epigenet. 2014;6(1):8.
Imboden M, Wielscher M, Rezwan FI, Amaral AFS, Schaffner E, Jeong A, et al. Epigenome-wide association study of lung function level and its change. Eur Respir J. 2019;54:1900457.
Lepeule J, Baccarelli A, Motta V, Cantone L, Litonjua AA, Sparrow D, et al. Gene promoter methylation is associated with lung function in the elderly: the Normative Aging Study. Epigenetics. 2012;7(3):261–9.
Qiu W, Baccarelli A, Carey VJ, Boutaoui N, Bacherman H, Klanderman B, et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med. 2012;185(4):373–81.
Duijts L, Reiss IK, Brusselle G, de Jongste JC. Early origins of chronic obstructive lung diseases across the life course. Eur J Epidemiol. 2014;29(12):871–85.
Kohansal R, Martinez-Camblor P, Agusti A, Buist AS, Mannino DM, Soriano JB. The natural history of chronic airflow obstruction revisited: an analysis of the Framingham offspring cohort. Am J Respir Crit Care Med. 2009;180(1):3–10.
Becklake MR, Kauffmann F. Gender differences in airway behaviour over the human life span. Thorax. 1999;54(12):1119–38.
Carey MA, Card JW, Voltz JW, Arbes SJ Jr, Germolec DR, Korach KS, et al. It’s all about sex: gender, lung development and lung disease. Trends Endocrinol Metab. 2007;18(8):308–13.
LoMauro A, Aliverti A. Sex differences in respiratory function. Breathe (Sheff). 2018;14(2):131–40.
Sunny SK, Zhang H, Rezwan FI, Relton CL, Henderson AJ, Merid SK, et al. Changes of DNA methylation are associated with changes in lung function during adolescence. Respir Res. 2020;21(1):80.
Kolosova I, Nethery D, Kern JA. Role of Smad2/3 and p38 MAP kinase in TGF-β1-induced epithelial–mesenchymal transition of pulmonary epithelial cells. J Cell Physiol. 2011;226(5):1248–54.
Morrow JD, Cho MH, Platig J, Zhou X, DeMeo DL, Qiu W, et al. Ensemble genomic analysis in human lung tissue identifies novel genes for chronic obstructive pulmonary disease. Hum Genomics. 2018;12(1):1.
Jones LC, Moussa L, Fulcher ML, Zhu Y, Hudson EJ, O’neal WK, et al. VAMP8 is a vesicle SNARE that regulates mucin secretion in airway goblet cells. J Physiol. 2012;590(3):545–62.
Kean MJ, Williams KC, Skalski M, Myers D, Burtnik A, Foster D, et al. VAMP3, syntaxin-13 and SNAP23 are involved in secretion of matrix metalloproteinases, degradation of the extracellular matrix and cell invasion. J Cell Sci. 2009;122(22):4089–98.
Gibbs JE, Beesley S, Plumb J, Singh D, Farrow S, Ray DW, et al. Circadian timing in the lung; a specific role for bronchiolar epithelial cells. Endocrinology. 2009;150(1):268–76.
Hwang J-W, Sundar IK, Yao H, Sellix MT, Rahman I. Circadian clock function is disrupted by environmental tobacco/cigarette smoke, leading to lung inflammation and injury via a SIRT1-BMAL1 pathway. FASEB J. 2014;28(1):176–94.
Osawa Y, Xu D, Sternberg D, Sonett JR, D’Armiento J, Panettieri RA, et al. Functional expression of the GABAB receptor in human airway smooth muscle. Am J Physiol-Lung Cell Mol Physiol. 2006;291(5):L923–31.
Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol. 1995;268(4):C799–822.
Baarsma HA, Skronska-Wasek W, Mutze K, Ciolek F, Wagner DE, John-Schuster G, et al. Noncanonical WNT-5A signaling impairs endogenous lung repair in COPD. J Exp Med. 2016;214(1):143–63.
Hu Y, Skronska-Wasek WA, Ota C, Mutze KIA, Baarsma H, Wagner DE, et al. The progenitor cell marker LGR5 is reduced in epithelial cells in emphysema. In: B61 epithelial cell biology in respiratory disease. American Thoracic Society International Conference Abstracts: American Thoracic Society; 2018. p. A3827-A.
den Dekker HT, Burrows K, Felix JF, Salas LA, Nedeljkovic I, Yao J, et al. Newborn DNA-methylation, childhood lung function, and the risks of asthma and COPD across the life course. Eur Respir J. 2019;53(4):1801795.
Lin Y-Z, Zhong X-N, Chen X, Liang Y, Zhang H, Zhu D-L. Roundabout signaling pathway involved in the pathogenesis of COPD by integrative bioinformatics analysis. Int J Chron Obstruct Pulmon Dis. 2019;14:2145–62.
Arshad SH, Holloway JW, Karmaus W, Zhang H, Ewart S, Mansfield L, et al. Cohort profile: the Isle Of Wight whole population birth cohort (IOWBC). Int J Epidemiol. 2018;47(4):1043–4.
Crapo R. Guidelines for methacholine and exercise challenge testing-1999. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. Am J Respir Crit Care Med. 2000;161:309–29.
Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–38.
McClelland M, Hanish J, Nelson M, Patel Y. KGB: a single buffer for all restriction endonucleases. Nucleic Acids Res. 1988;16(1):364.
Bibikova M, Fan J-B. GoldenGate® assay for DNA methylation profiling. DNA methylation. Berlin: Springer; 2009. p. 149–63.
Lehne B, Drong AW, Loh M, Zhang W, Scott WR, Tan ST, et al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol. 2015;16:37.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–9.
Du P, Zhang X, Huang C-C, Jafari N. Kibbe Wa, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels analysis. BMC Bioinformatics. 2010;11:587.
Andrews S, FastQC. A quality control tool for high throughput sequence data 2010. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 10 Aug 2020.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9.
Ogbuanu IU, Karmaus W, Arshad SH, Kurukulaaratchy RJ, Ewart S. Effect of breastfeeding duration on lung function at age 10 years: a prospective birth cohort study. Thorax. 2009;64(1):62–6.
Boyd A, Golding J, Macleod J, Lawlor DA, Fraser A, Henderson J, et al. Cohort Profile: the ’children of the 90s’—the index offspring of the Avon Longitudinal Study of Parents and Children. Int J Epidemiol. 2013;42(1):111–27.
Fraser A, Macdonald-Wallis C, Tilling K, Boyd A, Golding J, Davey Smith G, et al. Cohort Profile: the Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. Int J Epidemiol. 2013;42(1):97–110.
Sonnenschein van der Voort AM, Howe LD, Granell R, Duijts L, Sterne JA, Tilling K, et al. Influence of childhood growth on asthma and lung function in adolescence. J Allergy Clin Immunol. 2015;135(6):1435–43.
Northstone K, Lewcock M, Groom A, Boyd A, Macleod J, Timpson N, Wells N. The Avon Longitudinal Study of Parents and Children (ALSPAC): an update on the enrolled sample of index children in 2019. Welcome Open Res. 2019;4:51. https://doi.org/10.12688/wellcomeopenres.15132.1.
Relton CL, Gaunt T, McArdle W, Ho K, Duggirala A, Shihab H, et al. Data resource profile: accessible resource for integrated epigenomic studies (ARIES). Int J Epidemiol. 2015;44(4):1181–90.
Min JL, Hemani G, Davey Smith G, Relton C, Suderman M. Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics. 2018;34(23):3983–9.
Jones BL, Nagin DS, Roeder K. A SAS procedure based on mixture models for estimating developmental trajectories. Sociol Methods Res. 2001;29:74–393.
Nagin D. Group-based modeling of development. Cambridge: Harvard University Press; 2005.
Liang J, Xu X, Bennett JM, Ye W, Quinones AR. Ethnicity and changing functional health in middle and late life: a person-centered approach. J Gerontol B Psychol Sci Soc Sci. 2010;65(4):470–81.
Nagin DS, Tremblay RE. Analyzing developmental trajectories of distinct but related behaviors: a group-based method. Psychol Methods. 2001;6(1):18–34.
Nagin DS, Odgers CL. Group-based trajectory modeling in clinical research. Ann Rev Clin Psychol. 2010;6:109–38.
Nagin DS. Analyzing developmental trajectories: a semiparametric, group-based approach. Psychol Methods. 1999;4(2):139.
Li X, Hawkins GA, Ampleford EJ, Moore WC, Li H, Hastie AT, et al. Genome-wide association study identifies TH1 pathway genes associated with lung function in asthmatic patients. J Allergy Clin Immunol. 2013;132(2):313–20.
Ray MA, Tong X, Lockett GA, Zhang H, Karmaus WJ. An efficient approach to screening epigenome-wide data. Biomed Res Int. 2016;2016:2615348.
Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, Lord RV, et al. De novo identification of differentially methylated regions in the human genome. Epigenet Chromatin. 2015;8(1):6.
Chen J, Aronow BJ, Jegga AG. Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinform. 2009;10(1):73.
Acknowledgements
The authors gratefully acknowledge the cooperation of the children and parents who participated in this study and appreciate the hard work of the Isle of Wight research team in collecting data. We thank the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust Grant Reference 090532/Z/09/Z and MRC Hub Grant G0900747 91070) for the generation of the methylation data. The authors are thankful to the High-Performance Computing facility at the University of Memphis. For the ALSPAC cohort, we are extremely grateful to all the families who took part in this study, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, and nurses.
Funding
The study conveyed in this publication was supported by the National Institute of Allergy and Infectious Diseases under Award Number R01 AI121226 (MPI: Hongmei Zhang and John Holloway). The 10-year follow-up of IOW cohort was funded by National Asthma Campaign, UK (Grant No 364), and the 18-year follow-up by a grant from the National Heart and Blood Institute (R01 HL082925, PI, SH Arshad). The UK Medical Research Council (MRC) and Wellcome (Grant ref: 102215/2/13/2) and the University of Bristol provide core support for ALSPAC. A comprehensive list of grants funding is available on the ALSPAC website (http://www.bristol.ac.uk/alspac/external/documents/grant-acknowledgements.pdf). Generation of methylation array data was specifically funded by NIH R01AI121226, R01AI091905, BBSRC BBI025751/1, and BB/I025263/1, MRC MC_UU_12013/1, MC_UU_12013/2, MC_UU_12013/8. Lung function measurements were funded by grants from the MRC (G0401540/73080 and MR/M022501/1).
Author information
Authors and Affiliations
Contributions
SKS carried out the study, conducted all the statistical analysis, interpreted the data, and drafted the manuscript. HZ designed the study, guided the analysis, and involved in drafting and revision of the manuscript. FM contributed to the conception and critically revised the manuscript. JWH and SE supervised the DNA methylation measurement in IOW cohort and revised the manuscript. SHA was involved in data acquisition, DNA-M arraying, and study design in IOW cohort and reviewed the manuscript. AJH, CLR, and SR were involved in the ALSPAC study design and provided the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approvals for the IOW study were obtained from the Isle of Wight Local Research Ethics Committee (recruitment, 1, 2, and 4 years) and National Research Ethics Service, NRES Committee South Central—Southampton B (10 and 18 years) (06/Q1701/34). Written informed consent was obtained from parents to enrol newborns, and at subsequent follow-up, written informed consent was obtained from parents, participants, or both. At the University of Memphis, the internal review board first approved the project (FWA00006815) in 2015 (IRB ID: 3917). For ALSPAC, ethical approval for the study was obtained from the ALSPAC Ethics and Law Committee and the Local Research Ethics Committees. Consent for biological samples has been collected in accordance with the Human Tissue Act (2004). Informed consent for the use of data collected via questionnaires and clinics was obtained from participants following the recommendations of the ALSPAC Ethics and Law Committee at the time. Full details of ethical approvals are available at http://www.bristol.ac.uk/alspac/researchers/research-ethics/.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no potential competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Table S1: List of CpGs (k = 96) showing in both consistent and opposite direction of associations of DNA-M at childhood with lung function trajectories childhood-to-young adulthood in males and females between the IOW cohort and ALSPAC. Table S2: DMRs (k = 33) for lung function trajectory in relation to childhood DNA-M identified by DMRcate (FDR < 0.05) method. Figure S1: Circular plots of CpGs identified in DMRs (A) for males (B) for females.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sunny, S.K., Zhang, H., Mzayek, F. et al. Pre-adolescence DNA methylation is associated with lung function trajectories from pre-adolescence to adulthood. Clin Epigenet 13, 5 (2021). https://doi.org/10.1186/s13148-020-00992-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-020-00992-5