
Abstract
Background
Tocopherols, which are vitamin E compounds, play an important role in maintaining human health. Compared with other staple foods, maize grains contain high level of tocopherols.
Results
Two F2 populations (K22/CI7 and K22/Dan340, referred to as POP-1 and POP-2, respectively), which share a common parent (K22), were developed and genotyped using a GoldenGate assay containing 1,536 single nucleotide polymorphism (SNP) markers. An integrated genetic linkage map was constructed using 619 SNP markers, spanning a total of 1649.03 cM of the maize genome with an average interval of 2.67 cM. Seventeen quantitative trait loci (QTLs) for all the traits were detected in the first map and 13 in the second. In these two maps, QTLs for different traits were localized to the same genomic regions and some were co-located with candidate genes in the tocopherol biosynthesis pathway. Single QTL was responsible for 3.03% to 52.75% of the phenotypic variation and the QTLs in sum explained23.4% to 66.52% of the total phenotypic variation. A major QTL (qc5-1/qd5-1) affecting α-tocopherol (αT) was identified on chromosome 5 between the PZA03161.1 and PZA02068.1 in the POP-2. The QTL region was narrowed down from 18.7 Mb to 5.4 Mb by estimating the recombination using high-density markers of the QTL region. This allowed the identification of the candidate gene VTE4 which encodes γ-tocopherol methyltransferase, an enzyme that transforms γ-tocopherol (γT)to αT.
Conclusions
These results demonstrate that a few QTLs with major effects and several QTLs with medium to minor effects might contribute to the natural variation of tocopherols in maize grain. The high-density markers will help to fine map and identify the QTLs with major effects even in the preliminary segregating populations. Furthermore, this study provides a simple guide line for the breeders to improve traits that minimize the risk of malnutrition, especially in developing countries.
Similar content being viewed by others

Background
Vitamin E is the common name that describes eight naturally occurring compounds having tocopherol activity [1]. The eight compounds are lipid-soluble antioxidants with two distinct groups, tocopherols and tocotrienols. The two groups differ in the saturation of the side chain and vary in the number and location of methyl groups [2], and are classified according to the location of the methyl group: α-tocopherol (αT), β-tocopherol (βT), δ-tocopherol (δT), γ-tocopherol (γT), α-tocotrienol, β-tocotrienol, δ-tocotrienol and γ-tocotrienol [3, 4]. Vitamin E plays an important role in plants development and can protect cell membranes from oxidation. Vitamin E can prevent oxidation of polyunsaturated fatty acid by absorbing the superfluous free radicals produced in the lipid peroxidation chain reaction [5, 6]. This serves to remove the free radical intermediates, thereby preventing continuity of the oxidation reaction. Vitamin E can prevent several diseases in humans and other animals, such as cardiovascular disease, Alzheimer's disease, neurological disorders, cancer, cataracts, inflammatory diseases and age-related macular degeneration [1, 7]. Food and nutrition guidelines recommend 15 mg/day of vitamin E for both adults and teenagers [8]. Individuals in developed nations can easily fulfill their daily requirement of vitamin E, but vitamin E deficiency (VED) in the developing countries is more common in premature infants and elderly people [9]. Furthermore, VED that is not immediately treated can lead to other serious diseases such as muscle weakness, ataxia, blindness, dementia, and eventually spinocerebellar degeneration [9–12].
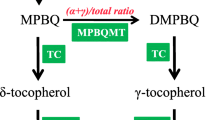
The tocopherol biosynthesis pathway has been well studied in the model plant – Arabidopsis (Figure 1) [4]. Several enzymes that participate in the biosynthetic pathway have been characterized and annotated in Arabidopsis and Synechocystis PCC6803 including 4-hydoxyphenyl-pyruvate dioxygenase (HPPD/PDS) [13, 14], tocopherol cyclase (VTE1/SXD1) [15, 16], homogentisic acid phytyltrasferase (VTE2/HPT)[17–20], 4-benzoquinol methyltransferase (VTE3)[21, 22], tocopherol methyltransferase (VTE4/γ-TMT) [23, 24], and phytol kinase (VTE5) [25]. In Arabidopsis, the first step in the biosynthetic pathway is to form the – homogentisic acid (HGA) and phytyldiphosphate (PDP). HGA can be synthesized from 4-hydroxyphenyl-pyruvate by HPPD/PDS and PDP can be translated from phytol by VTE5. Through the function of geranylgeranyl reductase (GGR), PDP can also be synthesized from geranylgeranyl diphosphate (GGPP), which is also a precursor for carotenoid and tocotrienol biosynthesis. The second step in the pathway involves forming the immediate precursor, 2-methyl-6-phytyl-1,4-benzoqiunol (MBPQ) from HGA and PDP through VTE2/HPT. Third, VTE3 translates MBPQ into 2, 3-dimethyl-5-phytyl-1,4-benzoqiunol (DMBPQ), and then converts to γT. The last step involves the formation of different tocopherols using VTE1 and VTE4/γ-TMT enzymes. Many of the genes involves in the tocopherol synthesis pathways are similar in different plants and have been cloned in several plants, such as tomato and rice [26–28].

Vitamin E synthesis pathway. The three tocopherol compounds measured in this study are in red. Key enzymes in the pathway are labeled in bold type and the blue bold type indicates enzymes used for co-localization analysis. HPPD: 4-hydoxyphenyl-pyruvate dioxygenase; DXS: 1-deoxy-D-xylulose-5-phophate synthase; DXR: 1-deoxy-D-xylulose-5-phosphate reductase; GGPS: geranylgeranyl diphosphate synthase; VTE1: tocopherol cyclase; VTE2: HPT, homogentisic acid phytyltransferase; VTE3: 2-methyl-6-phytyl-1, 4-benzoquinol methyltransferase; VTE4: tocopherol methyltransferase; VTE5: phytol kinase; HGGT: homogentisate geranylgeranyl transferase; PSY: phytoene synthase.
In addition to several other quality traits (such as oil and carotenoid), tocopherol content has been a focus of modern agriculture and several QTLs for tocopherol content has been mapped in different plant species [29–32]. Marwede et al. [29] used a double haploid (DH) population to locate several QTLs for γT, αT, TT and α/γ in winter oilseed rape. Elsa M and Vera-Ruiz et al. [30] performed fine mapping for the Tph1 gene, which can lead to a sharp reduction in βT in sunflower F2 and F3 populations. To date, two maize QTL mapping studies of tocopherols have been conducted. Wong et al. [31] mapped several QTLs for tocopherols using an F2:4 segregating population (W64a × A632) and one test-cross population with AE335 using 123 simple sequence repeat (SSR) markers, and identified QTLs for γT, αT, TT and α/γ on chromosomes 1 and 5. Chander et al. [32] identified 31 QTLs in 16 regions covering all chromosomes except chromosome 4 by using a recombinant inbred line (RIL) population with 208 SSR markers. In these studies, the confident interval of identified QTLs was >10 cM because of the limited number of markers used.
Recently, a new genotyping technique, using the third generation marker system with SNP markers has been developed [33, 34]. Compared with SSR markers, SNP markers are an ideal marker system because they are evenly distributed across the genome, are co-dominant, and accurate, and can be generated in a high-throughput and cost-effective manner. In this study, we used a maize GoldenGate assay containing 1536 SNPs to construct high-density linkage maps for two segregating populations with one common parent. The goals of this study were: (1) to identify QTLs affecting tocopherol content by analyzing two segregating populations with one common parent across the whole genome; (2) to fine map the major QTLs using the high-density markers; (3) and to explore the genetic architecture of tocopherol biosynthesis in different maize genetic backgrounds which could provide valuable information for further research.
Results
Phenotypic variation in tocopherols
Significant phenotypic variation was observed among the three parent lines in the traits of interest. For both γT and TT, the content was lowest in the common parent K22 (γT: 3.95 ± 0.15μg/g and TT: 9.18 ± 0.43μg/g), followed by CI7 (γT: 11.27 ± 0.68μg/g and TT: 18.09 ± 1.06μg/g) and was highest Dan340 (γT: 31.46 ± 0.42μg/g and TT: 34.51 ± 0.29μg/g). The αT content was highest in CI7 (6.76 ± 0.65μg/g), followed by the common parent K22 (5.13 ± 0.29μg/g), and was lowest in Dan340 (2.98 ± 0.21μg/g) which contains half the αT level of CI7. In contrast, α/γ ratio was highest in K22 (1.30 ± 0.03), followed by CI7 (0.60 ± 0.0) and was lowest in Dan340 (0.09 ± 0.01). The distribution of different tocopherols in the three parents also varied. In K22, the content of γT (3.95 ± 0.15μg/g)was just half as that of αT (5.13 ± 0.29μg/g) where as γT content in CI7 (11.27 ± 0.68μg/g) was nearly twice as that of αT (6.76 ± 0.65μg/g), even in Dan340 γT content (31.46 ± 0.42μg/g) was more than ten times as that of αT (2.98 ± 0.21μg/g) in Beijing location in 2009. And similar trends was also observed in Hainan location in 2009 (Table 1).
For different traits, the level of phenotypic variation varied by several folds (αT in POP-1) to nearly hundred folds (α/γ in POP-2) changes. The mean for γT in POP-1(11.02 ± 0.42μg/g) was less than that in POP-2 (22.46 ± 0.76μg/g) and the range was also larger in POP-2.the similar phenomenon was observed in TT. However the mean value for αT and α/γ in POP-1 was higher than that in POP-2 and was also consistent with the observed value for the respective parents of each line (Table 1).
The broad sense heritability (h2) was estimated from the F2:3 data and the two corresponding F2:4 populations. The heritability for each trait was high but varied for different traits. Heritability was highest (0.81) for γT in POP-2 and lowest (0.59) was divulged for αT in POP-2 (Table 1). Pearson correlation coefficients for each trait in every generation showed that γT was significantly positively correlated (r=0.82-0.96) with the secondary trait-TT but showed little or even no correlated with αT (r= (−0.07)-0.47) (Additional file 1: Table S1). The correlation coefficients between the F2:3 populations and two F2:4 populations ranged from 0.45 to 0.57 in POP-1 and from 0.26 to 0.77 in POP-2 (Additional file 1: Table S2).
Genetic linkage map
Among 1536 SNPs, 468 markers were polymorphic in POP-1 and 357 markers in POP-2. After deleting markers that were located in the same position or were unlinked, only 429 (POP-1) and 344 (POP-2) markers were used for linkage map construction (Table 2). Because of the common parent K22, the two maps had many common markers whereas, 619 unique markers were used to construct a consensus map following the method of Wu et al. (Additional file 1: Figure S1) [35]. The POP-1 map covered a 1389.3-cM region, smaller than that of POP-2 1567.5-cM region. The average interval distance between two markers was shorter in POP-1 (3.25 cM) than the POP-2 (4.57 cM). The integrated map covered a much larger region (1649.03 cM) than the two separate maps, with 619 SNP markers and a narrow interval distance of 2.67 cM (Table 2).
QTL mapping
After 1,000 permutation tests, the threshold logarithm of odds (LOD) scores were defined as 3.7 whereas 17 QTLs were detected in the POP-1 F2:3 and 13 QTLs in POP-2 F2:3 (Tables 3 and 4). In the Hainan environment, only seven QTLs were detected in POP-1 F2:4 and five in POP-2 F2:4. In Hubei, ten putative QTLs were observed for POP-1 and 12 for POP-2 (Tables 3 and 4). Seven QTLs for each of the POP-1 and POP-2 populations were detected in at least two environments. The major QTLs were confirmed in the three environments for the respective maps. In both maps the total QTLs detected were 30 and including 17 in POP-1 and 13 in POP-2. Most of the detected QTLs showed additive effects rather than dominate effects.
Among the 17 QTLs in the POP-1 F2:3, γT and αT had four each, whereas TT had six and α/γ had three (Table 3). These QTLs were mainly located in 11 regions on different chromosomes, with one each on chromosomes 1, 6, 7 and 8, two on chromosome 2 and four on chromosome 5. No QTLs were detected on chromosomes 3, 4, 9 and 10. Each QTL explained from 3.88% (αT on chromosome 1) to 29.63% (αT on chromosome 5) of the phenotypic variation. All the QTLs explained 37.59% (γT), 44.36% (αT), 51.73% (TT) and 23.40% (α/γ) of the total phenotypic variation for each trait. Two QTLs with a > 7 LOD were detected in the POP-1 map. The qc5-1 was between the markers PZA03161.1 and PZA02068.1 (134.1-159.7 cM in the POP-1 map) affecting the content of γT, αT, TT and α/γ it explained 5.37% to 29.63% of the total phenotypic variation for each trait. The other QTL, defined as qc5-2 were located between the markers PZA01327.1 and PHM1870.20 (55.1-76.7 cM in the POP-1 map), and affected the content of γT, αT, TT and α/γ; it explained 6.11% to 26.51% of the phenotypic variation for each trait (Figure 2A). The K22 allele of qc5-1 on the long arm of chromosome 5 was responsible for decreasing the content of αT, TT and α/γ and for increasing γT. While to qc5-2 at the short arm of chromosome 5, the favorable allele was from CI7 for γT, αT, TT, and K22 for α/γ (Table 3).

Distribution of major QTLs in the two maps (A), The two major QTLs on chromosome 5 in the POP-1 map (40–190 cM genetic distance); (B) The major QTL on chromosome 1 in the POP-2 map (130–200 cM genetic distance); (C) The major QTL on chromosome 5 in the POP-2 map (70 to 140 cM genetic distance). The symbol color of relates to the corresponding trait in each location beside the figure. Gray solid lines indicated the permutated threshold.
The 13 putative QTLs in POP-2 F2:3 were distributed as follows: four (γT), two (αT), four (TT), three (α/γ) (Table 4). These QTLs were located in five regions on different chromosomes; five QTLs each on chromosome 1 and 5, and three QTLs on chromosome 8. Three regions were on chromosome 1, and one each on chromosome 5 and 8. No QTL was observed on chromosomes 2, 3, 4, 6, 7, 9 and 10. Each QTL could explain the phenotypic variation from 3.03% (γT on chromosome 1; qd1-2) to 52.75% for αT on chromosome 5 (qd5-1, between PZA03161.1 and PZA02068.1, 144.60 – 169.78 cM; Table 4). All detected QTLs could explain 50.34% (γT), 57.76% (αT), 66.52% (TT) and 59.17% (α/γ) of the total variation for each trait. In POP-2 the qd1-1 on chromosome 1 between PHM3690.23 and PHM4926.16 (158.5-179.76 cM in the POP-2 map) could explain 50.67% variation for TT, –whereas qd5-1 could explain 52.75% phenotypic variation for αT (Table 4, Figure 2B and C). The Dan340 alleles at qd1-1 were associated with increasing γT, αT and TT contents but did not affect α/γ, whereas the qd5-1 alleles from K22 on the short arm of chromosome 5, had increasing (αT and α/γ) or decreasing (γT and TT) effects.
Fine mapping of qd5-1in the POP-2 map
To dissect the large qc5-1/qd5-1 QTL detected in both maps, the polymorphic markers at the threshold LOD near the peak were selected. The QTL contributed 29.63% (09BJF2:3), 20.06% (09HNF2:4), and 9.84% (10HBF2:4) of the phenotypic variation for αT in the POP-1, respectively. In POP-2 phenotypic variation of 52.75% (09BJF2:3), 12.44% (09HNF2:4) and 39.09% (10HBF2:4) for αT were observed. The significant effects indicated that the qd5-1 QTL could be a qualitative gene and can be fine mapped by analyzing the recombinants among the segregating populations.
The POP-2 was first selected to analyze recombination within the qd5-1 region because of its wide phenotypic variation for αT. From the initial mapping, the PZA03161.1 and PZA02058.1 markers were located at either end of the region separately by 25.2cM genetic distance and 18.7 Mb (186.4-205.1 Mb) physical distances. There were four additional markers within this region (Figure 3A). In total, five recombinant combinations were identified, dividing the 180 RILs (after deleting some ambiguous RILs) into 17 haplotypes (Figure 3C and D). The 17 haplotypes could be divided into 3 obvious groups based on 09BJF2:3 data: group 1 for haplotypes IV, V, VI, XIII, and XIV (Figure 3C; 4.8 - 7.6 ug/g, αT), group 2 for haplotypes I, II, III, VII, VIII and IV (8.9 - 11.8 ug/g, αT) and group 3 for the rest haplotypes (− 1.4 - 2.9 ug/g, αT). Similar trends were also observed in the F2:4 populations from the other two locations (Figure 3C and D). Comparing the phenotypes of type 1 (haplotype I from the parent K22 I in Figure 3C), type 3 (haplotype V from the K22/Dan340 F1 in Figure 3C) and type 2 (haplotype X from the parent Dan340; Figure 3C) indicated that the parent K22 had the favorable allele for αT in the QTL region (Figure 3B). The phenotype of group 1 was similar to type 3, the group 2 was similar with type 1 and group 3 phenotype was similar with type 2. Based on this information, the QTL region was narrowed down between markers PZA02751.1 and PZA01265.1 to around 10.8 Mb region. There are 39 bacterial artificial chromosomes (BACs) identified within this target region and included an annotation for the VTE4 tocopherol biosynthesis gene.

Haplotype analyses and fine-mapping of qd5-1 in the early generation in POP-2 (A) Map position of qd5-1 in three environments. (B) Summary of genotype and phenotypic effects in the genomic regions that contains qd5-1. (C, D) The detailed haplotype analyses between types 1 and 3 (C) or types 2 and 3 (D) as in Figure 3B. The bars indicate the missing data. (E) The distribution of BACs (black lines) and candidate genes (red line indicates VTE4) in the genomic regions after fine mapping. The black lines mean each BAC and the red line represent the BAC that the candidate gene VTE4 located, all the BACs information is from web site (http://www.maizesequence.org/%20B73_version%205a.60),.
Using the same approach, another QTL-qd1-1 in POP-2, which explained 30.81% of γT content, was analyzed. All the families were divided into 19 haplotypes and the haplotypes of I, II, III. IX, X, XI, and XII had the lowest γT content. Consistent with our expectation, the interval could narrowed from 21.2 cM (23.1 Mb, 218.1-241.2Mb) to ~6.5 cM (~5.2 Mb) in POP-2 map (Additional file 1: Figure S2).
Discussion
The genetic basis for tocopherol biosynthesis in maize grain
In the present study, 30 QTLs were detected in two F2:3 populations, with one to six QTLs for each trait. These QTLs can explain the phenotypic variation of each trait from 23.4% for α/γ in POP-1 to 66.5% for TT in POP-2. Two major QTLs (qc5-1 and qc5-2) that affect all four target traits were identified in POP-1. Both qc5-1 and qc5-2 were located on chromosome 5 and explained a maximum phenotypic variation of 30% and 25% for αT, respectively (Table 3). In POP2, the major QTLs one (qd1-1) on chromosome 1 and the qd5-1 QTL on chromosome 5 explained 51% (TT) and53% (αT) phenotypic variation respectively (Table 4). More than 80% of the QTLs detected in this study contributed in an additive manner. Hence, vitamin E biosynthesis in maize grain may be controlled by several major and a number of minor QTLs. This phenomenon is different with other agronomic traits, such as flowering time [36] and leaf architecture [37]. These traits are controlled by many minor QTLs and to data no major QTLs have been reported. Vitamin E is beneficial for human and animal health, but the absence of a selection index makes it difficult to breed for this trait, which is not as important compared as other economic traits such as yield. This type of trait may not be under strong selection pressure in the breeding and farming community, which is why major QTLs were easily identified. With the advent of such major additive QTLs the total tocopherols content of maize grains can be easily increased using marker assistant selection (MAS), which can be a simple guide for breeders to improve such traits. Furthermore, fine mapping of these major QTLs will explore the phenomenon in a comprehensive way because only a few candidate genes were detected in each QTL region, which distinguishes this pathway in maize from the extensively studied corresponding one in Arabidopsis. Hence, the study of tocopherol compounds in maize grains will help to increase the economic and nutritional value of maize crops which could account for more than half of the increased worldwide consumer demand for cereals [38].
When a QTL with a large effect is identified, determining the causal gene is a tedious and time-consuming task [39], although the involvement of a few major genes, facilitates the identification and fine mapping of the candidate gene. In addition, a single large-effect QTL often has multiple, closely linked QTLs with smaller, and sometimes opposite, effects on the phenotype [40, 41]. The qc5-1/qd5-1 QTL has a large effect on the overall phenotype [31, 32]. Table 5 shows some QTLs that were identified in previous study [32]. A comparison of the results shows that qd1-1 was only in POP-2, which is similar to the previous studies of chromosome 8 [32]. The populations used in this study are of different genetic background as compared with those in the previous studies, so the source may be different across the different genetic material. However, our analysis has resulted in better coverage of the maize genome by using a large number of markers to determine more possible QTLs playing any kind of major or minor role in this phenomenon.
It is interesting to note that the two candidate genes VTE4 and HPPD-5 from the tocopherol biosynthesis pathway were both located within one corresponding QTL region in this study, it suggested that the genetic system controlling the biosynthesis of tocopherol may be more complex in maize as compared with that in Arabidopsis. Hence, the tocopherol biosynthesis pathway should be explored in more maize populations with different genetic backgrounds. Further studies will also help to identify the exact number of QTLs with minor or major roles, as a large population size and high number of markers are required to provide a solid basis for further improvement. Finally, positional cloning of major QTLs is extremely important to validate the results described herein and to improve the overall performance of maize.
Regulation of tocopherol synthesis
Many biosynthetic genes control the rate of synthesis of tocopherol. Nine of the downstream genes have been thoroughly studied in Arabidopsis. VTE1, VTE2, VTE3, VTE4, VTE5 and HPPD are involved in the formation of the end-product, whereas GGPS, DXS, DXR and VTE5 are involved in synthesis of the tocopherol precursor (Figure 1). Maize homologous genes of the first six genes were identified through bioinformatics, with some genes having more than one copy in maize. The physical positions and abbreviated names of the enzyme and chromosome location of these candidate genes are given in B73 (Additional file 1: Table S3, http://www.maizesequence.org/,%20B73_version%205a.60).
Several QTLs co-localized with corresponding candidate genes, and just one QTL and candidate gene was found in more than one location (Tables 3 and 4). On chromosome 5, VTE4 were co-located in both maps, whereas HPPD-5 was found in only one population. The large QTL on chromosome 1 in POP-2 had no candidate gene, suggesting that the presence of additional unknown genes controlling tocopherol content have not been identified by comparative genomics.
How to mine genes with large QTLs in the future
The rapid development of the high-throughput SNP genotyping technique enables the easy construction of simple high-density linkage maps. In the present study, the linkage map constructed by SNP markers was 1389.3 cM in POP-1 and 1567.5 cM in POP-2 with an average interval distance of 3.25 and 4.57 cM, respectively (average interval distance was 2.67 cM in the consensus map). Previously, linkage maps were constructed by SSR markers with an interval distance of 10–30 cM in maize and varying distance for different organism [42]. High-density maps more precisely localize major QTLs to smaller region.
Fine-mapping requires the construction of advanced backcrossing populations and high-density markers to narrow down the QTL region to one gene or even a single SNP [43]. Fine mapping and functional validation are usually more costly, laborious and time consuming. Recently, the rapid development of association mapping has enabled the identification of a single gene within a year of collecting phenotypic and genotypic data. It provides a new tool for analyzing quantitative traits. Li et al. [44] combined traditional fine mapping and association mapping to identify the functional gene fatb and validated the functional sequence variation using in vivo gene expression profiling and in vitro complementation studies. They developed markers based on the predicted gene sequences and increased marker density to narrow down the region of interest, and also performed an association analysis with these markers in 74 lines. The combined linkage and association mapping is therefore a beneficial tool for identifying novel genes for different qualitative and quantitative traits [44].
We performed fine mapping for the large qd5-1 and qd1-1 by haplotype analysis in an early generation and obtained good results without constructing a large backcross population. This method allowed the large QTL qd5-1 region to be narrowed from 18.7 Mb to 5.4 Mb, assuming that the recombination occurred in the middle of the markers. There were 39 BACs in this region, with VTE4 – GRMZM2G035213 found in BAC209363 using bioinformatic analysis (Figure 3E, http://www.maizesequence.org/,%20B73_version%205a.60). Recently, VTE4 was identified as the underlying gene of this QTL using genome-wide and candidate gene association analyses [45]. Two functional polymorphisms (InDel7 and InDel118) were significantly associated with αT. InDel7 segregates in the parents of the two populations. InDel118 segregates in K22 and Dan340 populations. Hence, CI7 has the best haplotype (7/118), DAN340 has the worst haplotype (0/0), and the haplotype of K22 (7/0) is intermediate, which is also consistent with the phenotype of the three parents and QTL effects in the two populations. The cloned gene VTE4 provides an excellent sample of the high-density markers for QTL fine mapping and cloning.
The size of the qd1-1, QTL diminished from 23.1 Mb to 2.6 Mb, and there were 22 BACs in the 2.6 Mb region without any known candidate genes (Additional file 1: Figure S2E). Further research is needed to validate the functional site of these QTLs. There might be more genes controlling the tocopherol degradation pathway than the genes known to be involved in the synthesis pathway. Hence, much more in-depth work is needed for dissecting the metabolic pathway of tocopherols in maize grain including the synthesis and degradation pathways.
Conclusion
This study identified different major QTLs in different populations compared with previous studies [32]. Thoroughly understanding the genetic architecture of tocopherol biosynthetic and degradation pathway is required to construct more populations with different genetic backgrounds. According to the recent methodology of combining linkage and association mapping [44], dissecting the tocopherol pathway can be performed in a short period of time with maximum validations, thus providing the scientific community with a base for MAS. MAS may be a useful and cost-effective tool for improving the nutritional value of the world’s leading cereal. In the preceding decades many major QTLs for different traits were applied in breeding programs by developing some functional markers [46, 47]. Similarly, the three major QTLs in this study (qd1-1, qc5-1/qd5-1 and qc5-2) can provide a guide for the development of molecular markers for breeding program or further detailed and deep research.
Methods
Genetic materials
An elite Chinese inbred line K22 was chosen to cross with two other elite lines, CI7 and Dan340, which have significantly different tocopherol contents [48]. Four hundred kernels of each F2 population were planted to develop the F2:3 population by self-pollinating at the Changping experiment field of China Agricultural University in Beijing (spring, 2009). Thirteen individuals were grown in a 3-meters row with 0.5-meter spacing within the row. Ears were harvested after 40–45 days of pollination, and 237 F2:3 families of POP-1 and 218 F2:3 families of POP-2 were obtained and phenotyped, diseased and contaminated ears were excluded from analysis. These F2:3 families were used for phenotyping and validation in offspring. Trials were conducted at two locations with two replications per location. At one location, the F2:3 families were planted in the Nanbin farm in Yacheng of Hainan province with 11 plants in each 3-meters row (winter, 2009). Another trial was carried out in Hubei Academy of Agricultural Sciences with the same field design as that in Beijing in 2009 (spring, 2010). Pooled pollen from the line was used to pollinate at least five plants so as to harvest good ears for phenotyping. Finally, 189 (POP-1) and 198 (POP-2)F2:4 families were measured in Hainan, and 213 (POP-1) and 177 (POP-2) F2:4 families were measured in Hubei because of asynchronized flowering and additional developmental problems.
Reagents
The standards for γT, αT, δT and other chromatography-grade chemicals were purchased from Sigma (St Louis, MO, USA) and all other chemicals from Beijing Chemical Reagent Factory (Sinopharm Group Chemical Reagent Co., Ltd, Beijing, China).
Measurement of tocopherols
At physiological maturity the ears were harvested and shelled manually, and a sample of 50 well performed was taken for phenotyping. These kernels were selected from the middle of each F2:3 or F2:4 ears, by bulk-pollinated in each family, respectively. All the kernels used for phenotypic analysis were dried for 60 hours at 45°C, kept in the dark at 4°C and ground into powder for tocopherol extraction and measurement. Tocopherols were extracted with the modified method as described in previous studies [32, 48, 49]. Three metabolites γT, αT and δT were measured separately. In addition to αT and γT, two derived traits, TT (the sum of γT, αT and δT) and α/γ (the ratio of α-/γ-tocopherol) were also calculated.
The tocopherol content was determined by high performance lipid chromatography (HPLC) as described [32, 48, 49]. External standard curves were constructed with eight serial dilutions and with repeats for each dilute (R2 ≥ 0.99). The three tocopherols (γT, αT and δT) were separated on a reverse-phase C30 column (YMC CT99S05-2546WT C30, 4.6nm × 25cm, 5μm; Waters) at 30°C at 1.8 ml/min for the mobile phase (v/v/v, 75:20:5; acetonitrile/methanol /dichloromethane) by scanning at 295 nm without a reference wave and were identified by the retention time of the standards. The peak times for δT, γT and αT were 4.56 min, 5.15 min and 6.07 min, respectively. All phenotypic data were generated on ChemStation software (Agilent Technologies).
Phenotypic data analysis
The variance of traits and the correlation coefficients between traits were analyzed using the “PROC GLM” procedure of SAS 8.02 (SAS Institute 1999). The broad sense heritability was estimated as h2 =σg2 / (σg2 +σgy2+σe2/y) with SAS 8.02. Here, σg2 is the genetic variance, σgy2 is the interaction of genotype with year, σe2 is the residual error, and y is the number of years [50]. All the traits were analyzed with the same method.
Genotyping and linkage map construction
Genomic DNA was extracted from all 455 F2 single plants (237 lines of POP-1 and 218 lines of POP-2) and from their parental genotypes using the modified procedure of Murry and Thompson [51]. All the families and parents were genotyped using the GoldenGate assays (Illumina, San Diego, CA, USA) containing 1,536 SNPs [34]. The SNP genotyping was performed on an Illumina BeadStation 500G at Cornell University Life Sciences Core Laboratories Center using the protocol supported by Illumina Company [52]. The details of the SNP genotyping procedure and allele scoring have been described [34]. The data from polymorphic SNPs were used to construct a genetic linkage map using Mapmaker 3.0 for each population [53]. The threshold LOD score for the test of independence of marker pairs was set at 3.0, and the marker order with the highest LOD score was then selected. The Kosambi mapping function was used for calculating map distances. The two individual maps were then combined to form a consensus map using merge map [35].
QTL analysis
For QTL detection the whole genome was scanned using composite interval mapping (CIM) with 2 cM scanning intervals between markers and a windows size of 10 cM. We used Model 6 in the Zmapqtl module of Wincartographer 2.5 [54]. The threshold LOD values for putative QTLs for tocopherol content and composition were estimated after 1,000 permutations at a significant level of p < 0.05 [55]. The number of marker cofactors for the background control was set by forward–backward stepwise regression with five controlling markers.
Abbreviations
- γT:
-
γ-tocopherol
- αT:
-
α-tocopherol
- α/γ:
-
α-tocopherol/γ-tocopherol
- TT:
-
Total tocopherol
- SSR:
-
Simple sequence repeat
- SNP:
-
Single nucleotide polymorphism
- QTL:
-
Quantitative trait loci
- POP-2:
-
K22/Dan340 population
- POP-1:
-
K22/CI7 population
- MAS:
-
Marker assistant selection
- LOD:
-
Logarithm of odds
- HPLC:
-
High performance lipid chromatography
- h 2 :
-
The broad sense heritability
- chr:
-
Chromosome
- 10HBF2:
-
4: F2:4 populations in Hubei 2010
- 09HNF2:
-
4: F2:4 populations in Hainan 2009
- 09BJF2:
-
3: F2:3 populations in Beijing 2009.
References
Bramley PM, Elmadfa I, Kafatos A, Kelly FJ, Manios Y, Roxborough HE, Schuch W, Sheehy PJA, Wagner K-H: Critical reviews produced within the EU Concerted Action 'Nutritional enhancement of plant-based food in European trade' (Neodiet) – Vitamin E. Sci Food Agri. 2000, 80: 913-938.
Rocheford TR, Wong JC, Egesel CO, Lambert RJ: Enhancement of Vitamin E Levels in Corn. J Amer Coll Nutr. 2002, 21: 191-198.
Moss GP: Nomenclature of tocopherols and related compounds. Recommendations 1981. Eur J Biochem. 1981, 123: 473-475.
DellaPenna D: A decade of progress in understanding vitamin E synthesis in plants. J Plant Physiol. 2005, 162: 729-737.
Traber MG, Atkinson J: Vitamin E, antioxidant and nothing more. Free Rad Biol & Med. 2007, 43 (1): 4-15.
Herrera E, Barbas C: Vitamin E: action, metabolism and perspectives. J Physio and Biochem. 2001, 57 (2): 43-56.
Traber MG, Sies H: Vitamin in humans: demand and delivery. Annual Review Nutr. 1996, 16: 321-347.
Institute of Medicine: Food and Nutrition Board: Dietary reference intakes: Applications in dietary assessment. 2000, National Academy Press, Washington, DC, 289.
Sokol RJ: Vitamin E deficiency and neurologic disease. Annual Review Nutr. 1988, 8: 351-373.
Aicardi J: Diseases of the nervous system in childhood. Mac Keith Press. 1992, 108-202.
Eggermont E: Recent advances in vitamin E metabolism and deficiency. Eur J Pediatr. 2006, 165: 429-434.
Muller DPR: Vitamin E and neurological function. Mol Nutr Food Res. 2010, 54: 710-718.
Norris SR, Barrette TR, DellaPenna D: Genetic dissection of carotenoid synthesis in Arabidopsis defines plastoquinone as an essential component of phytoene desaturation. Plant Cell. 1995, 7: 2139-2149.
Norris SR, Shen XH, DellaPenna D: Complementation of the Arabidopsis pds1 mutation with the gene encoding phydroxyphenylpyruvate dioxygenase. Plant Physiol. 1998, 117: 1317-1323.
Porfirova S, Bergmüller E, Tropf S, Lemke R, Dörmann P: Isolation of an Arabidopsis mutant lacking vitamin E and identification of a cyclase essential for all tocopherol biosynthesis. Proc Natl Acad Sci. USA. 2002, 99: 12495-12500.
Sattler SE, Cahoon EB, Coughlan SJ, DellaPenna D: Characterization of tocopherol cyclases from higher plants and cyanobacteria. Evolutionary implications for tocopherol synthesis and function. Plant Physiol. 2003, 132: 2184-2195.
Collakova E, DellaPenna D: Isolation and functional analysis of homogentisate phytyltransferase from Synechocystis sp. PCC 6803 and Arabidopsis. Plant Physiol. 2001, 127: 1113-1124.
Schledz M, Seidler A, Beyer P, Neuhaus G: A novel phytyltransferase from Synechocystis sp. PCC 6803 involved in tocopherol biosynthesis. FEBS Lett. 2001, 499: 15-20.
Savidge B, Weiss JD, Wong YHH, Lassner MW, Mitsky TA, Shewmaker CK, Post-Beittenmiller D, Valentin HE: Isolation and characterization of homogentisate phytyltransferase genes from Synechocystis sp. PCC 6803 and Arabidopsis. Plant Physiol. 2002, 129: 321-332.
Sattler SE, Gilliland LU, Magallanes-Lundback M, Pollard M, DellaPenna D: Vitamin E is essential for seed longevity and for preventing lipid peroxidation during germination. Plant Cell. 2004, 16: 1419-1432.
Cheng Z, Sattler S, Maeda H, Sakuragi Y, Bryant DA, DellaPenna D: Highly divergent methyltransferases catalyze a conserved reaction in tocopherol and plastoquinone synthesis in cyanobacteria and photosynthetic eukaryotes. Plant Cell. 2003, 15: 2343-2356.
Motohashi R, Ito T, Kobayashi M, Taji T, Nagata N, Asami T, Yoshida S, Yamaguchi-Shinozaki K, Shinozaki K: Functional analysis of the 37 kDa inner envelope membrane polypeptide in chloroplast biogenesis using a Ds-tagged Arabidopsis pale-green mutant. Plant J. 2003, 34: 719-731.
Shintani D, DellaPenna D: Elevating the vitamin E content of plants through metabolic engineering. Science. 1998, 282: 2098-2100.
Ischebeck T, Zbierzak AM, Kanwischer M, Dörmann P: A salvage pathway for phytol metabolism in Arabidopsis. J Biol Chem. 2006, 281: 2470-2477.
Valentin HE, Lincoln K, Moshiri F, Jensen PK, Qi Q, Venkatesh TV, Karunanandaa B, Baszis R, Norris SR, Savidge B, Gruys KJ, Last RL: The Arabidopsis vitamin E pathway gene5-1 Mutant Reveals a Critical Role for Phytol Kinase in Seed Tocopherol Biosynthesis. Plant Cell. 2006, 18: 212-224.
Zou LP: Rice γ-tocopherol methyltransferase gene cloning and analysis of full-length cDNA. Hubei Agri Sci. 2008, 47 (11): 1221-1224.
Zou LP: Cloning and sequence analysis γ-tocopherol methyltransferase (γ-TMT) gene in tomato. J Anhui Agri Sci. 2008, 36 (2): 437-439.
Shintani DK, Cheng ZG, DellaPenna D: The role of 2-methyl-6-phytylbenzoquinone methyltransferase determining tocopherol composition in Synechocystis sp. PCC6803. FEBS Lett. 2002, 511: 1-5.
Marwede V, Gui MK, Becker HC, Ecke W: Mapping of QTL controlling tocopherol content in winter oilseed rape. Plant Breeding. 2005, 124: 20-26.
Vera-Ruiz EM, Velasco L, Leon AJ, Fernández-Martínez JM, Pérez-Vich B: Genetic mapping of the Tph1 gene controlling beta-tocopherol accumulation in sunflower seeds. Molecular Breeding. 2006, 17: 291-296.
Wong JC, Lambert RJ, Tadmor Y, Rocheford TR: QTL associated with accumulation of tocopherols in maize. Crop Sci. 2003, 3: 2257-2266.
Chander S, Guo YQ, Yang XH, Yan JB, Zhang YR, Song TM, Li JS: Genetic dissection of tocopherol content and composition in maize grain using quantitative trait loci analysis and the candidate gene approach. Mol Breeding. 2008, 22: 353-365.
Yan JB, Shah T, Warburton ML, Buckler ES, McMullen MD, Crouch JH: Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS One. 2009, 4 (12): e8451.
Yan JB, Yang XH, Shah T, Sa’nchez-Villeda H, Li JS, Warburton ML, Zhou Y, Crouch JH, Xu YB: High-throughput SNP genotyping with the GoldenGate assay in maize. Mol Breeding. 2010, 25: 441-451.
Wu YH, Bhat PR, Close TJ, Lonardi S: On the Accurate Construction of Consensus Genetic Maps CSB. 2008, Stanford, CA, http://alumni.cs.ucr.edu/~yonghui/mgmap.html, - Computational Systems Bioinformatics Conference.
Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint-Garcia S, Garcia A, Glaubitz JC, Goodman MM, Harjes C, Guill K, Kroon DE, Larsson S, Lepak NK, Li HH, Mitchell SE, Pressoir G, Peiffer JA, Rosas MO, Rocheford TR, Romay MC, Romero S, Salvo S, Villeda HS, Silva HS, Sun Q, Tian F, Upadyayula N, Ware D, Yates H, Yu JM, Zhang ZW, Kresovich S, Michael D, McMullen MD: The genetic architecture of maize flowering time. Science. 2009, 325 (5941): 714-718.
Tian F, Bradbury PJ, Brown PJ, Hung H, Sun Q, Flint-Garcia S, Rocheford TR, McMullen MD, Holland JB, Buckler ES: Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat Genet. 2011, 43: 159-162.
Yan JB, Warburton ML, Crouch J: Association mapping for enhancing maize genetic improvement. Crop Sci. 2011, 51: 433-449.
Ingvarsson PK, Street NR: Association genetics of complex traits in plant. New Phytol. 2011, 189: 909-922.
Doerge RW: Mapping and analysis of quantitative trait loci in experimental populations. Nat Rev Genets. 2002, 3: 43-52.
Mackay TFC, Stone EA, Ayroles JF: The genetics of quantitative traits: challenges and prospects. Nat Rev Genet. 2009, 10: 565-577.
Salvi S, Tuberosa R: To clone or not to clone plant QTLs, present and future challenges. Trends Plant Sci. 2005, 10: 297-304.
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Sherry R, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES: Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci. 2001, 98: 11479-11484.
Li L, Li L, Li Q, Yang XH, Zheng DB, Warburton ML, Chai YC, Zhang P, Guo YQ, Yan JB, Li JS: An 11-bp Insertion in Zea mays fatb reduces the palmitic acid content of fatty acids in maize grain. PLoS One. 2011, 6 (9): e24699.
Li Q, Yang XH, Xu ST, Cai Y, Zhang DL, Han YJ, Li L, Zhang ZX, Gao SB, Li JS, Yan JB: Genome-wide association studies identified three independent polymorphisms associated with α-tocopherol content in maize kernels. PLoS One. 2012, 7 (5): e36807.
Cao SL: Development of application with marker assisted-selection in maize breeding. Crops. 2008, 6: 107-109.
Yang H, Yang JP, Rong TZ, Wang FG, Tan J, Qiu ZG: Developing the maize lines based on selections of phi116 and umc1044 markers which are resistant to sheath blight. Mol Plant Breeding. 2007, 5 (3): 347-352.
Zhou Y, Fu ZY, Li Q, Xu ST, Chander S, Yang XH, Li JS, Yan JB: Comparative analysis of carotenoid and tocopherol compositions in high-oil and normal maize (Zea mays L.) inbreds. Acta Agronomica Sinica. 2009, 35: 2073-2084.
Kurilich AC, Juvik JA: Quantification of carotenoid and tocopherol antioxidants in Zea mays. J Agric Food Chem. 1999, 47: 1948-1955.
Knapp SJ, Stroup WW, Ross WM: Exact confidence intervals for heritability on a progeny mean basis. Crop Science. 1985, 25: 192-194.
Murry MG, Thompson WF: Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8: 4321-4325.
Fan JB, Gunderson KL, Bibikova M, Yeakley JM, Chen J, Wickham Garcia E, Lebruska LL, Laurent M, Shen R, Barker D: Illumina universal bead arrays. Methods Enzymol. 2006, 410: 57-73.
Lincoln SE, Daly MJ, Lander ES: Construction genetic maps with MAPMAKER/EXP 3.0. Whitehead Institute Technical Report, White-head Institute, Cambridge, Massachusetts. 1993, 3
Zeng ZB: Precision mapping of quantitative trait loci. Genetics. 1994, 136: 1457-1468.
Churchill GA, Doerge RW: Empirical threshold values for quantitative trait mapping. Genetics. 1994, 138: 963-971.
Acknowledgements
We would like to thank Abhishek Rathore for help with the construction of the linkage maps. We would also like to thank Santosh Deshpande and Weiwei Wen for their valuable comments in improving this manuscript. This research was supported by the National Hi-Tech Research and Development Program of China (2012AA10A307) and the National Natural Science Foundation of China (31222041).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no completing financial interests.
Authors’ contributions
XST and ZDL prepared the materials; XST, ZDL LQ and CY performed the analyses of the genotype and phenotype; LZG and WWD participated to determine the phenotype; ST constructed the linkage maps; YJB, LJS and YXH designed the experiments and analyzed the chip raw data; XST, FA and YJB wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12870_2012_1134_MOESM1_ESM.rtf
Additional file 1: Table S1. Correlation coefficients of trait pairs for tocopherol related traits in two segregating populations in three environments. Table S2. Correlation coefficients of each trait among three locations. Table S3. List of candidate genes related to tocopherols in maize grains compared with that in Arabidopsis. Figure S1. The entire linkage map of chromosome 1-chromosome 10. Figure S2. Haplotype analyses and fine mapping of qd1-1 in the early generation of K22/Dan340 segregation population A: Map position of qd1-1 in three environments, 2009 Beijing, 2009 Hainan and 2010 Hubei. B: Summary of genotype and phenotype in the genomic regions harboring qd1-1. C: The detailed haplotype analyses between type 1 and type 3 as Additional file 1: Figure S2B. The bar means the missing data. D: The detailed haplotype analyses between type 2 and type 3 as Additional file 1: Figure S2B. The bar means the missing data. The bar means the missing data. E: The distribution of BACs and candidate genes in the genomic regions after fine mapping. The black lines mean each BAC. All the BACs information is from web site (http://www.maizesequence.org/i%20B73RefGen_v2). (RTF 15 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Shutu, X., Dalong, Z., Ye, C. et al. Dissecting tocopherols content in maize (Zea mays L.), using two segregating populations and high-density single nucleotide polymorphism markers. BMC Plant Biol 12, 201 (2012). https://doi.org/10.1186/1471-2229-12-201
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2229-12-201