
Abstract
The cancer stem cell (CSC) hypothesis suggests that only a subpopulation of cells within a tumour is responsible for the initiation and progression of neoplasia. The original and best evidence for the existence of CSCs came from advances in the field of haematological malignancies. Thus far, putative CSCs have been isolated from various solid and non-solid tumours and shown to possess self-renewal, differentiation, and cancer regeneration properties. Although research in the field is progressing extremely fast, proof of concept for the CSC hypothesis is still lacking and key questions remain unanswered, e.g. the cell of origin for these cells. Nevertheless, it is undisputed that neoplastic transformation is associated with genetic and epigenetic alterations of normal cells, and a better understanding of these complex processes is of utmost importance for developing new anti-cancer therapies. In the present review, we discuss the CSC hypothesis with special emphasis on age-associated alterations that govern carcinogenesis, at least in some types of tumours. We present evidence from the scientific literature for age-related genetic and epigenetic alterations leading to cancer and discuss the main challenges in the field.
Similar content being viewed by others

Introduction
The cancer stem cell (CSCs) paradigm has started a new era in cancer research with significant implications for the formulation of future clinical therapeutics [recently reviewed in [1, 2]]. This hierarchical model for cancer, unlike the classical stochastic model, supports the existence of CSCs, or tumour-initiating cells, that are responsible for tumour formation, maintenance, growth and metastasis [3, 4]. Although CSCs were identified in solid and non-solid tumours, our current knowledge concerning the origin of these cells and the processes leading to their formation is limited due to the complexity of the experimental approaches that will be required to provide data of sufficient substance to support this hypothesis [5, 6]. Given the stem cell-like characteristics of CSCs, it has been proposed that CSCs originate from adult stem cells, progenitor cells or differentiated cells that have acquired ‘stemness’ properties [3, 6–8]. Although all three cell sources have the capacity to be genuine CSCs, in our view, adult stem cells appear to be the most probable target for malignant transformation, generating cells with stem-like properties and tumorigenic potential [6, 9, 10] and accounting for cancer as a ‘stem cell disease’ [9]. Unlike adult stem cells, progenitor cells and somatic cells are more lineage-committed and comprise reduced proliferation potential, thus requiring additional alterations to re-acquire the self-renewal potential [3, 6, 9].
One of the major drivers of malignant transformation is aging [11–15]. With age, all cells, including stem cells, accumulate genetic and/or epigenetic alterations, which affect the cellular, molecular and physiological functionality of tissues [13–16]. The consecutive deterioration of tissues is an important risk factor for the origin of age-related chronic diseases in humans, such as diabetes, cardiovascular and neurodegenerative diseases, etc. Age-associated effects may also lead to a higher risk of tumorigenesis, when in concomitant with deregulated cell signalling and changes in the microenvironment of stem cells. These effects increase the resistance to cellular senescence and programmed cell death and may ultimately lead to the transformation of stem cells into CSCs. Although direct evidence for this concept is missing, it is consistent with the notion that CSCs originate from adult stem or progenitor cells. Further research is needed to better understand the age-associated alterations involved in the generation of CSCs and advances in this area are expected to shed new light on cancer diagnosis and treatment. The present review focuses on the relationship between aging and cancer stem cells and aims to explore the latest advances in the field.
The CSCs model
In the last few decades, the concept that tumours may comprise a rare population of cells with self-renewal, proliferation and differentiation capacities, resembling stem cells, has emerged. The first evidence for CSCs came from the identification of leukemic stem cells (LSCs), which are the cancer-initiating cells of the hematopoietic system [17]. LSCs were identified as a population of cells expressing the CD34+CD38- cell surface markers with a uniquely oncogenic phenotype characterized by their ability to initiate leukaemia in non-obese diabetic mice with severe combined immunodeficiency disease (NOD/SCID mice) [18]. The development of fluorescence-activated cell sorting (FACS), together with the establishment of specific cell surface markers, permitted the subsequent identification of CSCs in solid tumours [7]: breast (CD44+CD24-/low) [5], prostate and ovarian (CD44+) [19, 20], brain and lung (CD133+) cancers [21, 22], and others [23–25]. In addition to malignant tumours, tumour stem cells have been identified in benign tumours, as demonstrated recently by Xu and colleagues (2009). These cells were isolated from pituitary adenoma and exhibited similar characteristics to multipotent stem-like cells, such as self-renewal capacity, multi-lineage differentiation, sphere formation, resistance to chemotherapy, and tumour formation in NOD/SCID mice [26]. The presence of CSCs in both benign and malignant tumours demonstrates the ability of these cells to facilitate tumour formation and support carcinogenesis.
The identification of CSCs led to the characterization of their stem cell-like properties (Figure 1), consistent with their ability to cause tumour formation and recurrence [27, 28]. By definition, normal stem cells give rise to the cellular components of an organ by asymmetric division mediating their self-renewal and differentiation potential [6]. Similarly, CSCs mimic this process by promoting aberrant organogenesis in a hierarchical-mode, in which a phenotypically heterogeneous progeny at different levels of differentiation and proliferation is formed [10]. The stem-like characteristics of CSCs include the expression of pluripotent markers such as Sox2, Oct4, and/or Nanog [27, 28] and of functional markers, like ALDH1, CD133+ (as lung stem cells), or CD34+CD38- [as hematopoietic stem cells (HSCs)] [9, 29]. Furthermore, CSCs resemble normal stem cells by sharing active signalling pathways, such as Notch, Hedgehog, and/or Wnt [3, 30], similar genetic and epigenetic profiles [31], and aptitude to form spheres in vitro[27, 32].

Characteristics of CSCs. CSCs are tumour-initiating cells that may result from malignant transformation of stem/progenitor cells, instigating the tumorigenic process. CSCs have been described to possess stem-like properties, such as self-renewal, proliferation and differentiation abilities, expression of pluripotent (e.g. Sox2, Oct4, Nanog) and functional (e.g. ALDH1, CD133+, CD34+CD38-) markers, active signalling pathways (e.g. Notch, Hedgehog, Wnt), genetic and epigenetic profiles similar to stem cells, and capacity to form spheres in vitro. CSCs can be efficiently detected when injected in immunocompromised mice, as these cells, through their self-renewal and differentiation potential, give rise to a tumour with phenotypic heterogeneity. Tumorigenesis is followed by angiogenesis and by the invasion and metastatic stages, as part of the disease progression. Indeed, CSCs have been associated with the induction of tumour vascularisation through the expression of vascular-related factors and by their contribution to metastasis through the induction of the EMT program. Their resistance to chemo and radiotherapies is clinically important as most anticancer agents target the tumour bulk but not the CSC population. The resistance ability of these cells may be associated with their slow-cycling phenotype, and/or expression of efflux transporters, anti-apoptotic proteins, DNA-repair mechanisms, or of free radicals scavengers.
The isolation and maintenance of CSCs advanced our understanding of cancer initiation and progression, resulting in in vitro models to characterize these cells, model cancer transformation and progression, study the effect of the microenvironment [33], screen for CSC-specific drugs [34, 35], and identify biomarkers for the onset, progression of cancer and its recurrence after therapy [36] (Figure 2). CSCs can be isolated from cancer cell lines or primary tumours based on the i) expression of surface markers [37, 38], ii) detection of the side population [39], iii) anoikis resistance [40], or iv) drug resistance [41]. However, the low frequency of CSCs in primary tumours and the difficulty to stably maintain these cells in vitro makes some of these systems difficult to use. To overcome these issues, in vitro models of cancer stem-like cells have been developed recently. Chen and colleagues (2012) developed a CSC model from mouse induced pluripotent stem cells (miPSC) cultured in a medium simulating the tumour microenvironment [35]. Sachlos et al (2012) established a valuable screening assay for CSCs-targeting drugs using neoplastic human pluripotent stem cells (hPSCs) [34]. Additionally, several reports demonstrated that cancer stem-like cells can be obtained by the reprogramming of cancer cells [42, 43] and primary tumours [36] to iPSC-like induced pluripotent cancer cells (iPCs). Unfortunately, this process is time-consuming and its efficiency is even lower than the reprogramming of non-tumorigenic somatic cells. The stem-like characteristics of iPCs were validated through the expression of pluripotent markers, such as Oct3/4, Sox2, or Nanog, as well as SSEA-4, Tra-1-60, or Tra-1-81; and the capacity of iPCs to form the three germ layers via embryoid bodies in vitro and teratomas in vivo[4, 27, 28, 42–44]. Furthermore, iPCs are resistant to several anticancer drugs [43], express CSCs markers, like CD133 [28], and demonstrate tumorigenic and metastatic properties in vivo[28, 36].

In vitro models of CSCs and their applications. Different in vitro models of CSCs have been created in an attempt to allow a better understanding of the properties of these cells but also of the cancer biology. In addition, these models have been employed in drug screening assays but also in the identification of biomarkers associated with different stages of neoplasia and its recurrence after therapy. Generally, CSCs can be isolated from primary tumours and cancer cell lines based on definite properties, such as expression of specific cell surface markers (e.g. CD44+, CD133+, CD34+CD38-), resistance to anoikis or to drugs, or possess of a side population phenotype. Furthermore, recent reports have demonstrated the generation of CSC-like cells through the reprogramming of cancer cells from both primary tumours and cancer cell lines.
Based on the tumorigenic potential and self-renewal properties of CSCs, these cells can be easily detected by serial transplantation in immunocompromised mice, while the progeny tumour represents the phenotypic heterogeneity of the parental tumour [10] (Figure 1). Conversely, non-tumorigenic cells have lower proliferative and anti-apoptotic capacities, as confirmed by their decreased Hoechst dye efflux or aldehyde dehydrogenase activities and do not form tumours in vivo[39]. This notion is consistent with the positive correlation between the higher incidence of differentiated cancer cells in a given tumour and favourable prognosis [3, 6, 7]. Differentiation of CSCs, or so called “differentiation therapy”, was therefore proposed as a novel approach to eliminate CSCs [45, 46] using different compounds, such as all-trans retinoic acid (ATRA), an inducer of the promyelocytic differentiation used for the treatment of acute promyelocytic leukaemia [47], and suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor and differentiation inducer of human breast and cancer cells [48].
CSCs are involved in cancer progression and metastasis [49] (Figure 1). These cells induce tumour vascularisation by promoting angiogenesis, through the expression of vascular-related factors, e.g. VEGF and PD-ECGF [50], and by stimulating the incorporation of CSC-derived endothelial cells into newly formed capillaries [51]. Tumour vasculature is not only important for supplying blood to the tumour but also for the proliferation and maintenance of CSCs [51], resulting in a “vicious cycle” contributing to cancer progression. Furthermore, CSCs may contribute to invasion and metastasis by acquiring migratory properties through the induction of the epithelial to mesenchymal transition (EMT) program [14, 49]. This concept was supported by the activation of β-catenin and the low levels of E-cadherin in stem-like tumour cells at the tumour-host interface [52] and the induction of mesenchymal markers in CSCs, such as vimentin and N-cadherin [28]. Moreover, tumours can become metastatic as a result of the accumulation of extra mutations and/or epigenetic modifications within the CSCs [2, 46].
The formation of CSCs in tumours and their maintenance, angiogenic support and subsequent metastatic potential are clinically important especially since most current antitumor therapies target the bulk tumour and not CSCs. Moreover, the resistance of CSCs to chemotherapy and radiotherapy supports their association in tumour recurrence after therapy [35, 53–55] (Figure 1). This is consistent with the increased expression of ABC efflux transporters [56, 57], like P-glycoprotein and/or ABCG2, and/or anti-apoptotic proteins (e.g. Bcl-2, survivin, and NF-κB) [3, 14], and with the higher DNA-repair capacity [14], elevated free radical scavenger properties [58], and/or slower proliferation potential related to a slow-cycling state (G0 phase) of these cells [53]. Overall, it is critical to develop novel therapies that are adjusted to effectively eliminate CSCs and tumour recurrence. However, considering that normal stem cells and CSCs are very similar, effective chemotherapeutic agents must selectively target CSCs but not normal stem cells [34, 53].
Origin of CSCs: Adult stem cells vsprogenitor cells
Do CSCs originate from adult stem or progenitor cells? Given that these cells represent a rare population within a tissue, similarly to CSCs in the tumour, makes them difficult to study [10]. Furthermore, the process in which an adult/progenitor cell undergoes malignant transformation into a CSC is very complex and may involve multiple stages. Nevertheless, strong evidence suggests that most tumours originate from CSCs through neoplastic alterations of adult stem or progenitor cells [2, 9, 59].
Adult stem cells constitute small populations within the tissues that are important for tissue homeostasis and regeneration by replacing senescent cells and those lost as a consequence of tissue injury [11]. Through asymmetric division, stem cells support their self-renewal while maintaining their tissue-specific differentiation capacity [13]. Although HSCs were the first adult stem cells to be described, the existence of adult stem cells have been confirmed in other tissues, such as heart [60], lung [61], brain [62], skeletal muscle [63], kidney [64], and others [65–67].
Adult stem cells have a longer lifespan than progenitor and somatic cells; long enough to allow the accumulation of age-associated genetic and/or epigenetic alterations responsible for malignant transformation into CSCs [2, 3, 10, 14, 15, 68, 69]. For this reason, during chronological aging, adult stem cells are more likely to be the target of alterations that may lead to the formation of CSCs. This notion is further supported by the observation that progenitor cells lose their self-renewal property during commitment, an important capacity that should be re-acquired in order to undergo transformation [70]. Adult stem cells can self-renew and thus require fewer mutations and/or epigenetic modifications to undergo neoplastic transformation than progenitor cells [3]. Progenitor cells may gain stem cell-like characteristics through the activation of self-renewal-related genes through de novo mutations [70] or gain of mutations of adult stem cell’s origin [3], or via EMT induction [2, 71].
Since hematopoietic lineage markers are well known, leukaemia has become an important model for the study of the cell origin of LSCs. Nevertheless, LSCs’ origin is still controversial [3, 6]. Many reports support that leukaemia arises from malignant HSCs [3]. A study on patients with acute myelogenous leukaemia (AML) detected different cell types of the myeloid and lymphoid lineages expressing the AML1/ETO transcript. The authors suggested that the acquisition of the genetic translocations had occurred at the stem cell stage, as these cells were able to differentiate into B cells and cells from the myeloid lineage, and some of which acquired additional mutations that ultimately resulted in AML [72]. These results are further supported by the observation that LSCs are CD34+CD38-, similarly to normal primitive cells, supporting that HSCs are a possible target for transformation into LSCs [73]. Chronic lymphocytic leukaemia (CLL), a malignancy involving mature B lymphocytes, was demonstrated to be caused by the accumulation of oncogenic episodes within the HSC population [74].
The acquisition of self-renewal capability in progenitor cells may induce leukaemia, as observed by the expression of the MLL-ENL [75], MOZ-TIF2 [76], and MLL-AF9 oncogenes, or by the activation of β-catenin [77] in progenitor cells of the hematopoietic system. However, when the same oncogenes were expressed in HSCs, fewer transformed cells were required for leukaemia induction in vivo and the tumours induced by these cells were more heterogeneous [2]. These results indicate that progenitor cells require self-renewal properties in order to be transformed into LSCs, a mechanism that already exists in HSCs. In addition to leukaemia, CSC’s origin has been determined in solid tumours such as gliomas. When Nf1 and p53 gene mutations were introduced into neural stem cells (NSCs), no abnormalities were observed, except when these alterations were present in the oligodendrocyte precursor cells (OPCs), as confirmed by the occurrence of glioma [78]. Instead, when telomerase expression was induced in adult mesenchymal stem cells, neoplastic transformation occurred and tumour initiation was observed [79]. Furthermore, evidence has shown that the induction of oncogene expression or abrogation of tumour suppression genes induces the transformation of stem cells into CSCs, leading to tumour development [80–84]. Morrison and colleagues (2008) studied the tumours of the peripheral nervous system (PNS) and demonstrated that the absence of Nf1-deficient neural crest stem cells in postnatal mice, supporting that the PNS tumours did not originate from these stem cells but rather from differentiated glia [8]. Another study showed that the lack of INK4A and ARF genes and the activation of the EGFR pathway in both NSCs and differentiated astrocytes induced gliomagenesis [85].
Further studies are necessary to improve our knowledge regarding the origin of CSCs, as these cells seem to result from the transformation of adult stem cells, progenitor and/or differentiated cells. Due to the longer lifespan of adult stem cells, these cells are more likely to be the target for the accumulation of genetic and/or epigenetic events that may induce the first steps of tumorigenesis and carcinogenesis. Furthermore, the self-renewal and differentiation properties of stem cells allow the accumulation of mutations and other alterations to the downstream progeny during the lifespan of an organism [68], explaining why some types of progenitor and differentiated cells can give rise to CSCs.
Aging and transformation of stem/progenitor cells into CSCs?
Mammalian aging is associated with a reduction in organismal functionality and this is accompanied by an age-related decline in tissue regeneration and homoeostasis [11, 13, 14, 68]. Due to its complexity, the biological mechanisms underlying the aging process are still not well known [68]. Diverse theories have been proposed in an attempt to explain this issue [11], including the mutation accumulation theory, suggested in 1952 by Medawar [86], and the antagonistic pleiotropy theory, suggested by Williams in 1957 [87]. Later on in 1977, Kirkwood published the disposable soma theory, suggesting that an organism has a limited amount of energy that should be divided between the non-reproductive part of the organism, or soma, and the reproductive part, or germ line. Lifespan is controlled by an energetic balance needed to allow the organism to repair age-related damage with minimal impact on reproductive capacity [88]. Immortal germ cells must resist stress to be able to transmit genetic information to subsequent generations with high accuracy and reliability [11]. In this context, adult stem cells can be considered part of the somatic content of an organism as they are not immortal and have a finite replicative lifespan, in contrast to embryonic stem cells (ESCs) and germ cells [89]. Thus, this theory predicts that adult stem cells may be subjected to age-related alterations, such as telomeric DNA reduction, DNA repair deficiency, chromosome rearrangements, genotoxic effects, and accumulation of genetic/epigenetic alterations [14] (Figure 3). Indeed, it is widely accepted that aging is associated with stem cell pool depletion, as a consequence of cellular senescence or apoptotic death, and/or their reduced functionality, leading to reduced tissue regeneration potential and impaired homeostasis [11, 89, 90]. These effects are associated with the onset of age-related human diseases, such as neurodegenerative disorders, heart failure, arthritis, diabetes, etc. [13, 68, 91] (Figure 3). Furthermore, the transformation of stem/progenitor cells into CSCs can be induced with time, leading to tumour formation and carcinogenesis [2, 3, 10, 14, 15, 68]. In fact, cell senescence and apoptosis are important mechanisms for the organism as a defence from tumour formation [7, 11, 13, 14, 92, 93]. This review has focused on aging-associated tumorigenesis.

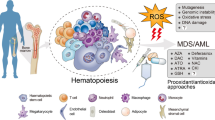
Age-related effects in the stem cell population. Stem cell populations, and their progeny, are subjected to age-effects that are related to the decline in tissue regeneration and homeostasis and consecutive onset of chronic human diseases, such as neurodegenerative disorders, heart failure, and diabetes. Deregulation of stem cell function or decrease of their population, due to cell senescence and apoptotic death, are the main factors for these age-related observations. Furthermore, malignant transformation of stem/progenitor cells may occur, leading to tumour formation and carcinogenesis. Indeed, the aging process is associated with the accumulation of genetic/epigenetic alterations within the stem/progenitor cell populations, together with telomeric DNA reduction, DNA repair deficiency, and chromosome rearrangements that may induce this process. During time, endogenous (e.g. ROS, RNS, spontaneous hydrolysis, alkylation, replication errors, telomere shortening, etc.) and exogenous (e.g. UV light, chemical, chemotherapeutic and radioactive compounds, X-rays, ionizing radiation, etc.) damage may occur, thus affecting the stem cell population.
Some types of cancer can be viewed as age-related disorders associated with genetic and/or epigenetic alterations of stem/progenitor cells along with the deregulation of their microenvironment [14, 68, 94–99]. In the case of HSCs, a direct link was established between the age-related loss of lineage specificity and regenerative capacity [95], and the onset of leukaemia [94–97]. This relationship is supported by the expression of leukaemia-related genetic [95] and epigenetic alterations [100] in aged HSCs, associated with an aged bone marrow microenvironment that allows the expansion of LSCs [97]. In addition to leukaemia, adult stem/progenitor cells deficiency and loss of their function during aging have been associated with the onset of non-hematopoietic tumours, such as skin and brain cancers. For example, the resistance of stem cells to cell death and senescence was shown to be mediated by age-associated self-renewal deregulation and genetic and/or epigenetic alterations, inducing their transformation into CSCs and skin tumour [98]. Moreover, aged NSCs have been associated with tumour formation and neurodegenerative disorders, as a consequence of their impairment and inability to perform tissue homeostasis [99].
Age-associated genetic events and generation of CSCs
During the lifespan of an organism, stem cells are subjected to DNA damage, such as base modifications, inter or intrastrand crosslinks, DNA strand breaks, etc. Such damage can result from the effect of endogenous [e.g. reactive oxygen species (ROS), reactive nitrogen species (RNS), spontaneous hydrolysis, alkylation, replication errors, etc.] and exogenous (e.g. UV light, chemical, chemotherapeutic and radioactive compounds, X-rays, ionizing radiation, etc.) genotoxic agents, telomere shortening, and spontaneous degradation of DNA, leading to genome instability [68, 101, 102] (Figure 3). Unlike damaged proteins, lipids and RNA, which can be replaced, damaged DNA cannot be substituted and it is easily heritable to daughter cells. To ensure that the genomic integrity of stem cells is well preserved throughout the lifespan of an organism, these cells must resist damage from noxious external stimuli and injuries. DNA damage response pathways are highly important for stem cells, as shown by the exceptional capability of stem cells to repair DNA, in contrast to other cell types [101], and by studies with DNA repair-deficient mice that showed a senescence- and apoptosis-related depletion of the stem cell population [103]. Nonetheless, age-dependent reduction in DNA damage repair pathways occurs in stem cells and may contribute to their dysfunction as well as malignant transformation and tumorigenesis [104, 105].
Stem cells may possess an additional cytoprotective system to protect against the acquisition of genetic and epigenetic modifications in their genomes. The immortal strand hypothesis states that, during the asymmetric division of a stem cell, the segregation of its DNA takes place in a non-random manner as it depends on the DNA strand’s age. In fact, the new stem cell acquires the oldest template from the DNA replication and the other daughter cell that undergoes differentiation obtains the newly synthesized strand. The stem cell pool includes the DNA strands with fewer mutations acquired during DNA replication and the most similar to the original cell population generated during development [106]. Although evidence exists to support this theory in many types of stem cells, the biochemical mechanisms responsible for this event are still under investigation [106–108].
Although stem cells possess efficient systems for protection from DNA damage, these systems are not perfect and may be overwhelmed. Therefore genetic/epigenetic alterations can elude and accumulate with time within cells [68]. This observation was confirmed by a study from Welch et al (2012), revealing that HSCs and progenitor cells from healthy individuals accumulate mutations with age, 0.13 exonic mutations per year of life [15]. Though most of these background mutations are not relevant for the initiation of leukaemia, few relevant mutations may occur and give advantage to the cell, which then undergoes transformation into a LSC and initiates leukaemia. These observations are consistent with the model of pre-tumour progression proposed by Calabrese et al (2004) that during the period preceding tumorigenesis, neutral mutations accumulate in stem cells without apparent effects on cell morphology or tumorigenesis. Since the stem cell population is very small, benign mutations can be maintained and accumulated over time due to genetic drift. This “pre-tumorigenesis process” may start from birth and proceed until the tumour formation, a process that can take years or even decades. The shift from the pre-tumour progression towards the tumour progression is marked by a combination of mutations that allows the stem/progenitor cells to undergo transformation and induce tumour formation in a multistep process [69]. Therefore, the early stage of this process is characterized by the formation of a pre-CSC. Shlush et al (2014) and Corces-Zimmerman et al (2014) have recently identified pre-leukaemic HSCs as chemoresistant HSCs containing mutations arising early in the evolution of AML [109, 110]. Such cells are distinct from HSCs but antecedent to leukemic cells and contain some, but not all, leukaemia-specific mutations where the earliest are in “landscaping” genes, genes implicated in the regulation of the epigenetic processes, and the latest are in “proliferative” genes [109, 110]. These stem/progenitor cells can acquire extra genetic/epigenetic alterations that can lead to genome instability [15, 69].
Telomere attrition or gaining of oncogenic events (e.g. BCR-ABL or AML1-ETO translocations in HSCs) are examples of such events that allow the selection of these cells among others [14, 15, 68]. Genome instability can lead to additional alterations, such as increased telomerase activity, and inactivation of tumour suppressor genes (e.g. p16INK4A, pRb, p53 and/or PTEN), which allow cells to resist to senescence and apoptosis signals and to become immortal [14, 68]. These events may lead to the malignant transformation of stem/progenitor cells into CSCs and tumorigenesis. Furthermore, the self-renewal properties of stem cells allow the propagation of these genetic and epigenetic events to their progeny, explaining why non-stem cells (e.g. progenitor cells) can undergo transformation into CSCs [14, 68]. Taken together, telomerase activity and/or activation of oncogenes and inactivation of tumour suppressor genes are important factors for the transformation of stem/progenitor cells into CSCs.
Telomerase expression is not homogeneous in all cells within an organism. Indeed, adult stem cells, as ESCs, germ cells and cancer cells, express telomerase, otherwise absent in somatic cells [111, 112]. Telomeric DNA extension and maintenance are ruled by telomerase, a ribonucleoprotein that uses an internal RNA subunit as a template, avoiding telomeres shortness and related chromosome instability and aneuploidy associated with loss of cell viability through cell cycle arrest and apoptosis or necrosis [12, 113]. Unlike germ cells, telomerase expression in adult stem cells decreases with age, causing telomere shortening with each cell division [114]. When telomeres become too short, a p53-mediated DNA-damage response becomes activated, leading to cell cycle arrest or apoptosis. In fact, downregulation of the tumour suppressor protein p53 in telomerase-deficient mice leads to carcinogenesis [115]. Therefore, telomerase has an important protective activity from genome instability and cancer. On the contrary, these defence mechanisms can lead to organ aging and failure as a consequence of stem cells loss, especially in tissues with high turnover. Tissues with high potential for longevity possess stem cells with longer telomeres and this influences the number of cell divisions a stem cell can undergo [116]. CSCs can therefore be generated from stem cells with short telomeres resulting in genomic instability and accumulation of mutations in tumour suppressor genes or in oncogenes. Consequently, telomerase activity can be increased in CSCs, allowing telomere maintenance and elongation in CSCs and tumorigenesis [111]. Telomerase has being studied in different mouse models of tumorigenesis and studies during aging. These studies showed an onset of age-related diseases related with telomerase-deficiency, for the exception of carcinogenesis. In fact, tumour occurrence was reduced even in the absence of the main tumour-suppressor pathways, except when p53 was downregulated. Higher occurrence of cancer was described when telomerase was overexpressed [12]. Telomerase activity is reactivated in most cancers (85-90%), but when this system is inhibited or is lacked (10-15% of the tumours), tumours express a telomerase-independent system, the alternative lengthening of telomeres (ALT) [117, 118]. ALT system is based on the copy of a telomeric DNA template through a homologous recombination and was demonstrated to be expressed in many tumours, being rare in carcinomas [118].
As discussed earlier, inactivation of tumour suppressor genes (e.g. p53, PTEN) together with activation of oncogenes (e.g. Ras, Bmi1, and c-myc) play an important step in the formation of CSCs and in tumorigenesis [7, 11, 13, 14, 68, 92, 93]. Tumour suppressor genes are well-known DNA damage-induced pathways’ components that promote cell senescence, apoptosis, or transient cell cycle inhibition through the upregulation of specific genes [119]. Downregulation of p53 has been observed in many types of cancer [68]. When its activation is stimulated by stress (e.g. damaged DNA, UV, and oncogenic events), this transcription factor induces the expression of various target genes implicated in several cellular pathways, such as the ones associated with cycle regulation, genome stability, and induction of cell differentiation [120]. Moreover, when activated in stem/progenitor cells, p53 may induce their apoptotic death by stimulating pro-apoptotic proteins (like Bak proteins) or by the down-regulating anti-apoptotic factors (such as Bcl-2 and Bcl-xL) [14]. Instead, when down-regulated due to inactivating mutations, deletion of the p14ARF gene, or through MDM2 gene multiplication in the genome, p53 provides an advantage to cells as they resist to apoptosis, leading to their immortality by unlimited cell divisions [120]. It has been demonstrated that p53 suppression may induce CSC expansion and tumour formation. Therefore, therapies that enhance p53 may lead to the elimination of this type of cells and the inhibition of cancer progression [119].
In addition to p53, PTEN and the corresponding abnormality activated PTEN/PI3K/AKT signalling pathway have been related with cancer. PTEN, a tumour suppressor gene that controls cell growth, migration, death and differentiation through AKT regulation [121], can undergo a decrease of its activity through mutations, deletions or methylation silencing of its promoter [122]. Indeed, when depleted in HSCs, NSCs or prostate stem cells, PTEN can lead to leukaemia [123], brain or prostate tumorigenesis, respectively [121]. Mutations in PI3K and AKT were reported in breast CSCs, but also in other cancers, and might be related with tumour proliferation [124].
The activation of oncogenes has been widely demonstrated in many cancers. The oncogenes c-myc and Bmi1 are such examples [125–127]. Recently, c-myc has been used together with other transcription factors for the generation of iPSCs from differentiated cells [128]. This fact demonstrates its importance for stem cell maintenance, although a role in regulating the balance between self-renewal and differentiation of stem cells has also been described [129]. Indeed, cell proliferation can be promoted via c-myc overexpression through its amplification, translocation or activation of downstream genes [126]. In CSCs, c-myc oncogene expression is higher than in other cancer cells and it was essential for CSCs proliferation and survival [130]. Another oncoprotein reported to be implicated in the self-renewal regulation of both normal and cancer stem cells is Bmi1 [131, 132]. Bmi1 is usually upregulated in cancer [127] and its expression was shown to be important for cancer initiation and progression, and maintenance of the CSC compartment [133, 134]. Another example of oncogenes expressing activating mutations in cancer is the Ras family [135]. This family of GTPases is located at the cell membrane and regulates signal transmission from hormones, growth factors and cytokines receptors. These proteins can regulate cell proliferation, differentiation and death through their influence in some signal transduction pathways (e.g. MAPK and PI3K/AKT) [135]. Activating missense mutations in Ras are found in many cancers [125]. Interestingly, different expression levels of K-Ras have diverse effects. For instance, when Ras is activated at endogenous levels, cell proliferation is enhanced, but its overexpression induces cell cycle arrest and senescence [125]. Instead, when K-Ras is absent in ES cells, these cells undergo aberrant transformation and acquire an abnormal self-renewal capacity, demonstrating the tumour suppression protein-like behaviour by promoting tumour formation when absent in stem cells [136].
After CSC formation followed by tumorigenesis, the progression of the disease may go through invasion stages such as metastasis. For this step, unlimited self-renewal properties of CSCs through its deregulation are fundamental [14, 68] and may be acquired through the activation of Wnt/β-catenin, EGFR, NOTCH, and/or Hedgehog signalling pathways, leading to cell survival, maintenance, and metastasis [14]. Wnt/β-catenin is an important signalling pathway that regulates stem cell potency of embryonic and adult stem cells but also their commitment and differentiation [137]. If Wnt/β-catenin functionality is decreased, the progenitor compartment is compromised due to a decline of the stem cell self-renewal ability [138]. In contrast, abnormal activation of this pathway through β-catenin expression is related with CSC maintenance and tumour development [77, 139–141]. Equally, upregulation of the oncoprotein PLAGL-2 in normal and malignant NSC promotes their self-renewal and proliferation through activation of Wnt/β-catenin signalling [142]. This fact is confirmed by the higher copy numbers of PLAGL-2 in human malignant gliomas and colon cancers [142].
Age-associated epigenetic events and generation of CSCs
Epigenetics is the study of molecular factors and processes that regulate gene function independently of alterations in the DNA sequence [143]. This process is important for the proper function of different cells in a multicellular organism by controlling the activation or silencing of specific genes. Epigenetic changes occur at multiple levels, such as DNA methylation and histone modifications, both affecting chromatin folding, and non-coding microRNAs [144]. The functions of DNA methylation, or the de novo addition or maintenance of methyl groups to CpG sites by DNA methyltransferases (DNMTs), include regulation of gene transcription, preservation of parental imprinting, X-chromosome inactivation, and prevention of homologous recombination and chromosomal instability [145, 146]. In addition, the posttranslational modifications of histones, such as acetylation and methylation of lysine residues of histones H3 and H4, control chromatin structure and therefore transcriptional activity [147].
Aging is associated with epigenetic changes. This observation is related to an accumulation of epimutations due to the decrement of the epigenetic control with time [16]. In fact, errors may occur during the DNA methylation maintenance, resulting in genes or genomic regions that undergo hypomethylation or hypermethylation during aging [16, 148]. The epigenetic deregulation and consecutive genetic expression variation may therefore induce the onset of human disorders, like neurological diseases and cancer [149].
Generally, cancer epigenomes display global DNA hypomethylation associated with hypermethylation at definite promoters [150]. DNA hypomethylation occurs at repetitive sequences, coding regions and at the promoters of many genes, such as the oncogenes Ras and Maspin. Furthermore, DNA hypomethylation leads to genome instability through chromosomal reorganizations [151–153]. Age-associated hypermethylation has been observed to occur at CpG islands [154] and at promoters of the key developmental genes [155] and polycomb group target genes (PCGT) [156]. Teschendorff et al (2010) demonstrated that methylation at the PCGT promoters can lead to the silencing maintenance in stem cells of usually suppressed genes, which may drive to carcinogenesis as stem cell features are preserved [156]. Furthermore, silencing of tumour-suppressor genes, such as APC, p16INK4A, p14ARF, etc., has been associated with DNA hypermethylation at the promoters [157].
Feinberg et al (2005) proposed a model for the origin of CSCs and therefore carcinogenesis: the epigenetic progenitor model of human cancer [153]. According to this model, cancer arises from stem/progenitor cells by three steps: (1) an epigenetic alteration, (2) a mutation-induced oncogene activation or tumour-suppressor silencing, which can be substituted by epigenetic alterations, and (3) genetic and epigenetic instability. The model, first proposed by Feinberg (2005), is further supported by the observation that cancer cells and ESCs share highly similar epigenetic profiles. Importantly, the notion that epigenetic alterations in stem/progenitor cells are the major driving force for carcinogenesis can explain why most types of cancers arise in the elderly. The case of patients with specific cancer-causing mutations (such as in the APC gene in colon cancer patients) are especially interesting because probably age-associated epigenetic changes are required for the transformation process [158].
Age-associated macromolecule accumulation
Deregulation of proteostasis may be involved in cancer and other human diseases, including neurodegenerative diseases, cystic fibrosis, and type 2 diabetes [159, 160]. Proteostasis, or homeostasis of the proteome, is an essential mechanism for cell function and viability that coordinates proteome stability by regulating protein synthesis, localization, and folding, and the removal of misfolded, degraded, or aggregated proteins [161]. These processes require many components that are tightly regulated by the cell: i) ribosomes that govern protein translation [162], ii) molecular chaperones that control protein folding, localization, and prevent undesirable protein aggregation [163], and iii) the autophagy and proteasome systems that remove unneeded, misfolded, modified, damaged, and aggregated proteins, which are not rescued by chaperones or other systems [164, 165]. In situations of severe proteotoxic stress, senescence or apoptosis can be induced [166].
Many reports have demonstrated that the functionality and efficiency of proteostasis decline with age as shown by the accumulation of damaged proteins in aged tissues [159, 160, 166–168]. Such damaged proteins may result from protein misfolding, aggregation, or modification, translation errors, ROS, or from genetic alterations [166, 169]. As a consequence, damaged proteins can accumulate within cells with age, instigating cell’s malfunction and death due to membrane damage and formation of toxic aggregates [160, 166]. For instance, the occurrence of augmented protein modifications, e.g. oxidation, glycation, or carbonylation, with age may disturb several cellular processes, like energy metabolism and protein synthesis, folding and degradation pathways [170]. Therefore, proper maintenance of protein homeostasis in stem cells is of vital importance as it maintains the cell’s proteome and therefore its functionality [159]. Vilchez et al (2013) proposed a model to clarify the role of proteostasis in the different types of stem cells and differentiated progeny. According to this model, both long-term stem cells (e.g. HSCs) and differentiated cells (e.g. neurons or cardiomyocytes) possess high-quality proteostasis, decreasing the effects caused by damaged proteins and allowing their long-term survival [160].
Therefore, the temporal accumulation of damaged proteins may occur due to the deregulation of proteostasis [159, 160, 166, 167]. In such conditions, the transcription factor heat shock factor 1 (HSF1) becomes activated and induces the expression of heat shock proteins (Hsps) that undertake the folding of the misfolded proteins and prevent their aggregation [167]. HSF1 has been described to have an important role in carcinogenesis as shown by its role in tumour initiation and progression through the regulation of the expression of Hsps and other targets [171, 172]. Indeed, Hsf1-/- mice expressing a mutant p53 are incapable of forming tumours [171]. Furthermore, HSF1 expression is important for proliferation of cancer cells [173], as demonstrated by a study showing that higher HSF1 expression in breast tumours is associated a poorer prognosis in breast cancer [174]. The importance of HSF1 in carcinogenesis may be associated with its capacity to regulate additional transcriptional programs distinct from heat shock, and therefore protein folding-unrelated, like energy metabolism, DNA repair, apoptosis, etc. The activation of these pathways by HSF1 facilitates malignant transformation, cancer cell survival, and proliferation [172]. HSP90, a target of HSF1, is a molecular chaperone that supports protein folding and prevents their aggregation [175]. This proteostasis factor may support the acquisition of genetic diversity of proteins [159]. Indeed, HSP90 seems to protect and maintain functionality of misfolded proteins that result from destabilizing mutations [159]. Such proteins, like p53, Akt, Bcr-Abl, among others, have important roles within the cellular pathways, and were found to be mutated in cancers [176, 177]. HSP90 may have an important role in the survival of CSCs and cancer cells, as demonstrated by the development of cancer therapies targeting this molecular chaperone [178].
If chaperones cannot repair damaged or misfolded proteins, these proteins undergo degradation through autophagy or proteasome [160]. Autophagy, a lysosomal catabolic pathway important for the degradation of damaged organelles and proteins, is an essential physiological mechanism for self-renewal and differentiation of adult stem cells [164, 179, 180]. Indeed, the long lifespan and the quiescent state of these cells limit their capacity to dilute “cell waste” through their progeny [168]. When this mechanism was absent in stem cells, a block of the differentiation potential together with a loss of pluripotency and self-renewal was observed in these cells [179]. Importantly, autophagy can circumvent oncogenesis by eliminating damaged mitochondria, that would otherwise lead to bioenergetic deficiency, an escalation of ROS levels, and oncogenic proteins like p16/SQTM1 [168]. Autophagy can be suppressed by several oncogenes (e.g. Akt, PI3K, etc.) and be induced by tumour suppressor proteins, such as PTEN, DAPK1, etc.) [168].
The proteasome system and autophagy are important cellular mechanisms that regulate an appropriate protein concentration within the cell [160, 165]. Such proteins are involved in many cellular pathways, such as cell cycle, apoptosis, signal transduction, etc. [181]. Although the importance of these systems for hESC pluripotency are known, their role in adult stem cells still needs to be clarified [160].
Taken together, adult stem cells maintain cytoprotective mechanisms to prevent the accumulation of proteins that could otherwise lead to cellular damage. The deregulation of some of the proteostasis pathways, such as autophagy and molecular chaperones, might be related with tumorigenesis. It will be important to understand the role of proteostasis in stem cells during aging and their relation with the generation of CSCs. It is clear that proteostasis is an important process for both CSCs and cancer cells, in terms of CSC maintenance [182–184], chemoresistance [185], migration and invasion [186] and as demonstrated by its activation in cancer cells [171, 172, 187].
Conclusions
In the last decade, significant progress has been made in the field of CSC biology and we are gradually gaining a better understanding of the role of these cells in tumour initiation and progression. Indeed, CSCs are cells with stem cell-like properties that may play a central role in the process of carcinogenesis. They have been implicated in tumorigenesis, angiogenesis, invasion and metastasis, and tumour recurrence after therapy. Recent cancer therapies have been focused on CSCs in the hope that their elimination will allow the elimination of self-renewal tumour cells and therefore the bulk tumour, leading to a long-term suppression of disease recurrence.
Although our knowledge in terms of the origin of CSCs has been improved in recent years, there is still much that remains unknown. Different models have been developed supporting the emerging consensus that CSCs arise from a stem/progenitor through temporal accumulation of genetic/epigenetic alterations [14, 15, 69, 95, 153]. Indeed, the complex transformation process of stem/progenitor cells into CSCs may involve different steps and it may occur in parallel with a deregulated microenvironment, associated with telomere attrition, inactivation of tumour suppressor genes and upregulation of oncogenes together with genomic and epigenetic instability. At more advanced stages of malignant transformation CSCs may evade the immune system, induce angiogenesis, and finally acquire metastatic capacities.
Due to the fact that age-related effects on stem/progenitor cells are difficult to study experimentally, many aspects of this process are still not well known; especially with regards to the transformation mechanism leading to the generation of CSCs. Therefore, it is of special importance to fill-in the gap between age-associated effects on stem/progenitor cells and tumorigenesis. Likewise, further characterization of the mechanisms involving proteostasis in stem cells and their role during aging is needed, as well as their relation with the development of CSCs.
Therefore, in the context of aging, genetics, epigenetics and the microenvironment are all interconnected, permitting tumour initiation and progression. It is of enormous importance to study the biological properties of CSCs, particularly genotypic and phenotypic features that distinguish them from normal stem cells. Knowledge gained from such studies is a pre-requisite for the development of new therapies that selectively target CSCs, neutralizing their metastatic potential and thus eliminating one of the most lethal age-related diseases, cancer.
Abbreviations
- AML:
-
Acute myelogenous leukaemia
- ATRA:
-
All-trans retinoic acid
- ALT:
-
Alternative lengthening of telomeres
- CSC:
-
Cancer stem cell
- CLL:
-
Chronic lymphocytic leukaemia
- DNMT:
-
DNA methyltransferase
- ESC:
-
Embryonic stem cell
- EMT:
-
Epithelial to mesenchymal transition
- FACS:
-
Fluorescence-activated cell sorting
- HSF1:
-
Heat shock factor 1
- Hsp:
-
Heat shock protein
- HSC:
-
Hematopoietic stem cell
- hPSC:
-
Human pluripotent stem cell
- iPC:
-
Induced pluripotent cancer cell
- LSC:
-
Leukemic stem cell
- miPSC:
-
Mouse induced pluripotent stem cell
- NSC:
-
Neural stem cell
- NOD/SCID:
-
Non-obese diabetic/combined immunodeficiency disease
- OPC:
-
Oligodendrocyte precursor cell
- PNS:
-
Peripheral nervous system
- PCGT:
-
Polycomb group target
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- SAHA:
-
Suberoylanilide hydroxamic acid
References
O'Brien CA, Kreso A, Dick JE: Cancer Stem Cells in Solid Tumors: An Overview. Semin Radiat Oncol. 2009, 19 (2): 71-77.
Dick JE: Stem cell concepts renew cancer research. Blood. 2008, 112 (13): 4793-4807.
Reya T, Morrison SJ, Clarke MF, Weissman IL: Stem cells, cancer, and cancer stem cells. Nature. 2001, 414 (6859): 105-111.
Liu HG, Chen C, Yang H, Pan YF, Zhang XH: Cancer stem cell subsets and their relationships. J Transl Med. 2011, 9:
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF: Prospective identification of tumorigenic breast cancer cells. P Natl Acad Sci USA. 2003, 100 (7): 3983-3988.
Lobo NA, Shimono Y, Qian D, Clarke MF: The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007, 23: 675-699.
Dalerba P, Cho RW, Clarke MF: Cancer stem cells: Models and concepts. Annu Rev Med. 2007, 58: 267-284.
Joseph NM, Mosher JT, Buchstaller J, Snider P, McKeever PE, Lim M, Conway SJ, Parada LF, Zhu Y, Morrison SJ: The loss of Nf1 transiently promotes self-renewal but not tumorigenesis by neural crest stem cells. Cancer Cell. 2008, 13 (2): 129-140.
Zhao RC, Zhu YS, Shi Y: New hope for cancer treatment: exploring the distinction between normal adult stem cells and cancer stem cells. Pharmacol Ther. 2008, 119 (1): 74-82.
Nguyen LV, Vanner R, Dirks P, Eaves CJ: Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012, 12 (2): 133-143.
Rando TA: Stem cells, ageing and the quest for immortality. Nature. 2006, 441 (7097): 1080-1086.
Blasco MA: Telomeres and human disease: Ageing, cancer and beyond. Nat Rev Genet. 2005, 6 (8): 611-622.
Liu L, Rando TA: Manifestations and mechanisms of stem cell aging. J Cell Biol. 2011, 193 (2): 257-266.
Mimeault M, Batra SK: Recent insights into the molecular mechanisms involved in aging and the malignant transformation of adult stem/progenitor cells and their therapeutic implications. Ageing Res Rev. 2009, 8 (2): 94-112.
Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al: The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012, 150 (2): 264-278.
Gravina S, Vijg J: Epigenetic factors in aging and longevity. Pflugers Arch. 2010, 459 (2): 247-258.
Bonnet D, Dick JE: Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997, 3 (7): 730-737.
George AA, Franklin J, Kerkof K, Shah AJ, Price M, Tsark E, Bockstoce D, Yao D, Hart N, Carcich S, et al: Detection of leukemic cells in the CD34(+)CD38(-) bone marrow progenitor population in children with acute lymphoblastic leukemia. Blood. 2001, 97 (12): 3925-3930.
Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra D, Zhou J, Claypool K, et al: Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006, 25 (12): 1696-1708.
Wang YC, Yo YT, Lee HY, Liao YP, Chao TK, Su PH, Lai HC: ALDH1-Bright Epithelial Ovarian Cancer Cells Are Associated with CD44 Expression, Drug Resistance, and Poor Clinical Outcome. Am J Pathol. 2012, 180 (3): 1159-1169.
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB: Identification of human brain tumour initiating cells. Nature. 2004, 432 (7015): 396-401.
Tirino V, Camerlingo R, Franco R, Malanga D, La Rocca A, Viglietto G, Rocco G, Pirozzi G: The role of CD133 in the identification and characterisation of tumour-initiating cells in non-small-cell lung cancer. Eur J Cardio-Thorac. 2009, 36 (3): 446-453.
Lee CJ, Dosch J, Simeone DM: Pancreatic cancer stem cells. J Clin Oncol. 2008, 26 (17): 2806-2812.
Vermeulen L, Todaro M, Mello FD, Sprick MR, Kemper K, Alea MP, Richel DJ, Stassi G, Medema JP: Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. P Natl Acad Sci USA. 2008, 105 (36): 13427-13432.
Fujimoto K, Beauchamp RD, Whitehead RH: Identification and isolation of candidate human colonic clonogenic cells based on cell surface integrin expression. Gastroenterology. 2002, 123 (6): 1941-1948.
Xu Q, Yuan X, Tunici P, Liu G, Fan X, Xu M, Hu J, Hwang JY, Farkas DL, Black KL, et al: Isolation of tumour stem-like cells from benign tumours. Brit J Cancer. 2009, 101 (2): 303-311.
Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R, Martin AG: Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012, 31 (11): 1354-1365.
Chiou SH, Wang ML, Chou YT, Chen CJ, Hong CF, Hsieh WJ, Chang HT, Chen YS, Lin TW, Hsu HS, et al: Coexpression of Oct4 and Nanog Enhances Malignancy in Lung Adenocarcinoma by Inducing Cancer Stem Cell-Like Properties and Epithelial-Mesenchymal Transdifferentiation. Cancer Res. 2010, 70 (24): 10433-10444.
Karsten U, Goletz S: What makes cancer stem cell markers different?. Springerplus. 2013, 2 (1): 301-
Klonisch T, Wiechec E, Hombach-Kionisch S, Ande SR, Wesselborg S, Schulze-Osthoff K, Los M: Cancer stem cell markers in common cancers - therapeutic implications. Trends Mol Med. 2008, 14 (10): 450-460.
Bloushtain-Qimron N, Yao J, Snyder EL, Shipitsin M, Campbell LL, Mani SA, Hua M, Chen HY, Ustyansky V, Antosiewicz JE, et al: Cell type-specific DNA methylation patterns in the human breast. P Natl Acad Sci USA. 2008, 105 (37): 14076-14081.
Tirino V, Camerlingo R, Franco R, Malanga D, La Rocca A, Viglietto G, Rocco G, Pirozzi G: The role of CD133 in the identification and characterisation of tumour-initiating cells in non-small-cell lung cancer. Eur J Cardiothorac Surg. 2009, 36 (3): 446-453.
Mather JP: In vitro models. Stem Cells. 2012, 30 (2): 95-99.
Sachlos E, Risueno RM, Laronde S, Shapovalova Z, Lee JH, Russell J, Malig M, McNicol JD, Fiebig-Comyn A, Graham M, et al: Identification of Drugs Including a Dopamine Receptor Antagonist that Selectively Target Cancer Stem Cells. Cell. 2012, 149 (6): 1284-1297.
Chen L, Kasai T, Li YG, Sugii Y, Jin GL, Okada M, Vaidyanath A, Mizutani A, Satoh A, Kudoh T, et al: A Model of Cancer Stem Cells Derived from Mouse Induced Pluripotent Stem Cells. Plos One. 2012, 7 (4):
Kim J, Hoffman JP, Alpaugh RK, Rhim AD, Reichert M, Stanger BZ, Furth EE, Sepulveda AR, Yuan CX, Won KJ, et al: An iPSC Line from Human Pancreatic Ductal Adenocarcinoma Undergoes Early to Invasive Stages of Pancreatic Cancer Progression (vol 3, pg 2088, 2013). Cell Rep. 2013, 4 (2): 403-403.
Yenigun VB, Ozpolat B, Kose GT: Response of CD44+/CD24-/low breast cancer stem/progenitor cells to tamoxifen and doxorubicininduced autophagy. Int J Mol Med. 2013, 31 (6): 1477-1483.
Roberts PE: Isolation and establishment of human tumor stem cells. Methods Cell Biol. 2008, 86: 325-342.
Chiba T, Kita K, Zheng YW, Yokosuka O, Saisho H, Iwama A, Nakauchi H, Taniguchi H: Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology. 2006, 44 (1): 240-251.
Ablett MP, Brien CS, Sims AH, Farnie G, Clarke RB: A differential role for CXCR4 in the regulation of normal versus malignant breast stem cell activity. 2013, 4:
Lu S, Labhasetwar V: Drug Resistant Breast Cancer Cell Line Displays Cancer Stem Cell Phenotype and Responds Sensitively to Epigenetic Drug SAHA. Drug Deliv Transl Res. 2013, 3 (2): 183-194.
Miyoshi N, Ishii H, Nagai K, Hoshino H, Mimori K, Tanaka F, Nagano H, Sekimoto M, Doki Y, Mori M: Defined factors induce reprogramming of gastrointestinal cancer cells. P Natl Acad Sci USA. 2010, 107 (1): 40-45.
Carette JE, Pruszak J, Varadarajan M, Blomen VA, Gokhale S, Camargo FD, Wernig M, Jaenisch R, Brummelkamp TR: Generation of iPSCs from cultured human malignant cells. Blood. 2010, 115 (20): 4039-4042.
Bae KM, Su Z, Frye C, McClellan S, Allan RW, Andrejewski JT, Kelley V, Jorgensen M, Steindler DA, Vieweg J, et al: Expression of Pluripotent Stem Cell Reprogramming Factors by Prostate Tumor Initiating Cells. J Urology. 2010, 183 (5): 2045-2053.
O'Brien CA, Kreso A, Dick JE: Cancer stem cells in solid tumors: an overview. Semin Radiat Oncol. 2009, 19 (2): 71-77.
Magee JA, Piskounova E, Morrison SJ: Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012, 21 (3): 283-296.
Lo Coco F, Nervi C, Avvisati G, Mandelli F: Acute promyelocytic leukemia: a curable disease. Leukemia. 1998, 12 (12): 1866-1880.
Munster PN, Troso-Sandoval T, Rosen N, Rifkind R, Marks PA, Richon VM: The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer cells. Cancer Res. 2001, 61 (23): 8492-8497.
Shiozawa Y, Nie BA, Pienta KJ, Morgan TM, Taichman RS: Cancer stem cells and their role in metastasis. Pharmacol Therapeut. 2013, 138 (2): 285-293.
Zhao Y, Bao Q, Renner A, Camaj P, Eichhorn M, Ischenko I, Angele M, Kleespies A, Jauch KW, Bruns C: Cancer stem cells and angiogenesis. Int J Dev Biol. 2011, 55 (4-5): 477-482.
Folkins C, Kerbel R: Tumor Angiogenesis and the Cancer Stem Cell Model. Angiogenesis. Edited by: Figg W, Folkman J. 2008, Springer US, 249-258.
Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T: Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005, 5 (9): 744-749.
Moore N, Lyle S: Quiescent, slow-cycling stem cell populations in cancer: a review of the evidence and discussion of significance. J Oncol. 2011, 2011:
Dean M, Fojo T, Bates S: Tumour stem cells and drug resistance. Nat Rev Cancer. 2005, 5 (4): 275-284.
Pajonk F, Vlashi E, McBride WH: Radiation Resistance of Cancer Stem Cells: The 4 R's of Radiobiology Revisited. Stem Cells. 2010, 28 (4): 639-648.
Sparreboom A, Danesi R, Ando Y, Chan J, Figg WD: Pharmacogenomics of ABC transporters and its role in cancer chemotherapy. Drug Resist Update. 2003, 6 (2): 71-84.
Park S, Shimizu C, Shimoyama T, Takeda M, Ando M, Kohno T, Katsumata N, Kang YK, Nishio K, Fujiwara Y: Gene expression profiling of ATP-binding cassette (ABC) transporters as a predictor of the pathologic response to neoadjuvant chemotherapy in breast cancer patients. Breast Cancer Res Tr. 2006, 99 (1): 9-17.
Phillips TM, McBride WH, Pajonk F: The response of CD24(-/low)/CD44(+) breast cancer-initiating cells to radiation. J Natl Cancer I. 2006, 98 (24): 1777-1785.
Zhang M, Rosen JM: Stem cells in the etiology and treatment of cancer. Curr Opin Genet Dev. 2006, 16 (1): 60-64.
van Vliet P, Sluijter JP, Doevendans PA, Goumans MJ: Isolation and expansion of resident cardiac progenitor cells. Expert Rev Cardiovasc Ther. 2007, 5 (1): 33-43.
Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T: Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005, 121 (6): 823-835.
Goritz C, Frisen J: Neural Stem Cells and Neurogenesis in the Adult. Cell Stem Cell. 2012, 10 (6): 657-659.
Peault B, Rudnicki M, Torrente Y, Cossu G, Tremblay JP, Partridge T, Gussoni E, Kunkel LM, Huard J: Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol Ther. 2007, 15 (5): 867-877.
Bussolati B, Bruno S, Grange C, Buttiglieri S, Deregibus MC, Cantino D, Camussi G: Isolation of renal progenitor cells from adult human kidney. Am J Pathol. 2005, 166 (2): 545-555.
Brittan M, Wright NA: Gastrointestinal stem cells. J Pathol. 2002, 197 (4): 492-509.
Schaffler A, Buchler C: Concise review: Adipose tissue-derived stromal cells - Basic and clinical implications for novel cell-based therapies. Stem Cells. 2007, 25 (4): 818-827.
Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS: Stem cells in normal breast development and breast cancer. Cell Prolif. 2003, 36 (Suppl 1): 59-72.
Rossi DJ, Jamieson CH, Weissman IL: Stems cells and the pathways to aging and cancer. Cell. 2008, 132 (4): 681-696.
Calabrese P, Tavare S, Shibata D: Pretumor progression: clonal evolution of human stem cell populations. Am J Pathol. 2004, 164 (4): 1337-1346.
Wu XZ: Origin of cancer stem cells: the role of self-renewal and differentiation. Ann Surg Oncol. 2008, 15 (2): 407-414.
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008, 133 (4): 704-715.
Miyamoto T, Weissman IL, Akashi K: AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc Natl Acad Sci U S A. 2000, 97 (13): 7521-7526.
Bonnet D, Dick JE: Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997, 3 (7): 730-737.
Kikushige Y, Ishikawa F, Miyamoto T, Shima T, Urata S, Yoshimoto G, Mori Y, Iino T, Yamauchi T, Eto T, et al: Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011, 20 (2): 246-259.
Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL: Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003, 17 (24): 3029-3035.
Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR, et al: MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004, 6 (6): 587-596.
Jamieson CHM, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, et al: Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. New Engl J Med. 2004, 351 (7): 657-667.
Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L, et al: Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011, 146 (2): 209-221.
Serakinci N, Guldberg P, Burns JS, Abdallah B, Schrodder H, Jensen T, Kassem M: Adult human mesenchymal stem cell as a target for neoplastic transformation. Oncogene. 2004, 23 (29): 5095-5098.
Zhu LQ, Gibson P, Currle DS, Tong Y, Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW, Gilbertson RJ: Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature. 2009, 457 (7229): 603-U114.
Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H: Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009, 457 (7229): 608-U119.
Llaguno SA, Chen J, Kwon CH, Parada LF: Neural and Cancer Stem Cells in Tumor Suppressor Mouse Models of Malignant Astrocytoma. Cold Sh Q B. 2008, 73: 421-426.
Perez-Caro M, Cobaleda C, Gonzalez-Herrero I, Vicente-Duenas C, Bermejo-Rodriguez C, Sanchez-Beato M, Orfao A, Pintado B, Flores T, Sanchez-Martin M, et al: Cancer induction by restriction of oncogene expression to the stem cell compartment. Embo J. 2009, 28 (1): 8-20.
Holczbauer A, Factor VM, Andersen JB, Marquardt JU, Kleiner DE, Raggi C, Kitade M, Seo D, Akita H, Durkin ME, et al: Modeling Pathogenesis of Primary Liver Cancer in Lineage-Specific Mouse Cell Types. Gastroenterology. 2013, 145 (1): 221-231.
Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R, et al: Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002, 1 (3): 269-277.
An Unsolved Problem of Biology. Edited by: Medawar PB. 1952, London: H.K. Lewis
Williams GC: Pleiotropy, Natural Selection, and the Evolution of Senescence. Sci Aging Knowl Environ. 2001, 2001 (1): cp13-
Kirkwood TBL: Evolution of Aging. Nature. 1977, 270 (5635): 301-304.
Waterstrat A, Van Zant G: Effects of aging on hematopoietic stem and progenitor cells. Curr Opin Immunol. 2009, 21 (4): 408-413.
Raveh-Amit H, Berzsenyi S, Vas V, Ye D, Dinnyes A: Tissue resident stem cells: till death do us part. Biogerontology. 2013, 14 (6): 573-590.
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G: The Hallmarks of Aging. Cell. 2013, 153 (6): 1194-1217.
Mimeault M, Batra SK: Recent insights into the molecular mechanisms involved in aging and the malignant transformation of adult stem/progenitor cells and their therapeutic implications. Ageing Res Rev. 2009, 8 (2): 94-112.
Ben-Porath I, Weinberg RA: The signals and pathways activating cellular senescence. Int J Biochem Cell B. 2005, 37 (5): 961-976.
Sudo K, Ema H, Morita Y, Nakauchi H: Age-associated characteristics of murine hematopoietic stem cells. J Exp Med. 2000, 192 (9): 1273-1280.
Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, Weissman IL: Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005, 102 (26): 9194-9199.
Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL: The aging of hematopoietic stem cells. Nat Med. 1996, 2 (9): 1011-1016.
Vas V, Wandhoff C, Dorr K, Niebel A, Geiger H: Contribution of an aged microenvironment to aging-associated myeloproliferative disease. Plos One. 2012, 7 (2): e31523-
Mimeault M, Batra SK: Recent advances on skin-resident stem/progenitor cell functions in skin regeneration, aging and cancers and novel anti-aging and cancer therapies. J Cell Mol Med. 2010, 14 (1-2): 116-134.
Deleyrolle LP, Reynolds BA: Identifying and enumerating neural stem cells: application to aging and cancer. Prog Brain Res. 2009, 175: 43-51.
Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA: Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007, 5 (8): e201-
Blanpain C, Mohrin M, Sotiropoulou PA, Passegue E: DNA-Damage Response in Tissue-Specific and Cancer Stem Cells. Cell Stem Cell. 2011, 8 (1): 16-29.
Maugeri-Sacca M, Bartucci M, De Maria R: DNA Damage Repair Pathways in Cancer Stem Cells. Mol Cancer Ther. 2012, 11 (8): 1627-1636.
Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ: Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007, 1 (1): 113-126.
Kenyon J, Gerson SL: The role of DNA damage repair in aging of adult stem cells. Nucleic Acids Res. 2007, 35 (22): 7557-7565.
Park Y, Gerson SL: DNA repair defects in stem cell function and aging. Annu Rev Med. 2005, 56: 495-508.
Potten CS, Owen G, Booth D: Intestinal stem cells protect their genome by selective segregation of template DNA strands. J Cell Sci. 2002, 115 (11): 2381-2388.
Karpowicz P, Morshead C, Kam A, Jervis E, Ramuns J, Cheng V, van der Kooy D: Support for the immortal strand hypothesis: neural stem cells partition DNA asymmetrically in vitro. J Cell Biol. 2005, 170 (5): 721-732.
Shinin V, Gayraud-Morel B, Gomes D, Tajbakhsh S: Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat Cell Biol. 2006, 8 (7): 677-U669.
Corces-Zimmerman MR, Hong W-J, Weissman IL, Medeiros BC, Majeti R: Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proceedings of the National Academy of Sciences. 2014
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, et al: Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014, 506 (7488): 328-333.
Armanios M, Greider CW: Telomerase and cancer stem cells. Cold Spring Harb Symp Quant Biol. 2005, 70: 205-208.
Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PLC, Coviello GM, Wright WE, Weinrich SL, Shay JW: Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science. 1994, 266 (5193): 2011-2015.
Shay JW, Wright WE: Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis. 2005, 26 (5): 867-874.
Flores I, Canela A, Vera E, Tejera A, Cotsarelis G, Blasco MA: The longest telomeres: a general signature of adult stem cell compartments. Gene Dev. 2008, 22 (5): 654-667.
Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA: p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999, 97 (4): 527-538.
Blagoev KB: Organ aging and susceptibility to cancer may be related to the geometry of the stem cell niche. P Natl Acad Sci USA. 2011, 108 (48): 19216-19221.
Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E, Netto GJ, Epstein JI, Lotan TL, Westra WH, et al: Prevalence of the Alternative Lengthening of Telomeres Telomere Maintenance Mechanism in Human Cancer Subtypes. Am J Pathol. 2011, 179 (4): 1608-1615.
Shay JW, Reddel RR, Wright WE: Cancer. Cancer and telomeres--an ALTernative to telomerase. Science. 2012, 336 (6087): 1388-1390.
Bonizzi G, Cicalese A, Insinga A, Pelicci PG: The emerging role of p53 in stem cells. Trends Mol Med. 2012, 18 (1): 6-12.
Vogelstein B, Lane D, Levine AJ: Surfing the p53 network. Nature. 2000, 408 (6810): 307-310.
Hill R, Wu H: PTEN, stem cells, and cancer stem cells. J Biol Chem. 2009, 284 (18): 11755-11759.
Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JFM: The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Tar. 2008, 8 (3): 187-198.
Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ: Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006, 441 (7092): 475-482.
Donovan CA, Pommier RF, Schillace R, O'Neill S, Muller P, Alabran JL, Hansen JE, Murphy JA, Naik AM, Vetto JT, et al: Correlation of Breast Cancer Axillary Lymph Node Metastases With Stem Cell Mutations. Jama Surg. 2013, 148 (9): 873-878.
Lau KS, Haigis KM: Non-redundancy within the RAS oncogene family: Insights into mutational disparities in cancer. Mol Cells. 2009, 28 (4): 315-320.
Miller DM, Thomas SD, Islam A, Muench D, Sedoris K: c-Myc and Cancer Metabolism. Clin Cancer Res. 2012, 18 (20): 5546-5553.
Crea F, Paolicchi E, Marquez VE, Danesi R: Polycomb genes and cancer: Time for clinical application?. Crit Rev Oncol Hemat. 2012, 83 (2): 184-193.
Takahashi K, Yamanaka S: Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006, 126 (4): 663-676.
Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, Pasche AC, Knabenhans C, MacDonald HR, Trumpp A: c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Gene Dev. 2004, 18 (22): 2747-2763.
Wang JL, Wang H, Li ZZ, Wu QL, Lathia JD, McLendon RE, Hjelmeland AB, Rich JN: c-Myc Is Required for Maintenance of Glioma Cancer Stem Cells. Plos One. 2008, 3 (11):
Nakauchi H, Oguro H, Negishi M, Iwama A: Polycomb gene product Bmi-1 regulates stem cell self-renewal. Ernst Schering Res Found Workshop. 2005, 85-100. 54
Molofsky AV, He S, Bydon M, Morrison SJ, Pardal R: Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 2005, 19 (12): 1432-1437.
Proctor E, Waghray M, Lee CJ, Heidt DG, Yalamanchili M, Li CW, Bednar F, Simeone DM: Bmi1 Enhances Tumorigenicity and Cancer Stem Cell Function in Pancreatic Adenocarcinoma. Plos One. 2013, 8 (2):
Lukacs RU, Memarzadeh S, Wu H, Witte ON: Bmi-1 Is a Crucial Regulator of Prostate Stem Cell Self-Renewal and Malignant Transformation. Cell Stem Cell. 2010, 7 (6): 682-693.
Velho S, Haigis KM: Regulation of homeostasis and oncogenesis in the intestinal epithelium by Ras. Experimental Cell Research. 2011, 317 (19): 2732-2739.
James RM, Arends MJ, Plowman SJ, Brooks DG, Miles CG, West JD, Patek CE: K-ras proto-oncogene exhibits tumor suppressor activity as its absence promotes tumorigenesis in murine teratomas. Mol Cancer Res. 2003, 1 (11): 820-825.
Sokol SY: Maintaining embryonic stem cell pluripotency with Wnt signaling. Development. 2011, 138 (20): 4341-4350.
Reya T, Clevers H: Wnt signalling in stem cells and cancer. Nature. 2005, 434 (7035): 843-850.
Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, Huber M, Hohl D, Cano A, Birchmeier W, et al: Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature. 2008, 452 (7187): 650-U612.
Zurawel RH, Chiappa SA, Allen C, Raffel C: Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Res. 1998, 58 (5): 896-899.
Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM: Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. Embo J. 1999, 18 (21): 5931-5942.
Zheng HW, Ying HQ, Wiedemeyer R, Yan HY, Quayle SN, Ivanova EV, Paik JH, Zhang HL, Xiao YH, Perry SR, et al: PLAGL2 Regulates Wnt Signaling to Impede Differentiation in Neural Stem Cells and Gliomas. Cancer Cell. 2010, 17 (5): 497-509.
Rodenhiser D, Mann M: Epigenetics and human disease: translating basic biology into clinical applications. Canadian Medical Association Journal. 2006, 174 (3): 341-348.
Ducasse M, Brown MA: Epigenetic aberrations and cancer. Mol Cancer. 2006, 5: 60-
Lan J, Hua S, He X, Zhang Y: DNA methyltransferases and methyl-binding proteins of mammals. Acta Biochim Biophys Sin (Shanghai). 2010, 42 (4): 243-252.
Mishra A, Verma M: Epigenetics of solid cancer stem cells. Methods Mol Biol. 2012, 863: 15-31.
Arnaudo AM, Garcia BA: Proteomic characterization of novel histone post-translational modifications. Epigenetics Chromatin. 2013, 6 (1): 24-
Horvath S: DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14 (10): R115-
Egger G, Liang G, Aparicio A, Jones PA: Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004, 429 (6990): 457-463.
Jones PA, Baylin SB: The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002, 3 (6): 415-428.
Eden A, Gaudet F, Waghmare A, Jaenisch R: Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003, 300 (5618): 455-
Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, et al: Epigenetic stem cell signature in cancer. Nat Genet. 2007, 39 (2): 157-158.
Feinberg AP, Ohlsson R, Henikoff S: The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006, 7 (1): 21-33.
Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, et al: Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009, 5 (8): e1000602-
Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, et al: Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010, 20 (4): 434-439.
Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, et al: Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010, 20 (4): 440-446.
Kulis M, Esteller M: DNA methylation and cancer. Adv Genet. 2010, 70: 27-56.
Hiltunen MO, Alhonen L, Koistinaho J, Myohanen S, Paakkonen M, Marin S, Kosma VM, Janne J: Hypermethylation of the APC (adenomatous polyposis coli) gene promoter region in human colorectal carcinoma. Int J Cancer. 1997, 70 (6): 644-648.
Powers ET, Balch WE: Diversity in the origins of proteostasis networks--a driver for protein function in evolution. Nat Rev Mol Cell Biol. 2013, 14 (4): 237-248.
Vilchez D, Simic MS, Dillin A: Proteostasis and aging of stem cells. Trends Cell Biol. 2013
Balch WE, Morimoto RI, Dillin A, Kelly JW: Adapting proteostasis for disease intervention. Science. 2008, 319 (5865): 916-919.
Gebauer F, Hentze MW: Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004, 5 (10): 827-835.
Hartl FU, Bracher A, Hayer-Hartl M: Molecular chaperones in protein folding and proteostasis. Nature. 2011, 475 (7356): 324-332.
Oczypok EA, Oury TD, Chu CT: It's a cell-eat-cell world: autophagy and phagocytosis. Am J Pathol. 2013, 182 (3): 612-622.
Jung T, Catalgol B, Grune T: The proteasomal system. Mol Aspects Med. 2009, 30 (4): 191-296.
Signer RA, Morrison SJ: Mechanisms that regulate stem cell aging and life span. Cell Stem Cell. 2013, 12 (2): 152-165.
Anckar J, Sistonen L: Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011, 80: 1089-1115.
Rubinsztein DC, Marino G, Kroemer G: Autophagy and aging. Cell. 2011, 146 (5): 682-695.
Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM: Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003, 424 (6950): 805-808.
Baraibar MA, Liu L, Ahmed EK, Friguet B: Protein Oxidative Damage at the Crossroads of Cellular Senescence, Aging, and Age-Related Diseases. Oxid Med Cell Longev. 2012
Dai C, Whitesell L, Rogers AB, Lindquist S: Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007, 130 (6): 1005-1018.
Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, Lindquist S: HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012, 150 (3): 549-562.
Meng L, Gabai VL, Sherman MY: Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene. 2010, 29 (37): 5204-5213.
Santagata S, Hu R, Lin NU, Mendillo ML, Collins LC, Hankinson SE, Schnitt SJ, Whitesell L, Tamimi RM, Lindquist S, et al: High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. P Natl Acad Sci USA. 2011, 108 (45): 18378-18383.
Wiech H, Buchner J, Zimmermann R, Jakob U: Hsp90 chaperones protein folding in vitro. Nature. 1992, 358 (6382): 169-170.
Li YY, Zhang T, Schwartz SJ, Sun DX: New developments in Hsp90 inhibitors as anti-cancer therapeutics: Mechanisms, clinical perspective and more potential. Drug Resist Update. 2009, 12 (1-2): 17-27.
McClellan AJ, Xia Y, Deutschbauer AM, Davis RW, Gerstein M, Frydman J: Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell. 2007, 131 (1): 121-135.
Trepel J, Mollapour M, Giaccone G, Neckers L: Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010, 10 (8): 537-549.
Phadwal K, Watson AS, Simon AK: Tightrope act: autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell Mol Life Sci. 2013, 70 (1): 89-103.
Salemi S, Yousefi S, Constantinescu MA, Fey MF, Simon HU: Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res. 2012, 22 (2): 432-435.
Tanaka K: The proteasome: from basic mechanisms to emerging roles. Keio J Med. 2013, 62 (1): 1-12.
Wei L, Liu TT, Wang HH, Hong HM, Yu AL, Feng HP, Chang WW: Hsp27 participates in the maintenance of breast cancer stem cells through regulation of epithelial-mesenchymal transition and nuclear factor-kappa B. Breast Cancer Res. 2011, 13 (5):
Nishizawa S, Hirohashi Y, Torigoe T, Takahashi A, Tamura Y, Mori T, Kanaseki T, Kamiguchi K, Asanuma H, Morita R, et al: HSP DNAJB8 Controls Tumor-Initiating Ability in Renal Cancer Stem-like Cells. Cancer Res. 2012, 72 (11): 2844-2854.
Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V, Tharinger H, et al: Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2013, 32 (18): 2261-2272.
Lee CH, Hong HM, Chang YY, Chang WW: Inhibition of heat shock protein (Hsp) 27 potentiates the suppressive effect of Hsp90 inhibitors in targeting breast cancer stem-like cells. Biochimie. 2012, 94 (6): 1382-1389.
Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A, Dinsdale D, Condorelli F, Brandner S, Campanella M, et al: The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene. 2013, 32 (6): 699-712.
Tang D, Khaleque MA, Jones EL, Theriault JR, Li C, Wong WH, Stevenson MA, Calderwood SK: Expression of heat shock proteins and heat shock protein messenger ribonucleic acid in human prostate carcinoma in vitro and in tumors in vivo. Cell Stress Chaperones. 2005, 10 (1): 46-58.
Acknowledgments
This work was supported by grants from EU FP7 projects (D-BOARD, HEALTH-F2-2012-305815; Anistem, PIAPP-GA-2011-286264; EpiHealth, HEALTH-2012-F2-278418; EpiHealthNet, PITN-GA-2012-317146; IDPbyNMR, PITN-GA-2010-264257). MHA and AM gratefully acknowledge funding from King Abdulaziz City for Science and Technology (KACST) and King AbdulAziz University (KAU).
Declarations
Publication charges for this article have been funded by the Center of Excellence in Genomic Medicine Research (CEGMR), King Abdulaziz University, Jeddah, 21589, Kingdom of Saudi Arabia.
This article has been published as part of BMC Cancer Volume 15 Supplement 1, 2015: Selected articles from the 2nd International Genomic Medical Conference (IGMC 2013): Cancer. The full contents of the supplement are available online at http://www.biomedcentral.com/bmccancer/supplements/15/S1
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SSF wrote the manuscript. HR, JK, MHA, AM, and AD edited the final version.
All authors read and approved the final version.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Franco, S.S., Raveh-Amit, H., Kobolák, J. et al. The crossroads between cancer stem cells and aging. BMC Cancer 15 (Suppl 1), S1 (2015). https://doi.org/10.1186/1471-2407-15-S1-S1
Published:
DOI: https://doi.org/10.1186/1471-2407-15-S1-S1