
Abstract
Background
Maize rough dwarf disease (MRDD) is a devastating viral disease that results in considerable yield losses worldwide. Three major strains of virus cause MRDD, including maize rough dwarf virus in Europe, Mal de Río Cuarto virus in South America, and rice black-streaked dwarf virus in East Asia. These viral pathogens belong to the genus fijivirus in the family Reoviridae. Resistance against MRDD is a complex trait that involves a number of quantitative trait loci (QTL). The primary approach used to minimize yield losses from these viruses is to breed and deploy resistant maize hybrids.
Results
Of the 50 heterogeneous inbred families (HIFs), 24 showed consistent responses to MRDD across different years and locations, in which 9 were resistant and 15 were susceptible. We performed trait-marker association analysis on the 24 HIFs and found six chromosomal regions which were putatively associated with MRDD resistance. We then conducted QTL analysis and detected a major resistance QTL, qMrdd1, on chromosome 8. By applying recombinant-derived progeny testing to self-pollinated backcrossed families, we fine-mapped the qMrdd1 locus into a 1.2-Mb region flanked by markers M103-4 and M105-3. The qMrdd1 locus acted in a recessive manner to reduce the disease-severity index (DSI) by 24.2–39.3%. The genetic effect of qMrdd1 was validated using another F6 recombinant inbred line (RIL) population in which MRDD resistance was segregating and two genotypes at the qMrdd1 locus differed significantly in DSI values.
Conclusions
The qMrdd1 locus is a major resistance QTL, acting in a recessive manner to increase maize resistance to MRDD. We mapped qMrdd1 to a 1.2-Mb region, which will enable the introgression of qMrdd1-based resistance into elite maize hybrids and reduce MRDD-related crop losses.
Similar content being viewed by others
Background
Maize rough dwarf disease (MRDD) is a viral disease that results in substantial yield losses in Europe, East Asia, and South America [1–4]. MRDD was discovered in 1954 in China (South Xinjiang and West Gansu) and has posed a grave threat to maize production during the last two decades, especially in the Yellow-Huai-Hai River plain [5]. Between 2008 and 2011, MRDD has affected over three million hm2 of crops each year. Yield losses are generally over 30% in affected areas and can reach 100% in regions of severe infection [6]. The virus that causes MRDD belongs to the genus Fijivirus in the family Reoviridae, but virus strains vary between continents. MRDD is caused by maize rough dwarf virus in Europe, Mal de Río Cuarto virus in South America, and rice black-streaked dwarf virus in East Asia [7]. These viruses are transmitted in a persistent manner by planthopper insect vectors [8].
MRDD symptoms include stunting, dark-green leaves, waxy enations on abaxial surfaces of leaves and sheaths, malformed tassels and upper leaves, suppressed flowering, and a lack of ears (or nubbins). Current methods for controlling MRDD include pesticides, shifting the date(s) when seeds are planted (i.e., based on projected insect populations), and improving field management [9]. These methods limit the planthopper population and reduce, to some extent, MRDD severity, but always with high risk and low efficiency. The identification of MRDD-resistant strains, however, likely represents the most cost-effective and environmentally friendly way to minimize yield losses. It is therefore important to develop and deploy resistant hybrids by mapping and cloning genes and quantitative trait loci (QTLs) that confer resistance to MRDD.
Under natural-infection conditions, the maize germplasm displays variable resistance to MRDD [10–14]. The major source of resistance is derived from US hybrid P78599. Evaluation of 96 inbred lines and 136 hybrids suggests that MRDD resistance is a quantitative trait [15]. Wang et al. (2000) reported that maize resistance to MRDD is a quantitative trait controlled by many genes, each with a small effect [16]. In Argentina, a partially resistant line yielded moderate heritability of resistance to the Mal de Río Cuarto virus, ranging from 0.44 to 0.56 [4]. Using an F2:3 QTL-mapping strategy, two QTLs were identified on bins 1.03 and 8.03/4 that together explained 36.2% of the phenotypic variance [17]. A major QTL on chromosome 8 for MRDD resistance was identified in the Chinese maize inbred line, X178, based on 514 gene-derived single nucleotide polymorphisms (SNPs) [6]. Using an F2 population derived from the highly resistant line 90110 and the susceptible line Ye478, Luan (2012) found at least three QTLs within chromosome bins 6.02, 7.02, and 8.07 that confer MRDD resistance [18].
In this study, we applied trait-marker association to 24 heterogeneous inbred families (HIFs) and QTL analysis to segregating population derived from HIFs to identify regions of the maize genome that affect resistance to MRDD. We then fine-mapped the major QTL by subjecting self-pollinated backcrossed families to recombinant-derived progeny testing. Finally, an F6 recombinant inbred line (RIL) population was used to validate the effect of this QTL. These results provide valuable information concerning maize resistance to MRDD, and markers developed within the qMrdd1 region may prove useful in resistance breeding programs.
Results
Evaluation of HIFs in resistance to MRDD
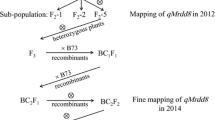
The 50 HIFs developed from a maize hybrid CL1165 were evaluated for their resistance to MRDD in Taian for three years (from 2008 to 2010) and another two locations (Feicheng and Jining) in 2010. Of them, 9 displayed consistent resistance regardless of the year or location; while 15 showed high susceptibility in every location across three years (Additional file 1). These 24 HIFs were therefore used for subsequent trait-marker analysis (Figure 1).

Experimental flow chart for QTL identification and fine-mapping. Twenty-four HIFs were selected for trait-marker association analysis. Associated regions were validated using two BC1 populations, and a major QTL, qMrdd1, was identified and mapped. Recombinants identified within the BC1F4 population were genotyped and phenotyped to fine-map the QTL.
Trait-marker association analysis in HIFs
Genotyping 56,110 SNPs on the 24 HIFs generated 48,384 successful calls based on a cluster-separation score of ≥0.3 and <50% missing data. Of these 48,384 SNPs, 8,668 were polymorphic with a minor allelic frequency > 0.05 among the 24 HIFs. Importantly, 105 SNPs co-segregated with MRDD resistance. After projecting onto the maize accessioned golden path (AGPv2), 103 co-segregating SNPs were found to cluster within six chromosomal regions (Table 1). The remaining two SNPs likely resulted from random error and were excluded from further analysis. The six genomic regions represent candidate MRDD-resistant loci.
QTL analysis of maize resistance to MRDD
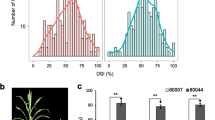
Based on both genotypes and phenotypes, four HIFs, NT401 (susceptible), NT399 (resistant), NT409 (susceptible), and NT411 (resistant), were selected to prepare segregating populations. The F1 hybrid between NT401 and NT399 was backcrossed twice to the susceptible NT401 to generate BC2F1 populations. The F1 hybrid between NT409 and NT411 was backcrossed to the susceptible NT409 and then self-pollinated to generate BC1F2 populations (Figure 1). The totally 485 BC2F1 and 211 BC1F2 families were then evaluated for MRDD susceptibility at four experimental stations: Taian, Feicheng, Heze, and Jining. The disease-severity index (DSI) values were estimated for BC1 individuals based on their BC2F1 or BC1F2 families. The resultant DSI values displayed continuous distributions, ranging from 66.7–100% in Taian (BC1/BC2F1), 39.7–100% in Feicheng (BC1/BC2F1), 51.9–100% in Taian (BC1/BC1F2), and 67.9–100% in Jining (BC1/BC1F2), respectively (Figure 2B), implying the quantitative nature of maize resistance to MRDD. Intriguingly, DSI distributions skewed toward the susceptible parent, suggesting that a major dominant resistance QTL was absent. In Taian, for example, BC2F1 individuals were extremely susceptible to MRDD, and BC1F2 individuals exhibited a segregation bias towards susceptibility (Figure 2B). In addition, location had a large effect on DSI. A population planted in Jining generally had a higher DSI than the same BC2F1 family in Taian. There was no MRDD outbreak in Heze, so nearly every family appeared MRDD resistant (even the susceptible line, NT409). Data from this location were therefore excluded from further analysis. As such, environmental conditions could fluctuate wildly in MRDD resistance (Figure 2B).

Phenotype of MRDD. A. Plants with MRDD scores of 1–9. Disease scores were primarily based on plant height. Here 1 = no symptom; 3 = slightly shorten superior internodes, about 4/5 plant height of healthy plant; 5 = dark-green leaves, waxy enations on abaxial surfaces of leaves and sheaths, obviously shorten superior internodes, about 2/3 plant height of healthy plant; 7 = severe shorten internodes, malformed tassels, about 1/2 plant height of healthy plant; 9 = severe stunning; suppressed flowering, and a lack of ears; plant height <1/3 of healthy plant. B. DSI distributions within segregating populations. Vertical arrows represent the DSI of parental lines.
Within the six candidate regions, 18 simple-sequence repeat (SSR) primer pairs were retrieved from the public database, and additional 81 primer pairs were developed. These 99 SSR markers were used to screen for polymorphisms between the parental lines, resulting in the identification of 14 polymorphic SSR markers (Table 2 and Additional file 2). Each BC1 individual was then genotyped using these 14 SSR markers to distinguish heterozygous from homozygous genotypes at the six candidate regions. The phenotype of a given BC1 individual was represented by DSI values calculated from its BC1F2 or BC2F1 progeny. Mean DSI value was calculated individually for either heterozygous or homozygous BC1 genotypes at each of the six candidate regions. Based on DSI values from BC1F2 progeny at Taian and Jining, three SSR markers within bins 8.04/05, bnlg162, umc1670, and umc1172, were significantly associated with MRDD resistance, as difference in DSI was significant between heterozygous and homozygous BC1 genotypes (P < 0.01). Another two SSR markers within bin 5.03, C5-5 and C5-24, were marginally associated with MRDD resistance in Taian (P = 0.04 and P = 0.06, respectively) (Table 3). For bins 8.04/05, the mean difference in DSI between homozygous and heterozygous BC1 genotypes differed by 9.6% and 7.0% in Taian and Jining, respectively. At the other five chromosomal regions, however, differences in DSI between these two BC1 genotypes were less than 4.0%. Based on DSI values from BC2F1 progeny at Taian and Feicheng, three SSR markers on bins 8.04/05 and two SSR markers on bin 5.03 were marginally associated with MRDD resistance (Table 3). The homozygous and heterozygous BC1 genotypes exhibited very similar DSI values (<2.0% difference). Taken together, the bins 8.04/05 region was most likely to contain a major MRDD resistance QTL.
The BC1F2 population was selected to map the resistance QTL since it showed stronger segregation in MRDD resistance than the BC2F1 population. Based on B73 reference sequence, 63 PCR-based markers were synthesized within bins 8.04/05 and nine of them were polymorphic between the parental lines, NT409 and NT411. Besides three previous SSR markers, these nine markers were also used to genotype the 211 BC1 individuals. Based on genotypic data and the DSI values from BC1F2 families, we performed linkage mapping to define the resistance QTL (in Jining and Taian). A resistance QTL was detected within a 15-Mb region flanked by the markers M103-4 and umc1172 (Figure 3A). This resistance QTL, designated qMrdd1, explained 33.7% and 41.3% of total phenotypic variation in Jining and Taian, respectively (Table 4).

Mapping qMrdd1. A. Diagram of QTL plots for qMrdd1 in 211 BC1F2 families. The logarithm of odds profile, the relative position of qMrdd1, and relevant markers are displayed using QTL cartographer version 2.5. B. Fine mapping of QTL-qMrdd1 based on recombinant progeny. One hundred and one BC1F3 recombinants fell into 23 types based on their genotype at 17 markers. The genetic structure of each type is shown. Black, gray, and white rectangles correspond to homozygous NT411 alleles, heterozygous NT409/NT411 alleles, and homozygous NT409 alleles, respectively. Self-pollinated progeny of these BC1F3 plants were genotyped using markers within the heterozygous region, resulting in three genotypes of progeny. Details concerning the number of recombinants that were planted, the number of progeny that were planted, and the DSI of all three genotypes of progeny are listed in the table. Significant differences (P < 0.05) among the three genotypes indicated that qMrdd1 localized to the heterozygous region and that their parental recombinant(s) was segregating (S). An insignificant difference (P ≥0.05) among the three genotypes suggested that qMrdd1 localized to the homozygous region and that their parental recombinant(s) was not segregating (NS). Analysis of both genotype and phenotype for all recombinant types enabled the positioning of qMrdd1 within a 1.2-Mb region between markers M103-4 and M105-3. DP: deduced phenotype, LOD: logarithm of odds, No. R: number of recombinants, No. P: number of progeny, NA: not available, 411: progeny with a homozygous genotype the same as the MRDD-resistant parental line NT411, H: progeny with a heterozygous genotype, 409: progeny with a homozygous genotype the same as the MRDD-susceptible parental line NT409.
Fine-mapping of qMrdd1
To fine-map qMrdd1, the flanking markers umc1172 and M103-4 were used to screen recombinants from BC1F2 families in Taian. Fifteen BC1F2 recombinants were identified and self-pollinated to generate BC1F3 families. Of 2,685 BC1F3 individuals, 237 recombinants were screened and self-pollinated to generate BC1F4 progeny. To resolve recombinants associated with BC1F4 progeny, 269 SSR primer pairs were designed within the qMrdd1 region, 34 of which were polymorphic. Finally, 15 SSR markers (M103-4, M104-3, M105-3, M106-15, M108-1, M109-12, bnlg1460, M112-5, umc1858, M113-2, M113-6, M114-3, M115-5, M117-2, and M117-5) that were evenly distributed (~1–2 Mb between adjacent markers) throughout the qMrdd1 region were used to resolve the 237 recombinants, resulting in 23 types (Figure 3B, Table 2).
Recombinant-derived BC1F4 progeny were selected and planted in three locations. In Taian, we grew 2,203 BC1F4 individuals derived from 33 recombinants that included all 23 types. In Feicheng, we grew 2,700 individuals derived from 37 recombinants that included 22 types. Finally, in Jining we grew 1,805 BC1F4 individuals derived from 31 recombinants that included 21 types. Self-pollinated BC1F4 progeny had three genotypes within the heterozygous portion of the qMrdd1 locus: homozygous NT409, homozygous NT411, and heterozygous. DSIs for these three genotypes were separately calculated for each BC1F4 family. For each of the 23 recombination types, the genotype matched the phenotype. Types I–VII (see Figure 3B) had the homozygous NT409 sequence upstream of the recombination breakpoint, and heterozygous sequences downstream. Types II–VII were highly susceptible to MRDD regardless of the genotypes, whereas types I exhibited a significant difference in MRDD resistance between the three genotypes. This indicated that qMrdd1 is located downstream of M103-4 and upstream of bnlg1460. Types VIII–XIII had heterozygous sequences upstream of the recombination breakpoint and homozygous NT409 sequences downstream. All Types showed a significant difference in MRDD resistance between the three genotypes, regardless of the experimental location. This clearly indicated that qMrdd1 is located within the heterozygous region. Types XIV and XV also showed segregation of the MRDD resistance trait and thus restricted qMrdd1 into the heterozygous region upstream of the breakpoint. The remaining types (XVI–XXIII) were resistant to MRDD regardless of the genotype or experimental location. This implied that qMrdd1 is located within the homozygous NT411 region but not in the heterozygous region. Only types XIII exhibited a phenotype that varied with experimental location. A significant difference (P < 0.05) in MRDD resistance between genotypes of types XIII BC1F4 progeny was detected in Taian and Jining but not in Feicheng (P = 0.06). This discrepancy may have resulted from the small number of BC1F4 progeny (43 individuals) in Feicheng. As such, the resistance phenotype for types XIII was considered to segregate, placing qMrdd1 within the heterozygous region upstream of M106-15. Recombination breakpoints associated with types I and XIV were closest to qMrdd1, allowing us to fine-map qMrdd1 into the region between M103-4 and M105-3, a physical distance of 1.2 Mb (Figure 3B).
Genetic model of qMrdd1resistance to MRDD
The genetic effect of qMrdd1 was investigated in BC1F4 families (three genotypes and three replications). Plants homozygous for the NT411 qMrdd1 region showed a significantly lower DSI than the other two genotypes (Figure 4). In Taian, resistance to MRDD was evaluated for 2,203 BC1F4 plants. Plants homozygous for NT409 qMrdd1 alleles had a DSI of 79.8%, which was very similar to heterozygous plants (NT411/NT409; 77.1%). In contrast, plants homozygous for NT411 qMrdd1 alleles had a significantly lower DSI (49.9%). In Jining, 1,805 BC1F4 plants were evaluated; although DSIs were generally higher, a similar genetic pattern was observed. DSIs were 95.8% and 96.4% for plants carrying homozygous NT409 or heterozygous qMrdd1 alleles, respectively. In contrast, plants carrying homozygous NT411 qMrdd1 alleles had a DSI of 71.7%. Of 2,700 BC1F4 plants grown in Feicheng, plants carrying qMrdd1 regions that were homozygous for NT411, heterozygous, or homozygous for NT409 had DSIs of 33.2%, 67.1%, and 72.5%, respectively. At all three test facilities there was no difference (P > 0.05) in DSI between heterozygous and homozygous NT409 genotypes at the qMrdd1 locus, indicating that the qMrdd1 QTL acts in a recessive manner to confer resistance to MRDD. Moreover, the strong genetic effect of homozygous NT411 qMrdd1 alleles, which reduce DSI by 24.2–39.3%, suggested that this QTL represents a viable tool for enhancing maize resistance to MRDD.

Effect of QTL- qMrdd1 on BC 1 F 4 populations across three experimental sites. BC1F4 populations were divided into three genotypes (NT409/NT409, NT409/NT411, and NT411/NT411) according to genotypes within the qMrdd1 region. Average DSI values are shown. Error bars indicate s.e.m. Multiple comparisons between genotypes were analyzed using SAS 9.1 PROC general linear model with Tukey’s adjustment.
Validation of qMrdd1 in F6RILs
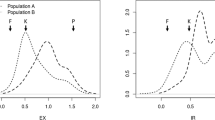
We next determined whether the qMrdd1 QTL is present in other MRDD-resistant lines of maize. Resistance was measured for 157 F6 RILs derived from the cross between X178 and HuangC. These studies were conducted in three locations: Taian, Feicheng, and Jining. SSR markers developed for the qMrdd1 region were used to screen parental lines for polymorphisms. Marker M103-7, which is located within the 1.2-Mb region of qMrdd1, was selected to genotype the RIL population. Of 157 RILs, 91 and 63 were homozygous for X178 and HuangC sequences at M103-7, respectively. Only three RILs were heterozygous at the M103-7 locus and thus were excluded from subsequent analyses. RILs homozygous for the X178 genotype had DSIs of 30.5%, 40.3%, and 29.3% in Taian, Jining, and Feicheng, respectively. In contrast, RILs homozygous for the HuangC genotype had DSIs of 56.1%, 74.3%, and 44.5% in Taian, Jining, and Feicheng, respectively. As such, there was a clear difference in MRDD resistance (P < 0.01) between two homozygous genotypes (Figure 5). These findings suggested that the qMrdd1 QTL could reduce DSI by 15.2–34.0% in the RIL population, which agrees with our previous fine-mapping result.

DSI in an F 6 RIL population. RILs were genotyped at marker M103-7. DSI distributions and mean values are shown for two homozygous genotypes, X178 and HuangC at three experimental sites.
Discussion
Accurate phenotypic evaluation is critical for marker-trait association analyses, especially for quantitative traits [19]. Because large-scale inoculation of plants with MRDD is unfeasible and uniform infection is unreliable, we relied on natural infection processes. Plants were raised in the cities of Jining, Feicheng, Taian, and Heze within Shandong province, where MRDD is prevalent. Because of poor performance in 2011, i.e., the lack of planthoppers, Heze was eliminated as a testing site during subsequent analyses. Subsequent fine-mapping tests were also performed in Taian, Jining, and Feicheng to avoid MRDD escape. The environment significantly influenced MRDD development, as the disease was more serious in Jining compared with Taian and Feicheng. This may have resulted from different numbers of planthoppers at these three test sites. Fortunately, the qMrdd1 locus had a stable genetic effect across these different environments, implying that the natural infection method was a valid approach and that our scoring system was appropriate for QTL analysis of MRDD resistance.
Viral resistance can be influenced by both genetic background and the environment [20, 21]. HIFs derived from the same cross share similar genetic backgrounds, making them ideal for analyzing quantitative traits. To identify the major QTL involved in MRDD resistance, 50 HIFs developed from our breeding program were selected for this study. For segregating populations prepared from two HIFs with very different levels of MRDD resistance, a continuous distribution of resistance was observed rather than two distinct classes. This may have resulted from residual genetic-background differences or environmental conditions. Whole-genome SNP analysis revealed that 14.2% of 20,278 called SNPs differed between susceptible (NT409) and resistant (NT411) plant lines. A smaller difference (3.1% of 51,628 called SNPs) was observed between NT401 and NT399, which are susceptible and resistant lines, respectively. Here we focused on the major QTL for MRDD resistance, but other chromosomal regions may also be involved. A region in chromosome 5, for example, showed a marginally significant correlation with MRDD resistance in both mapping populations. To generate populations for fine mapping we self-pollinated rather than backcrossed because most BC2F1 families were highly susceptible to MRDD, since different genotypes at qMrdd1 had similar phenotypes in BC2F1 plants in 2011.
The recombinant-derived progeny test is an efficient and powerful method for fine-mapping QTLs within a backcrossed population when a susceptible inbred line is used as the recurrent parent. This method can be used to accurately phenotype recombinants by analyzing trait-marker associations in progeny [22–24]. Here we expand the application of this method to include self-pollinated progeny. Compared with backcrossed progeny, progeny generated by self-pollination can capture the effect of all three genotypes and create more recombinants for fine-mapping. However, not all recombinants produced by self-pollination can be used for fine-mapping, as segregating genotypes within the targeted region are not created when recombinants that are homozygous at both flanking markers are self-pollinated. Crosses that involve heterozygous plants represent an effective way of solving this problem.
By applying the recombinant-derived progeny test to self-pollinated progeny, qMrdd1 was fine-mapped to a 1.2-Mb region. This region decreased DSI by 24.2–39.3%. Forty-three additional recombinants were identified from 8,047 BC1F5 for further fine-mapping (data not shown). Compared with previous reports, this represents significant progress towards cloning and applying qMrdd1. Consistent fine-mapping results between test sites suggest that the recombinant-derived progeny-test represents a powerful solution for fine-mapping a dominant QTL in backcrossed progeny or a partially dominant or recessive QTL in self-pollinated progeny. Moreover, mapping results from different years (2011 and 2012) indicated that the genetic effect of QTL-qMrdd1 is heritable. Finally, qMrdd1 decreased DSI by 15.2–34.0% in an F6 RIL population composed of 157 lines, suggesting that qMrdd1 confers a stable genetic effect in diverse genetic backgrounds.
The major QTL mapped in the current study overlaps with resistance regions reported by Di Renzo [17] and Shi [6], implying the same QTL functions across different mapping populations. However, this QTL has not been detected in the research conducted by Luan [18]. This may be due to a different scoring system used By Luan who adopted four indexes, including shorten superior internode, waxy enation, tassel type, and disease severity of MRDD, rather than an overall evaluation of MRDD symptom.
Most important agronomic traits are quantitative in nature and polygenic. Compared with monogenic or oligogenic traits, therefore, these polygenic traits are extremely difficult for breeders and pathologists to manage [21]. Isolation of QTLs, especially major QTLs, may simplify the analysis of quantitative traits and provide important resources for trait improvements. Before QTLs can be applied routinely to breeding programs, a number of challenges must be addressed. These include improving diagnostic assays to detect QTLs and identifying genetic markers for marker-assisted selection [25]. In the present study, we used a reliable scoring system and developed a number of high-density markers within and around the qMrdd1 region. These tools can be used for widespread marker-assisted selection to improve maize resistance to MRDD.
There are two categories of viral resistance in plants—passive resistance and positive resistance. Passive resistance is conferred by recessive plant factors, which are essential for the virus to complete the infection cycle. These typically involve protein forms that cannot be recognized by specific viral components. In contrast, positive resistance is conferred by dominant plant factors that trigger defense mechanisms in response to viral invasion [21]. The qMrdd1 QTL conferred MRDD resistance only in plants homozygous for NT411 alleles, indicating that it involves a recessive gene involved in passive resistance. This information will facilitate identification of a candidate gene for qMrdd1.
Conclusions
Breeding resistant maize hybrids is the most cost-effective way to minimize yield losses from MRDD. We have mapped qMrdd1 to a 1.2-Mb region and showed that it acts in a recessive manner across different genetic backgrounds. The discovery and fine-mapping of this major QTL involved in MRDD resistance lays the foundation for positional cloning of qMrdd1 and moves us closer to genetically controlling MRDD infestation during maize production.
Methods
Plant materials
Plant materials initially selected for QTL analysis were 50 F9 HIFs that were derived from two F5 plants of a single F4 individual from a hybrid, CL1165 (Figure 1). These 50 HIFs were evaluated for MRDD resistance in the summers of 2008, 2009, and 2010 at three locations, Taian, Feicheng, and Jining. In each location, the field test was conducted in randomized complete block design (RCBD) with locations as complete blocks. 25 seeds from each HIF were sown in a single row 0.6 m in width and 5 m in length. Of the 24 F9 families with steadily contrasting phenotype to MRDD, two resistant (NT399 and NT411) and two susceptible (NT401 and NT409) HIFs were selected to prepare two crosses, one between NT409 and NT411 and the other between NT399 and NT401. NT411 and NT409 shares 85.8% of 20,278 called SNPs; while NT399 and NT401 shares 96.9% of 51,628 called SNPs. In 2009, two crosses were established in the Hainan winter nursery. In 2010, F1 plants were backcrossed to the susceptible parental line in Taian. In the Hainan winter nursery, 211 BC1 plants derived from the NT409/NT411 cross were self-pollinated to produce BC1F2 families. In addition, 485 BC1 plants derived from the NT399/NT401 cross were backcrossed to NT401 to produce BC2F1 families. In 2011, the BC2F1 and BC1F2 families, together with parental lines, were planted in three locations: Heze, Taian, and Jining (or Feicheng) within Shandong province. In each location, the field test was conducted in RCBD with locations as complete blocks. 25 seeds from each family were sown in a single row 0.6 m in width and 5 m in length.
Based on QTL mapping, plants from the BC1F2 and BC1F3 families that contained recombination breakpoints within the target QTL region were selected for repeated self-pollination. In the summer of 2012, the resultant BC1F4 progeny were planted in three locations without duplication—Jining, Feicheng, and Taian. All BC1F4 plants were genotyped at the qMrdd1 locus and assayed for MRDD resistance.
Survey of MRDD symptoms in the field
Plants were grown in four locations, namely Jining, Feicheng, Heze, and Taian (Shandong province, China), and allowed to become infected with MRDD under natural conditions, i.e., without artificial inoculations. Seeds were sown at May 24 to 26 to coincide with planthopper infestation and to make viral inoculation more likely. MRDD resistance was evaluated at the grain-filling stage. For the 50 HIFs, we scored them as resistant, intermediate resistant, and susceptible across three years, since we just want to know which HIFs are consistent resistant or susceptible across different years at different locations. For QTL analysis, mapping, and fine-mapping efforts, a scoring system (1, 3, 5, 7, or 9) based on overall symptoms was adopted to evaluate MRDD resistance. Infected plants with different resistance scores are depicted in Figure 2A. The DSI was used to represent MRDD severity of families and was calculated as [26]:
All the phenotypic data across different replicates were assessed independently.
Genotyping
Leaf tissue was harvested for DNA extraction according to the SDS method [27]. SNPs were genotyped using the MaizeSNP50 DNA analysis kit (Illumina, San Diego, CA), which can survey 56,110 SNPs, using an Illumina BeadStation 500G at Cornell University Life Sciences Core Laboratories Center. Details concerning the SNP genotyping procedure and allele scoring have been described [28]. For PCR-based marker genotyping, amplicons were subjected to 1% agarose gel electrophoresis and visualized using a gel-imaging system (Bio-Rad Laboratories Inc.). Alternatively, amplicons were separated using 6% polyacrylamide-gel electrophoresis and visualized with silver-staining.
Trait-marker association analysis
SNPs with >50% missing data or a cluster-separation score of <0.3 were excluded from further analyses. TASSEL 2.0 was used to retrieve polymorphic SNPs with a minor-allelic frequency of >0.1, and the general linear model was used to analyze correlations between polymorphic SNPs and phenotype. Tightly linked SNPs were then mapped to B73 AGPv2 [29] through BLAST comparisons. Every region (<100 kb) that contained >2 co-segregating SNPs was considered a candidate region containing a QTL that conferred rice black-streaked dwarf virus resistance.
Validation and mapping of the major QTL
SSR primer pairs that covered all candidate regions were retrieved from public databases (http://www.maizegdb.org/) or developed based on B73 reference sequences as described by Zhang [23]. All primers were synthesized by Invitrogen (Beijing, China). SSR primers were first used to identify polymorphisms between the two parental lines. Polymorphic SSR markers were then used to genotype each plant in the BC1 populations. The phenotype of each BC1 individual was represented by DSI of corresponding BC1F2 families or BC2F1 families. Trait-marker correlations were analyzed using one-way ANOVA in SAS 9.1. For regions associated with MRDD resistance, more PCR-based markers were developed to map target QTLs. Linkage mapping of polymorphic SSRs was performed using MAPMAKER 3.0b [30]. Linkage groups were identified using the ‘Group’ command with a logarithm of odds score of ≥3.0. Recombination frequency was converted into centiMorgans using the Kosambi mapping function [31]. QTLs were detected using the composite interval mapping method [32] as with the QTL cartographer (Version 2.5) [33]. A significance threshold for identifying a putative QTL was obtained from 1,000 permutations at P < 0.05 for each dataset.
Fine-mapping of qMrdd1
The recombinant-derived progeny test [34] was used for QTL fine-mapping. Based on the QTL region mapped by winQTLcart, the BC1F2 population was screened for recombinants. This was followed by self-pollination in Shandong province in 2011. Progeny were planted in the Hainan winter nursery to screen for new recombinants. Newly-screened BC1F3 recombinants, which were heterozygous at one flanking marker and homozygous at the other flanking marker, were selected for further self-pollination. This produced a segregating population for fine-mapping. Based on developed markers, BC1F3 recombinants were classified into distinct types. In 2012, progeny of diverse BC1F3 recombinant types were planted in Taian, Feicheng, and Jining, with >100 kernels for every type in a single plot.
For each recombinant, the qMrdd1 region was separated into two segments, heterozygous and homozygous, that flanked the recombination breakpoint. Based on markers within heterozygous sequences, self-pollinated progeny were classified into three genotypes: homozygous NT409/NT409, homozygous NT411/NT411, and heterozygous NT409/NT411. Comparisons of score values between these genotypes were performed using one-way ANOVA in SAS 9.1. A significant (P < 0.05) or insignificant (P ≥ 0.05) difference in MRDD resistance between these genotypes indicated that the resistance QTL localized to heterozygous or homozygous segments within qMrdd1, respectively. The phenotypes for three genotypes within the same BC1F3 recombinant-derived progeny were represented by DSI values. If two or more BC1F3 individuals shared the same donor fragment, they can be grouped as one recombination type. The availability of both genotype and deduced phenotype for each recombinant type allowed for fine-mapping of the resistance QTL.
Validation of the qMrdd1locus in an RIL population
The effect of qMrdd1 was also investigated in an F6 RIL population derived from a cross between X178 (MRDD resistant) and HuangC (MRDD susceptible), a commercial hybrid (ND108) widely grown in China. In 2012, 157 F6 RILs, together with parental lines, were evaluated for MRDD resistance in three locations (namely Taian, Jining, and Feicheng) in RCBD with locations as complete blocks. A total of 25 seeds for each RIL were sown in a single row 0.6 m in width and 5 m in length. SSR markers generated during the fine-mapped process were used to genotype, i.e., screen for polymorphisms, among the 157 RILs. Correlations between genotype and MRDD resistance were analyzed using one-way ANOVA in SAS 9.1.
Abbreviations
- DSI:
-
Disease severity index
- HIFs:
-
Heterogeneous inbred families
- Mb:
-
Megabase pairs
- MRDD:
-
Maize rough dwarf disease
- PCR:
-
Polymerase chain reaction
- QTL:
-
Quantitative trait loci
- RIL:
-
Recombinant inbred line
- SNP:
-
Single nucleotide polymorphism
- SSR:
-
Simple sequence repeat.
References
Harpaz I: Needle transmission of a new maize virus. Nature. 1959, 184 (4688): 77-78.
Dovas CI, Eythymiou K, Katis NI: First report of maize rough dwarf virus (MRDV) on maize crops in Greece. Plant Pathol. 2004, 53 (2): 238-238. 10.1111/j.0032-0862.2004.00973.x.
Huth W, Maurath R, Imgraben H, Schroder M: Maize rough dwarf virus for the first time detected in Germany. Nachrichtenblatt des Deutschen Pflanzenschutzdienstes. 2007, 59 (8): 173-175.
Di Renzo MA, Bonamico NC, DÍaz DD, Salerno JC, IbaÑEz MM, Gesumaria JJ: Inheritance of resistance to Mal de Río Cuarto (MRC) disease in Zea mays (L.). J Agric Sci. 2002, 139 (01): 47-53.
Tao YF, Liu QC, Xu ML: The research progress on maize rough dwarf disease. J Maize Sci. 2013, 21 (1): 149-152.
Shi LY, Hao ZF, Weng JF, Xie CX, Liu CL, Zhang DG, Li MS, Bai L, Li XH, Zhang SH: Identification of a major quantitative trait locus for resistance to maize rough dwarf virus in a Chinese maize inbred line X178 using a linkage map based on 514 gene-derived single nucleotide polymorphisms. Mol Breeding. 2011, 30 (2): 615-625.
Milne RG, Lovisolo O: Maize rough dwarf and related viruses. Adv virus res. 1977, 21: 267-341.
Caciagli P, Casetta A: Maize rough dwarf virus (Reoviridae) in its planthopper vector Laodelphax striatellus in relation to vector infectivity. Ann Appl Biol. 1986, 109 (2): 337-344. 10.1111/j.1744-7348.1986.tb05325.x.
Wang AL, Wang JJ, Chen CH: Study on maize rough dwarf virus incidence law and its integrated control technique. J Maize Sci. 2005, 13 (4): 114-116.
Guo QT, Li Z, Dong Z: Observation and analysis of varietal resistance of maize rough dwarf virus disease (MRDV). Plant Prot. 1995, 1: 21-23.
Wang GY, Han HL, Zhao FC, Wang HD, Kong XM, Ye JR: Identification on disease resistance of maize varieties (lines) to maize rough dwarf virus. J Zhejiang Agric Sci. 2011, 23 (3): 564-567.
Shang YF, Zhao J, Du S, Lu X, Wang S, Sun H, Yang C: Identification and investigation on resistance to virus diseases of both maize commercial varieties and germplasm at seedling stage. Shandong Agric Sci. 2001, 4: 3-5.
Xue L, Zhang D, Xu L, Jin MM, Peng CJ, Xu CW: Mining and analyzing genetic diversity for maize rough dwarf disease resistant gerplasms and its application in maize breeding. Acta Agron Sin. 2011, 37 (12): 2123-2129.
Chen YK, Li XH, Xiao MJ, Li MS, Yuan SX, Wang XD, Zhang SH: Genetic variation in sixty-four maize inbred lines in relation to maize rough dwarf virus. Acta Agron Sin. 2006, 32 (12): 1848-1854.
Liu ZZ, Chi SM: Resistance of corn genotypes to maize rough dwarf virus. J Maize Sci. 1996, 4: 68-70.
Wang AL, Zhao DF, Chen ZH, Wang JJ, Shao XS, Wei GY: Studies on genetic basis and recurrent selection effect of inbred line maize resistance to MRDV. J Maize Sci. 2000, 8: 80-82.
Di Renzo MA, Bonamico NC, DÍaz DG, IbaÑez MA, Faricelli ME, Balzarini MG, Salerno JC: Microsatellite markers linked to QTL for resistance to Mal de Río Cuarto disease in Zea mays L. J Agric Sci. 2004, 142 (3): 289-295. 10.1017/S0021859604004307.
Luan JW, Wang F, Li YJ, Zhang B, Zhang JR: Mapping quantitative trait loci conferring resistance to rice black-streaked virus in maize (Zea mays L.). Theor Appl Genet. 2012, 125 (4): 781-791. 10.1007/s00122-012-1871-1.
Myles S, Peiffer J, Brown PJ, Ersoz ES, Zhang Z, Costich DE, Buckler ES: Association mapping: critical considerations shift from genotyping to experimental design. Plant Cell. 2009, 21 (8): 2194-2202. 10.1105/tpc.109.068437.
Fraser RSS: The genetics of resistance to plant viruses. Annu Rev of Phytopathol. 1990, 28: 179-200. 10.1146/annurev.py.28.090190.001143.
Gómez P, Rodríguez-Hernández AM, Moury B, Aranda MA: Genetic resistance for the sustainable control of plant virus diseases: breeding, mechanisms and durability. Eur J Plant Pathol. 2009, 125 (1): 1-22. 10.1007/s10658-009-9468-5.
Yang Q, Yin GM, Guo YL, Zhang DF, Chen SJ, Xu ML: A major QTL for resistance to Gibberella stalk rot in maize. Theor Appl Genet. 2010, 121 (4): 673-687. 10.1007/s00122-010-1339-0.
Zhang DF, Liu YJ, Guo YL, Yang Q, Ye JR, Chen SJ, Xu ML: Fine-mapping of qRfg2, a QTL for resistance to Gibberella stalk rot in maize. Theor Appl Genet. 2012, 124 (3): 585-596. 10.1007/s00122-011-1731-4.
Zhang Y, Xu L, Fan XM, Tan J, Chen W, Xu ML: QTL mapping of resistance to gray leaf spot in maize. Theor Appl Genet. 2012, 125 (8): 1797-1808. 10.1007/s00122-012-1954-z.
Maule AJ, Caranta C, Boulton MI: Sources of natural resistance to plant viruses: status and prospects. Mol Plant Pathol. 2007, 8 (2): 223-231. 10.1111/j.1364-3703.2007.00386.x.
Grau CR, Radke VL, Gillespie FL: Resistance of soybean cultivars to Sclerotinia sclerotiorum. Plant Dis. 1982, 66 (6): 506-508.
Murray MG, Thompson WF: Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8 (19): 4321-4325. 10.1093/nar/8.19.4321.
Yan J, Yang X, Shah T, Sanchez-Villeda H, Li J, Warburton M, Zhou Y, Crouch JH, Xu Y: High-throughput SNP genotyping with the GoldenGate assay in maize. Mol Breeding. 2010, 25 (3): 441-451. 10.1007/s11032-009-9343-2.
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al: The B73 maize genome: complexity, diversity, and dynamics. Science. 2009, 326 (5956): 1112-1115. 10.1126/science.1178534.
Lincoln S, Daly M, Lander E: Mapping genetic mapping with MAPMAKER/EXP3.0. Cambridge: Whitehead Institute Technical Report; 1992.
Kosambi DD: The estimation of map distances from recombination values. Ann Eugenics. 1944, 12 (3): 172-175.
Zeng ZB: Precision mapping of quantitative trait loci. Genetics. 1994, 136 (4): 1457-1468.
Basten CJ, Weir BS, Zeng ZB: QTL cartographer: a reference manual and tutorial for QTL mapping. Raleigh, NC: Department of Statistics North Carolina State University; 1997.
Yang Q, Zhang DF, Xu ML: A sequential quantitative trait locus fine-mapping strategy using recombinant-derived progeny. J of Integr Plant Biol. 2012, 54 (4): 228-237. 10.1111/j.1744-7909.2012.01108.x.
Acknowledgements
This study was financially supported by the Ministry of Agriculture of China (2011ZX08009-003-001), the National High-tech and development Program of China, (2012AA10A306 and 2012AA101104) and the National Basic Research ‘973’ program of China (2009CB118402). We thank Dr. Xiaohong Yang at the China Agricultural University for genotyping at 56,110 SNPs.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YFT and QCL carried out molecular genetic studies, performed statistical analysis and participated in phenotypic evaluation. MLX supervised the research, designed the experiments and involved in data analysis. BSL, HHW and YJZ performed the field maize cultivation and pollination, as well as phenotypic evaluation. XYH, BBW and JSL helped with the development and genotyping of the RILs population. YFT wrote the draft manuscript and MLX and JRY edited and revised the manuscript. All authors read and approved the final manuscript.
Yongfu Tao, Qingcai Liu contributed equally to this work.
Electronic supplementary material
12870_2013_1355_MOESM1_ESM.docx
Additional file 1: Evaluation of 50 HIFs in resistance to MRDD across different years and locations. R means resistant, IR means intermediate resistant, S means susceptible.(DOCX 18 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tao, Y., Liu, Q., Wang, H. et al. Identification and fine-mapping of a QTL, qMrdd1, that confers recessive resistance to maize rough dwarf disease. BMC Plant Biol 13, 145 (2013). https://doi.org/10.1186/1471-2229-13-145
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-13-145