
Abstract
Background
Flower colour variation is one of the most crucial selection criteria in the breeding of a flowering pot plant, as is also the case for azalea (Rhododendron simsii hybrids). Flavonoid biosynthesis was studied intensively in several species. In azalea, flower colour can be described by means of a 3-gene model. However, this model does not clarify pink-coloration. The last decade gene expression studies have been implemented widely for studying flower colour. However, the methods used were often only semi-quantitative or quantification was not done according to the MIQE-guidelines. We aimed to develop an accurate protocol for RT-qPCR and to validate the protocol to study flower colour in an azalea mapping population.
Results
An accurate RT-qPCR protocol had to be established. RNA quality was evaluated in a combined approach by means of different techniques e.g. SPUD-assay and Experion-analysis. We demonstrated the importance of testing noRT-samples for all genes under study to detect contaminating DNA. In spite of the limited sequence information available, we prepared a set of 11 reference genes which was validated in flower petals; a combination of three reference genes was most optimal. Finally we also used plasmids for the construction of standard curves. This allowed us to calculate gene-specific PCR efficiencies for every gene to assure an accurate quantification. The validity of the protocol was demonstrated by means of the study of six genes of the flavonoid biosynthesis pathway. No correlations were found between flower colour and the individual expression profiles. However, the combination of early pathway genes (CHS, F3H, F3'H and FLS) is clearly related to co-pigmentation with flavonols. The late pathway genes DFR and ANS are to a minor extent involved in differentiating between coloured and white flowers. Concerning pink coloration, we could demonstrate that the lower intensity in this type of flowers is correlated to the expression of F3'H.
Conclusions
Currently in plant research, validated and qualitative RT-qPCR protocols are still rare. The protocol in this study can be implemented on all plant species to assure accurate quantification of gene expression. We have been able to correlate flower colour to the combined regulation of structural genes, both in the early and late branch of the pathway. This allowed us to differentiate between flower colours in a broader genetic background as was done so far in flower colour studies. These data will now be used for eQTL mapping to comprehend even more the regulation of this pathway.
Similar content being viewed by others


Background
As for all flowering plants, flower characteristics and especially flower colour are among the most important features for pot azalea (Rhododendron simsii hybrids) breeding. Flavonoids account for this pigmentation in azalea [1, 2]. The flavonoid biosynthesis pathway is one of the best studied biochemical pathways in plants, especially in petunia and snapdragon [3–7]. Flavonoids are synthesized by a branched pathway that yields both coloured pigments (anthocyanins) and colourless co-pigments (flavonols). In De Cooman et al. [8], it was observed that the azalea co-pigment formation follows a slightly aberrant pathway compared to anthocyanin production (Figure 1). Anthocyanins tend to occur mainly as cyanidins, azaleatin is the most common flavonol in azalea [2]. Azalea flower colour ranges from purple through carmine red, red, pink and white. Furthermore, azalea flowers can also be picotee type, with a different-coloured centre and margin, or flecked. The latter is expected to be caused by transposon activities [9]. Flower colour segregation in azalea can be predicted by a Mendelian model encompassing 3 major genes (P, W & Q; [10]). Purple flower colour is dominant over all other colours and is encoded by P. In the absence of the allele for P, W differentiates between (red) coloured (W-) and white flowers (ww). Q encodes for co-pigmentation by means of flavonols; in combination with the allele for W it results in carmine red flowers. Red flowers are recessive for Q (qq). This model does not clarify the presence of pink flowers, but the authors suggested pink to be a gradation in pigment. Also Sasaki et al. [11] state that flower colour intensity is determined by the amount of anthocyanin present. By means of image analysis, De Keyser et al. [12] recently confirmed in azalea that pink can be seen as (carmine) red at a lower intensity level. Studying the gene expression levels of the flavonoid biosynthetic genes could be informative to shed a light on this pink mystery as well. By means of the transgenic approach, Nakamura et al. [13] created pink torenia plants by down regulation of flavonoid 3′-hydroxylase (F3′H) and flavonoid 3′,5′-hydroxylase (F3′5′H) genes and also Boase et al. [14] reported that the suppression of the latter gene resulted in reduced colour intensity. The past decade, genetic engineering is explored widely for the modification of floricultural plants (reviewed in [15]). Expression levels of the targeted genes were always determined in order to identify their correlation to the flower colour phenotype [13, 16–18]. The exploration of natural flower colour differences by means of gene expression studies is only done between a limited number of genotypes, e.g. in cyclamen [19], Ipomoea[20], Freesia hybrida[21], azalea [22, 23] or Oncidium[24]. No data are currently available on the consistent effect of the studied genes in other genotypes with the same flower colour. Moreover, the quantification methods used in the aforementioned studies are not the most accurate. Some studies still describe the use of Northern blots [18, 24] or semi-quantitative RT-PCR (reversed transcription PCR) [16, 19, 21, 23, 24], others do use quantitative RT-PCR (RT-qPCR) but limit themselves to the comparative Cq (quantification cycle) method [25] in combination with the use of only a single non-validated reference gene. However, multiple, assay-validated reference genes are considered to be an essential component of a consistent RT-qPCR assay [26–30]. mRNA quantification can potentially be a very powerful and reliable technique for investigating gene expression, but only if handled thoughtfully [26, 31]. Due to the sensitivity and in order to increase accuracy, the technique was optimised intensively the past decades at all crucial steps from RNA isolation up to the final quantification (reviewed in [31, 32]). MIQE-guidelines (M inimum I nformation for Publication of Q uantitative Real-time PCR E xperiments; [26]) were set in order to stimulate the scientific community to quantify in an accurate manner and also to provide all essential data when publishing gene expression studies. However, in plant science, still too many papers on gene expression are published with inaccurate quantification [27–29, 33], as was also illustrated for flower colour.

Proposed flavonoid biosynthesis pathway in azalea. The pathway only leads to the production of cyanidin pigments and is redrafted after [8, 34]. CHS: chalcone synthase; CHI: chalcone isomerase; F3H: flavanone 3-hydroxylase; F3'H: flavonoid 3'-hydroxylase; DFR: dihydroflavonol 4-reductase; ANS: anthocyanidin synthase; OMT: O-methyltransferase; UFGT: UDP-glucose:flavonoid 3-O-glucosyltransferase; RT: rhamnosyl transferase; FLS: flavonol synthase.
Hence, the aim of this paper is dual.
-
1.
The establishment of a reliable RT-qPCR protocol for transcriptional profiling that can be applied in all plant species, even when only limited transcriptomic data are available. Optimisation at crucial steps is described into detail, with a focus on RNA quality, reference gene validation, the use of noRT (no Reversed Transcriptase) samples and the implementation of plasmid-derived standard curves for PCR efficiency correction.
-
2.
Study of gene expression in relation to flower colour in an azalea mapping population to identify correlations that are not limited to specific genotypes but are consistent over the whole azalea gene pool. Ultimately, the idea is to use these gene expression data to study flower colour in a genetical genomics approach.
Results
Sampling
In azalea flowering, generally four developmental stages are considered: closed buds (stage 1), buds showing colour at the top but with the scales still present (stage 2), candle stage without any scales left (stage 3) and the opened flower (stage 4) [23]. Expression of both the early gene CHS (chalcone synthase) and the late gene DFR (dihydroflavonol 4-reductase) appeared to be highest in stage 3 [23], hence this stage was selected for the evaluation of flower colour gene expression. Nakatsuka et al. [34] report a higher expression in azalea for some of the early flavonoid biosynthesis genes in stage 2, but these are only 2-fold differences. We therefore preferred to quantify the expression profile of all genes on the same sample, which would allow us to correlate expression profiles of the different genes in our analysis.
RNA quality control
Azalea RNA concentration varied tremendously between samples and was for some samples too low (Additional file 1) to test all genes in one RT-qPCR experiment. Hence we decided to extract RNA in duplicate from each sample. These technical replicates were then pooled after DNase treatment and purified together as one sample. RNA purity was measured spectrophotometrically. Contaminating proteins are displayed at an absorbance optimum of 280 nm, an A260/280 ratio above 1.8 is considered of an acceptable RNA purity although 2 would be optimal [35]. Concerning polysaccharide and polyphenol contamination, A260/230 is measured. A value of 2.5 means free of contamination [36], 2 is acceptable. However, the absorbance ratio’s only reflect RNA purity [26, 37] but not RNA integrity [37]. Absorption ratios were satisfying, except for low-concentrated samples (<15 ng/μl) where both A260/230 and A260/280 were clearly decreased. The low absorption ratios could indicate the presence of potential inhibitors. However, the reliability of the measurement can also be questioned in case of low RNA concentrations.
Performing a SPUD assay is considered to be the method of choice to evaluate the influence of inhibitory components on the RT-qPCR performance [32, 38]. Therefore a subset of 14 randomly selected samples was used for a SPUD analysis. The difference in mean Cq-value between the SPUD control and RNA/cDNA samples did not exceed the variation within the SPUD control group (Figure 2) and remained below the proposed cut-off value of 1 Cq [39]. This confirmed that no PCR inhibitors were present in spite of the low absorption ratios in 3 samples (Additional file 1).

Results of the SPUD assay. Box plot of the Cq values obtained after the analysis of 3 sample types (RNA, cDNA and control) in a SPUD assay. For RNA/cDNA 14 different samples were measured in duplicate, 14 replicates were used for the SPUD control.
Finally, RNA integrity was checked on the same subset of samples. In order to see how degradation evolved in our own material, we constructed a degradation series. A decrease of the ribosomal peaks and a shift in the electropherogram towards the so-called fast region [40] is clearly noticed (Figure 3). A visible degradation was also spotted on the gel-view (Figure 3). For low-concentrated samples, gel views were even the only reliable indicator for quality since the signal was too weak to verify on the electropherogram. Based on the degradation series, RNA was considered to be degraded when the 25S/18S rRNA ratio was below 1; degradation also becomes very well noticeable in the virtual gel view at this point (lane 4 and 5, Figure 3). According to these settings, all tested RNA samples were graded as good quality. Consequently, the robustness of our RNA isolation procedure from flower petals was demonstrated; RNA samples could even be placed for 15 hours at room temperature, without any visible degradation (data not shown). Hence, RNA quality results were extrapolated to all cDNA samples isolated from azalea flower buds in this study.

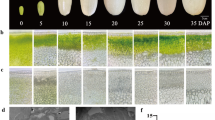
RNA quality control with the Experion (Bio-Rad). Electropherogram (right) and virtual gel-view (left) of an RNA degradation series that was constructed by heating an RNA sample for 0, 15, 30, 45 and 60 min at 80°C. The loading marker and small RNA band and cytoplasmic 18S and 25S as well as 16S and 23S chloroplast and mitochondrial ribosomal bands are indicated with arrows. Lanes: (L) size standard, (1) intact RNA, (2) 15 min, (3) 30 min, (4) 45 min, (5) 60 min. Intensity settings can vary between lanes.
Amplification specificity
Amplification of DNA in cDNA samples could result in an overestimation of the actual gene expression level of a gene or, even worse, in the false detection of expression. Developing primers spanning an intron or targeting exon-exon junctions can prevent co-amplification of DNA during RT-qPCR. Alignments with homologous sequences were made for all target genes (Table 1). No introns were present in CHS; intron-spanning primers were developed in ANS (anthocyanidin synthase) and DFR. In FLS (flavonol synthase) and F3′H (flavonoid 3′-hydroxylase) primers amplified a single exon but were located at the 3′ end of the sequence to reduce the influence of RNA degradation. The azalea F3H (flavanone 3-hydroxylase) fragment was too short and covered only a single exon. EST (Expressed Sequence Tags) sequences of the reference genes (Table 2) could not be evaluated for the presence of introns since their functional annotation was not specific enough. Hence, not all primers were intron-spanning and some introns were too small to prevent co-amplification of DNA [32]. Therefore DNA contamination had to be checked for after all. NoRTs were included for all samples and amplification was performed on these noRTs with all primer sets (both reference and target genes). In case of amplification of noRTs, contamination was considered to be negligible when the difference in Cq between the noRT and the sample was above seven cycles. In that case, at least 128-fold less contaminating DNA was present compared to cDNA. This is even above the five cycles that are the default setting for the same feature in qBase+ (Biogazelle), the software module that was developed by Hellemans et al. [41] for RT-qPCR data analysis. Only three samples amplified using the DFR primers and one sample using the F3′H primers were considered to be contaminated. Hence, these particular data were discarded from the dataset and only a single biological replicate was used instead for further calculations.
Reference genes
The possible conservation of gene expression stability across different plant species [27] was an opportunity to select conventionally used reference genes in azalea. However, in a crop with only little sequence information available, this required degenerate PCR, with a low success-rate. Only GAPDH (glyceraldehyde 3-phosphate dehydrogenase) could be isolated as such. Hence, 13 fragments were selected based on putative functions from an azalea EST database [42] as candidate reference genes (Table 2). Amplification patterns of two of these genes (HK134 and HK190) did not satisfy in flower petals (data not shown). The expression of the 11 remaining reference genes was determined in petals of eight azalea cultivars and standard-curve derived quantities were imported into geNorm [30]. With a pair wise variation V2/3 of 0.145, the use of two reference genes seems sufficient (see Additional file 2). However, this value is nearby the proposed cut-off value of 0.15 and with V3/4 being only 0.108, three reference genes appeared to be most favourable for normalisation of gene expression in azalea flower buds. These validated reference genes have an optimal M-value (for homogeneous tissues) below 0.5 (M = 0.368 [41]) and belong to different functional classes. Hence they are not likely to be co-regulated, what enforces their trustworthiness for combination into a normalisation factor [30]. Unfortunately, when analysing the second assay, quite some noRTs amplified with one of the selected reference genes (HK173). Therefore this gene had to be eliminated as a reference gene for the final analysis. Hence, normalisation was done with a normalisation factor based on two reference genes (HK5 and HK129). The normalisation factor had a less optimal M-value of 0.524 over the three assays, still this solution was preferred over using unreliable expression data for normalisation.
Standard curves
Plasmids containing the fragments of interest were used for the construction of a relative dilution series. Initially, reproducibility and stability of these dilution series was a major problem. However, this problem could be circumvented by linearization of the plasmids [43] and by diluting the linear fragments in a yeast tRNA solution. The addition of a carrier such as yeast tRNA prevents the loss of very little quantities in the smallest dilution steps [44]. In this way, the error on the linear regression of the dilution series was not worth mentioning. The SD(E) values (Additional file 3) were always below 0.01.
It is possible to analyze a standard curve only once for each gene and to apply the derived PCR efficiency in all further analysis. However, we preferred to work with run-specific amplification efficiencies to avoid the introduction of confounding technical variation. This was the best option, since amplification efficiencies of the individual standard curves clearly differ in time (Additional file 3), The PCR efficiency of e.g. HK129 varied between 0.94 and 0.81. The efficiencies for F3′H and certainly for DFR were far below the optimum, but by using the run-specific amplification efficiency, this difference in efficiency was accounted for and calculation errors were significantly reduced between assays.
Flower colour gene expression
We aimed at finding gene expression differences for six key genes of the flavonoid biosynthesis pathway between four flower colour groups: white, red, carmine red and pink in an azalea mapping population. Initially we selected five seedlings from each flower colour group in combination with the (pink-coloured) parents of the crossing population (assay 1; see Additional file 1). No significant correlations were found between the colour grouping and the gene expression levels of the individual genes (data not shown). Since these data were in due course to be used for eQTL (expression Quantitative Trait Locus) mapping, we gradually expanded the dataset in order to determine the minimal sample size with sufficient power in eQTL mapping. First 29 samples were added to the dataset (assay 2; see Additional file 1) and Kruskal-Wallis analysis was performed to determine the power of eQTL mapping. This yielded only highly significant (p < 0.001) correlations for CHS. Eventually, we needed a total of 70 siblings to obtain enough power to detect (preliminary) eQTLs for 50% of the genes (Figure 4). We therefore considered 70 samples (+2 parents) to be sufficient for our gene expression study.

Power analysis by means of Kruskal-Wallis eQTL mapping. Preliminary eQTL mapping by means of Kruskal-Wallis analysis was performed in MapQTL®5 [91]. Two population sizes were compared: 49 siblings (upper panel) and 70 siblings (lower panel). For each gene (ANS, CHS, DFR, F3H, F3′ H and FLS) the number of markers (vertical axis) that correlated at a certain significance level (horizontal axis) is given.
The results of all three assays were hence combined in a single dataset with 23 white flowers, 22 red, 19 carmine red and 8 pink ones. Due to the spread of the analysis over 3 different time points, inter-run calibration (IRC) was required to correct for potential run-to-run variation. Using (multiple) IRCs as advised by [41] was not feasible since these were not implemented consequently in every assay. Instead, the overall gene expression level per plate (and per gene) was used for inter-run calibration. The geometric mean was preferred over the arithmetic mean for calculating this IRC factor, as the former controls better for possible outlying values [30]. To verify whether our methodology did not introduce bias in the dataset, we decided to compare the outcome of both calculation methods. For this purpose, the samples of the total dataset were split up again after averaging the calibrated normalised relative quantities (CNRQ) of the biological replicates. All gene expression results, both CNRQ and NRQ (normalised relative quantities) per assay, are shown in Additional file 4. Mantel-analysis confirmed the consistency of the inter-run calibration method applied. The (C)NRQ values in both matrices were significantly correlated at the level of p = 0.001 for assay 2 and 3 and p = 0.004 for assay 1.
The mean difference in Cq values between technical replicates varied between 0.07 and 0.27 cycles. However, the variation in the technical replicates was considered negligible compared to biological variation. The fold differences of CNRQ values of some biological replicates varied noticeably (see Additional file 5). This was most pronounced for F3′H with a substantial higher mean and maximum fold difference. The latter is due to sample 234, which shows a lot of variation for the other genes as well. The biological variation in DFR expression is less pronounced, but with a mean/median of 1.76/1.38 still rather high.
No correlation could be found between the flower colour groups and gene expression levels (Table 3). Nevertheless, the expression of some genes appeared to be correlated to others, for CHS and FLS there was even a significant correlation with all other genes (Table 3). The flavonoid biosynthesis pathway can be partitioned among early and late pathway genes, but the breaking point differs between species [45, 46]. In azalea, F3H and F3′H are considered as early pathway genes together with CHS and FLS; ANS and DFR are some of the late pathway genes [8]. Taking different combinations of early or late pathway genes as an input for discriminant analysis, some of these combinations appeared to be able to distinguish to a minor extent between flower colour groups (Table 4). Combining the expression of all 4 early pathway genes could classify 51.4% of the samples in the correct colour group. Co-pigmentation of flavonols cannot be visualised in white flowers and therefore the interpretation of the expression profiles in this group can be misleading, certainly for FLS. When white flowers were omitted from the dataset, already 65.3% of the samples could be assigned to the correct flower colour group based on the same combination of early pathway genes. In case we classified samples according to flower colour intensity (pink versus (carmine) red), the expression levels of the early pathway genes could assign over 85% of the samples correctly. Even the combination of all genes performed very well for this purpose. Interestingly, when we compared the F3′H gene expression levels between both groups (Mann–Whitney U-test), a significant difference (p = 0.0425) was found. When [13] down regulated this gene in torenia, flower colour turned to pink as well. These results confirm that F3′H gene expression is an important factor for the establishment of flower colour intensity in azalea as well.
When samples were classified according to their co-pigmentation pattern (Q/q [12]), again the combined information of the early pathway genes could discriminate best between both classes (68.1% correct classifications, Table 4). Also the combination of all six genes scores quite well in grouping the samples (63.8%). The difference between coloured and white flowers (W/w [12]) can be evaluated most reliable based on the expression of CHS, F3H and F3′H. The addition of FLS gene expression slightly reduces the information content (58.3% versus 59.7%), most likely due to the fact that flavonols have no impact on the phenotypic classification of W. However, when we look at the effect of the late pathway genes ANS and DFR, we can conclude that the expression of these genes is mainly involved in differentiating between white and coloured flowers as well.
Discussion
Optimisation of the RT-qPCR protocol
A good RT-qPCR experiment should always be based on a well-thought sampling protocol. Gene expression experiments essentially reflect a snapshot of RNA at the moment of extraction. Therefore, biological replicates are a prerequisite [26]. In this study, biological replicates were gathered on different flowers of a single plant. Indeed, sampling on two independent plants would have been a better approach since any influence of the physiological condition of the plant onto the overall gene expression would have been taken into account. However, when evaluating gene expression in a crossing population with only one plant per genotype, this is not an option. Growing all plants together at optimal conditions and sampling in a standardized way was therefore expected to be sufficient to fade out this effect as much as possible.
RT-qPCR has become the method of choice for gene expression analysis, but it suffers from considerable pitfalls, e.g. when it comes to evaluation of the RNA quality. Reporting on RNA quality assessment is one of the key-elements of the MIQE-guidelines [26] but is currently not done in 3 out of 4 published gene expression studies in plants [33]. Moreover, the results of the quality assessments are often not shown in the other 25%, although this information is crucial for the significance of the published results. Nevertheless, this parameter has a major impact on RT-qPCR performance [33, 39, 47, 48], but there is no gold standard to define RNA quality and every method can have a different appreciation [39]. Absorption ratio’s only reflect RNA purity [37], whereas a SPUD-assay can evaluate for the inhibitory effect of these impurities [32, 38]. Our results demonstrate that only looking at the absorption ratios can lead to wrong assumptions concerning the RNA quality. In spite of the low absorption ratios of several samples, no PCR inhibition was seen in the SPUD assay, indicating the acceptable quality of our samples. Assessing PCR efficiency in a test sample by serial dilution of the sample can be an alternative method to identify inhibition [32] but is not so obvious in case of low concentrated samples. D’haene and Hellemans [49] demonstrate that inhibitors can be derived from the shape of the amplification curve, but this is not an objective method. Hence, we advise to perform a SPUD assay on a representative subset of the samples every time a new sample type, treatment and/or extraction protocol is used.
To assess RNA integrity as well, microfluidic capillary electrophoresis was implemented. This technology recently gained interest in the plant RNA community (reviewed in [33]), but is partly based on the ribosomal peak ratio (28S/18S). Since the relationship between this ratio and mRNA integrity appears to be unclear [40, 48, 50, 51], RIN (RNA Integrity Number [36]) and RQI (RNA Quality Indicator [52]) values that take into account the complete electropherogram were introduced as a more solid measure for RNA integrity. However, these values were initially assigned by using electropherograms of various mammalian tissues to train the software in an adaptive learning approach. In plants, no 28S rRNA is present, instead there is a 25S rRNA peak. In addition, total RNA in chloroplast-containing plant tissues also consists of 16S and 23S rRNA [53], adding 2 extra peaks. These rRNA peaks will be recognized as degradation peaks by the software, leading to a miscalculation of the RIN/RQI value and an underestimation of the true integrity of the material in plants. This is clearly seen in the result of Pico de Coana et al. [54]. Moreover, an optimal 28S/18S rRNA ratio of 2 is without any evidence extrapolated to plant 25S/18S rRNA [55]. These researchers rely on the software outputs, but they omit to look at the raw data to decide on the true quality of the RNA. Microfluidic capillary electrophoresis in plant science can be of great value (when the technology is available) but should always be restricted to a visual evaluation of the electropherograms and virtual gel views. The construction of a degradation series can then help to decide on the level of RNA integrity of specific samples.
Co-purification of traces of DNA during RNA extraction is inevitable, therefore noRT samples have been analysed in all cases. As is also asked for in the MIQE-guidelines [26], noRT results should always be given when gene expression data are published. However, far too often papers are published in which qPCR data are lacking results of the noRTs. How these authors (and the readers) can be sure that the so-called gene expression differences are not false positive signals? In the case the use of noRTs is described, it is not always clear what these noRTs exactly consist of. Some researchers just add RNA as a control in the RT-qPCR (e.g. [56, 57]). However, to control in addition for DNA contamination during the cDNA synthesis step, we handled the RNA for noRT samples in exactly the same way as the normal samples. The same compounds were added, except off course the RT enzyme, as advised by Nolan et al. [32]. Suppliers of reversed transcriptase enzymes should provide special kits with additional buffers and primers for this purpose and this is unfortunately not always feasible. As an alternative, one could indeed use diluted RNA as a noRT sample and add the RT-reaction mixture as an additional sample in the analysis to control for potential contamination in this mixture. Even more crucial, in our opinion, is the analysis of noRTs with all primers. Often only a single gene is used to control for genomic DNA contamination [22, 56–58]. The fact that in our dataset an individual sample was suffering from contamination when one specific gene was amplified, but not when the other genes were amplified, strengthens the need to test all primer sets on all noRT samples. Also Laurell et al. [59] state that the sensitivity towards genomic DNA contamination differs greatly between assays. These authors developed ValidPrime as an efficient alternative for the use of noRT controls, but currently no such assays are available for plant studies yet.
For normalisation of gene expression data, reference genes are indispensable [30]. The use of reference genes controls for variations in extraction yield, reverse-transcription and efficiency of amplification. It is without question that multiple, assay-validated reference genes are considered to be an essential component of a consistent qPCR assay [26], also in plant science [27–29]. In azalea, we aimed at developing a basic set of reference genes for application in all azalea gene expression studies. Czechowski et al. [60] demonstrated that the commonly used reference genes were not always the best candidates. Also GAPDH was not withdrawn as a reliable reference gene in our analysis. Therefore alternatives were looked for. Microarray data can be an ideal source of reference genes [61], but are lacking in azalea. Coker and Davies [62] took advantage of EST data for reference gene selection in tomato. Since a limited set of 62 ESTs was available in azalea [42], candidate reference genes were selected from this dataset. The proposed set of 11 azalea reference genes is a valuable toolbox for future qPCR research in azalea. However, each experimental condition demands a specific set of reference genes [63, 64] and even different lab protocols seem to have an influence on reference gene selection [65]. Therefore, validation of this set in the desired tissues and conditions will be essential to select the appropriate assay-specific reference genes.
Several quantification strategies with altered normalisation methods are available, all depending on the PCR efficiency (E) for their calculations [25, 41]. The quantification approach can have a serious impact on the final results [66]. Assuming an optimal PCR efficiency is not recommended [26, 41]. The use of sample-specific amplification efficiencies [67–70] has become more common in RT-qPCR studies [71] since it allows quantification without standard curves. However, the outcome of using sample-specific amplification efficiencies can vary drastically depending on the settings and is reported to increase the random error [72]. Recently, Regier and Frey [66] demonstrated that using the average target specific efficiency (based on sample specific efficiency estimations) can be an alternative to the standard curve method in case a reliable algorithm is used (e.g. LinReg). Nevertheless, the use of standard curves remains the most precise method [73, 74]. Based on the equation of a standard curve, the qPCR efficiency can be calculated. In our study, plasmid DNA was used for standard curve construction. Hellemans et al. [41] advise to make the dilution series with a sample that mimics as much as possible the samples to be analysed in qPCR [41], most often this is a mixture of representative cDNA samples [57, 75]. Plasmid DNA consists of a different sample matrix, what can result in altered efficiencies due to the presence of different kinds of inhibitory components [76]. However, the absence of PCR inhibitors was controlled for by means of the SPUD assay. Moreover, in absolute quantification studies the use of plasmid DNA to construct a dilution series is even preferred [77]. Especially in case of the limited availability of cDNA, plasmid DNA also has the advantage of being available plentiful and is therefore a valuable alternative for the construction of standard curves.
Flower colour gene expression
Optimisation at all stages of the RT-qPCR has resulted in a reliable protocol for quantification of gene expression in azalea. We also aimed at studying the correlation between flower colour and the expression of candidate genes of the flavonoid biosynthesis pathway in a broader genetic background in contrast with what is currently reported in other ornamentals [19–22, 24]. Moreover, we ultimately wanted to use flower colour as a model system for genetical genomics [78] in azalea. Most crucial was therefore the minimal required population size with sufficient power for eQTL mapping [79]. With 4 different flower colour groups, conventional power analysis [80] was not an option. But according to Shi et al. [81] even in small populations the power should already be sufficient to detect eQTLs. Therefore we started with a small subpopulation of 20 plants and gradually expanded to a final population of 70 siblings. This stepwise approach forced us to use an alternative method for inter-run calibration. The performance of a Mantel-test validated the approach for our assay. However, this method of inter-run calibration cannot automatically be considered to be trustworthy in other experiments. We believe that the rather small expression differences between our samples and genes had a significant impact here. Experiments in which large expression differences are measured are more likely to suffer from using the average gene expression as an inter-run calibrator and we therefore want to encourage the use of inter-run calibration as described in Hellemans et al. [41]. However, after validation with a Mantel-test, one could use the described methodology when lacking proper inter-run calibrators. The use of 3 biological replicates could have allowed to identify outlier values in some samples with high biological variation. However, these values do reflect the true variation present in the flower buds and can therefore not be neglected. These data clearly reinforce the substantial interest of using biological (rather than technical) replicates in every qPCR experiment.
The individual expression profiles were not discriminative enough to differentiate between colour groups. Also in other species, no such correlations have been reported since most studies limit themselves to the comparison of gene expression between few cultivars with different flower colours [19–22, 24]. The use of multiple genotypes in each flower colour group certainly complicates the analysis. When the biological variation within a genotype is already substantial, detecting differences between genotypes is even harder. Only when the expression of F3′H was compared between pink and (carmine) red flowers, a significant expression difference was found. This implicates that there clearly is a link between the flower colour intensity and the F3′H expression. Similar conclusions can be drawn from the combined effect of early pathway genes (so including F3′H) on flower colour intensity, with very high percentages of correctly assigned genotypes. With a transgenic approach in torenia, Nakamura et al. [13] also demonstrated that the regulation of F3′H is crucial to manipulate flower colour intensity. Also F3′5′H is reported to be involved in pink [13, 14] but this gene is only of interest for the production of dephinidin derivatives [82]. Delphidin pigments can be present in purple azalea flowers, but this colour was not present in the studied population. Therefore the expression of this gene was not determined. Besides these two flavonoid biosynthetic genes, pale-anthocyanin coloration can also be the result of a mutation in a putative glutathione S-tranferase gene that is responsible for the transport of pigments to the vacuole [83]. Therefore it would certainly be interesting to determine the expression of such transporter genes as well. HPLC measurements of the pigment types and concentrations could add even more to the elucidation of pink in azalea.
Also for the other genes, the combination of expression profiles was highly informative, since flower colour regulation is known to occur mainly via a coordinated transcriptional control of structural genes [5, 7]). Especially the early pathway genes CHS, F3H, F3′H and FLS can discriminate rather well between the colour groups when white flowers are omitted from the analysis and these genes are most suited to differentiate for co-pigmentation as well. This makes sense, since the early pathway is indeed responsible for the production of the flavonols as co-pigments. To be able to include white flowers in the analysis, HPLC data would be needed to score for the presence of flavonols. The late pathway genes ANS and DFR are less informative but are still helpful for the classification of coloration. This could implicate that the difference between white and coloured flowers is situated rather at the regulation of the late pathway gene expression. Also in potato, DFR is known to be involved in the difference between white and coloured tubers [84] and Jung et al. [85] reported that the regulation of white pigmentation in potato is situated at the transcriptional level.
Due to the actual presence of gene expression differences that are related to the transcriptional regulation of the flavonoid biosynthetic pathway, these data are well-suited for eQTL mapping. For this purpose, not only the expression profiles of the individual genes but also the discriminant functions will be used as a first step towards a priori eQTL mapping [86] on the genetic map of the population under study [87]. As such, the gene expression information will be used in a genetical genomics approach [78] to evaluate the impact of the entire pathway on the flower colour. This can confirm the existence of a co-regulation network and will help to understand more the observed variation in flower colour. Moreover, the presence of markers for myb-functional genes on the genetic map can be valuable candidate genes potentially co-localising with flower colour eQTLs.
Conclusions
To conclude, we are convinced that optimisation at crucial steps resulted in the development of a reliable protocol for gene expression analysis that is not only applicable to azalea, but can easily be used on other plant material as well. Currently in plant research, validated and qualitative RT-qPCR protocols are still rare. A pool of azalea reference genes was constructed, three of them are sufficient for normalisation of gene expression in flower petals, but the remaining genes can in the future also be used for normalisation in other azalea tissues, e.g. leaves and shoots. We also stressed on the importance of a multi-level RNA quality control, to evaluate both RNA purity and RNA integrity, with special attention for the bottlenecks for automated procedures on plant RNA. Furthermore, the co-amplification of contaminating DNA in few samples showed the importance of analysing noRT samples with all genes under study. Finally the advantages of using plasmid-derived standard curves in every analysis was demonstrated as well.
The accurate protocol resulted in the quantification of several flavonoid biosynthesis genes in a subset of 70 siblings of an azalea mapping population. The expression of F3′H could differentiate between pink and (carmine) red flower colour groups. The combined regulation of the early pathway genes clearly has an impact on the co-pigmentation and the late pathway genes ANS and DFR are to a minor extent involved in differentiating between white and coloured flower phenotypes. These gene expression profiles will now be used as eQTLs to study flower colour in a genetical genomics approach. This might help us to point-out the actual genes that are encompassed in W and Q. Providing more detailed data on pigment composition (HPLC) in the petals of the different genotypes could even add an additional level (mQTLs or metabolite QTLs) of information to this map-based approach.
Methods
RNA isolation
RNA was isolated from flower buds in the candle stage (25–30 mm) [23] of 70 siblings of the ‘GxH’ crossing population [87] and both parents (‘98-13-4’ and ‘Sima’). From each plant, two individual buds were sampled (a and b) as biological replicates. For reference gene selection, candle stage flower buds of eight azalea cultivars (‘Hellmut Vogel’ and seven of its flower colour sports: ‘Paloma’, ‘Hector’, ‘Mw. Troch’, ‘Nordlicht’, ‘Terra Nova’, ‘Zalm Vogel’ and ‘Super Nova’) displaying a range of colours were used. Approximately 70 mg of petal tissue (other bud organs were carefully removed) was weighed per sample in duplicate in pre-cooled 2 ml safe-lock tubes (Eppendorf). Three zirconium beads were added to the tubes and the plant material was crushed in a pre-cooled block of the Retsch Tissuelyser (Qiagen) for 2 times 30 s at 30 Hz. After a short centrifugation (30 s, 4°C, full speed), the tubes were placed on ice and RNA was isolated according to the protocol of the RNAqueous kit® (Ambion) in combination with the Plant RNA Isolation Aid (Ambion). Elution was done in three steps (40/25/25 μl) and eluents were pooled. DNase treatment occurred on 80 μl of RNA with the DNA-free kit (Ambion). 10 μl DNaseI buffer and 1.5 μl rDNaseI were added, followed by an incubation step of 30 min at 37°C. DNase Inactivation Reagent (10 μl) was added and samples were incubated for 2 min at room temperature. After centrifugation (90 s, 10000 g) the supernatant was transferred to a new tube. Duplicate samples were finally pooled and purified [88] using 0.3 M Sodium Acetate pH5.5 (Ambion). Two and a half volumes of 100% EtOH was added and samples were incubated for at least 15 min at −80°C or overnight at −20°C. Supernatant was removed after 25 min centrifugation (14000 rpm, 4°C) and 1 ml 70% EtOH was added. Again tubes were centrifuged for 20 min at the same conditions and supernatant was discarded. The RNA pellet was dried in a vacuum-desiccator and resolved in 25 μl of RNase-free water. Samples were stored at −80°C until cDNA synthesis.
RNA quantity/quality
RNA was quantified by means of the NanoDrop spectrophotometer (Isogen). The presence of inhibitory components was evaluated (on a subset of 14 samples, Additional file 1) by means of the SPUD-assay developed by [30, 32]. A stock solution of 5 μM of the 101 bp SPUD amplicon (Sigma) was diluted 1/108 in yeast tRNA (50 ng/μl; Invitrogen). 0.5 μl of the diluted amplicon, 0.48 μM of both forward and reverse SPUD primers (Invitrogen), 0.1 μM of the dual-labelled (Fam-Tamra) SPUD probe (MWG-Biotech) and 1× LightCycler480 Probes Master Mix (Roche) was combined in a total volume of 10 μl in a white 384-well plate (Roche). For each sample, 1 μl of RNA or 2 μl of cDNA was added and all samples were analysed in duplicate. In the SPUD control samples, no RNA or cDNA was added; NTCs (No Template Control) were included as well. Plates were sealed with an adhesive film. Cycling conditions in the LightCycler480 (Roche) were 10 min at 95°C, followed by 45 cycles of 10s 95°C, 30 s 60°C and 1s 72°C. Fluorescence data were recorded every cycle at the end of the annealing/elongation step at 60°C. Data were analysed using the LightCycler480 software version 1.5 (Roche). Cq-values were exported to Microsoft Excel for further calculations. Finally, RNA quality and quantity was also determined on the same subset of samples using the Experion microfluidic capillary electrophoresis system (Bio-Rad) in combination with the RNA StdSens Chips (Bio-Rad). A degradation series was prepared by heating an RNA sample for 15, 30, 45 and 60 min at 80°C in a PCR machine.
Reverse transcription
First strand cDNA synthesis was performed with the SuperScript III First-Strand Synthesis SuperMix (Invitrogen) according to the manufacturers protocol and starting from 100 ng of RNA or 6 μl for low-concentrated samples (< 17 ng/μl). Oligo(dT)20 was used for priming and all incubations occurred in a Perkin Elmer 2720 (Applied Biosystems). As a control for DNA contamination, noRTs were created in the same way as samples, except for the SuperScript III/RnaseOUT Enzyme Mix that was omitted in these cases. Both cDNA and noRT samples were diluted 1/3 and stored at −20°C.
Reference genes
Homolog’s of commonly used reference genes (ubiquitin, GAPDH, β-actin, α-6-tubulin, TATA-box binding protein, elongation factor α) were searched for in azalea with degenerate primers; gene-isolation was only doing well for GAPDH. The fragment was cloned using the TOPO TA Cloning Kit (Invitrogen) and sequenced in order to develop specific RT-qPCR primers (Table 2). Twelve candidate reference genes were selected out of 62 annotated genes from a Rhododendron simsii hybrid ‘Flamenco’ EST library [42] and qPCR primers were developed with melting temperatures 58-60°C, primer lengths 20–24 bp and amplicon lengths 151–165 bp. (Primer Express 2.0, Table 2). Primers were at first tested on the EST containing plasmids. Primer pairs that amplified the proper fragment were, together with GAPDH primers, tested in duplo in a RT-qPCR assay on cDNA from flower petals of 8 azalea cultivars. PCR analysis was carried out in an ABI7000 thermocycler (Applied Biosystems). Amplification mixture consisted of 12.5 μl of SYBR Green I Master Mix (Applied Biosystems), 7.5 pmol of both primers and 2 μl cDNA in a total volume of 25 μl. Cycling conditions were 2 min 50°C, 10 min 95°C and 40 cycles of 15 s 95°C and 1 min 60°C. For melting curve analysis, cycling conditions were 15 s 95°C, 15 s 60°C followed by ramping from 60°C to 95°C with a ramp speed of 2% and a final step of 15 s 95°C. Cq-values were averaged and transformed to quantities using standard curves. These data were used for reference gene selection using geNorm software [30].
Standard curves
Amplified fragments of both reference and target genes were cloned using the TOPO TA Cloning Kit (Invitrogen) containing TOP10F’ chemically competent cells and the pCR2.1-TOPO cloning vector. For CHS and DFR, full length cDNA sequences were previously cloned [23]. Plasmid DNA was purified (GFX Micro Plasmid Prep Kit, Amersham) and linearised using 10 U of Hind III (Invitrogen) for 2 h at 37°C, followed by an enzyme inactivation step for 10 min at 70°C. The stock concentration of plasmids was diluted to a working solution of 1 ng/μl in 50 ng/μl yeast tRNA (Invitrogen). Standard curves were constructed as six log10 dilutions of this working solution in yeast tRNA (50 ng/μl). To prevent extrapolation, the range of the standard curve was set to cover Cq values of the cDNA samples. It must also be strengthened that the diluted aliquots were never stored longer as 24 h at 4°C to preserve quality [89] were and prepared newly from the same stock of plasmid DNA stored at −20°C if needed again later. Standard curves were used for calculation of PCR efficiencies (E = 10(−1/slope) -1).
Quantification
Six RT-qPCR primer sets were developed in azalea for genes coding for key enzymes in the flavonoid biosynthesis pathway: chalcone synthase (CHS), flavanone 3-hydroxylase (F3H), flavonoid 3′-hydroxylase (F3′H), anthocyanidin synthase (ANS), dihydroflavonol 4-reductase (DFR) and flavonol synthase (FLS) (Table 1). CHS and DFR were R. simsii hybrid sequences [9], the others from R. Xpulchrum[34]. Primers were designed using Primer Express 2.0 (Applied Biosystems). Primers were targeted to the 3′ end and preferably spanning an intron. Intron/exon positions were predicted based on homologies with poplar or Arabidopsis sequences. Small amplicon sizes were preferred because this gives more consistent results [48]. All samples, noRTs, NTCs and standard curves were measured in duplicate in a LightCycler480 (Roche). In a white 384-well plate (Roche), 375 nM of each primer and 5 μl of LightCycler480 SYBR Green I Master (Roche) was used with 2 μl of sample in a total volume of 10 μl. Plates were sealed with an adhesive film. Cycling conditions were 5 min at 95°C, followed by 40 cycles of 10 s 95°C, 12 s 60°C and 10 s 72°C. Data acquisition was done at the end of every cycle. Melting curve analysis was performed as follows: 5 s 95°C, 1 min 65°C and heating to 97°C with a ramp rate of 0.06°C/s. Data acquisition occurred 10 times for every °C. Data were analysed using the LightCycler480 software version 1.5 (Roche). We started with gene expression analysis on 20 siblings and both parent plants. In a second phase, 29 new siblings were analysed and finally a third assay was run with 21 seedlings for gene expression analysis (See Additional file 1). Within an assay, the sample-maximisation method was preferred and samples were analysed in a single plate per gene. The 2nd derivative method of Luu-The et al. [90] was selected for Cq determination in every run. Cq-values were exported to Microsoft Excel; technical replicates were averaged geometrically. For combining the 3 assays, the overall gene expression level per plate and per gene (geometric mean) was used for inter-run calibration. Gene specific amplification efficiencies derived from standard curves and a normalisation factor [30] based on two validated reference genes (HK5 and HK129) was used for calculation of (calibrated) normalised relative quantities ((C)NRQ). Biological replicates were averaged geometrically as well.
Data analysis
Log-transformed data were used as an input for statistics. SPSS Statistics 19 software package was used for all statistical data analysis. Kruskal-Wallis (in MapQTL®5 [91]) was used as an alternative for power analysis to determine the required population size. Power was sufficient when at least half of the genes correlated with markers at the level of p < 0.001. To verify the inter-run calibration method, two calculation methods were compared for each assay: standard quantification in the individual assay (NRQ-values) and the same subset of samples calculated within the global dataset of 72 samples (CNRQ-values). Bivariate spearman correlation coefficients were calculated between log-transformed values of all samples for every gene, resulting in assay-specific correlation matrices. Correlation matrices of comparable datasets were used as an input for Mantel analysis [92] by means of the Mantel nonparametric test calculator [93].
References
De Loose R: Flavonoid glycosides in the petals of some Rhododendron species and hybrids. Phytochem. 1968, 9: 875-879.
De Loose R: The flower pigments of the Belgian hybrids of Rhododendron simsii and other species and varieties from Rhododendron subseries obtusum. Phytochem. 1969, 88: 253-259.
Gerats AGM, Martin C: Flavonoid synthesis in Petunia hybrida: genetics and molecular biology of flower colour. Recent advances in phytochemistry. Edited by: Stafford HA, Ibrahim RK. 1992, 165-200. New York: Plenum Press,
Holton TA, Cornish EC: Genetics and biochemistry of anthocyanin biosynthesis. Plant Cell. 1995, 7: 1071-1083.
Mol J, Grotewold R, Koes RE: How genes paint flowers and seeds. Trends Plant Sci. 1998, 3: 212-217. 10.1016/S1360-1385(98)01242-4
Winkel-Shirley B: Biosynthesis of flavonoids and effects of stress. Curr Opin Plant Biol. 2002, 5: 218-223. 10.1016/S1369-5266(02)00256-X
Schwinn K, Venail J, Shang Y, Mackay S, Alm V, Butelli E, Oyama R, Bailey P, Davies K, Martin C: A small family of MYB-regulatory genes controls floral pigmentation intensity and patterning in the genus Antirrhinum. Plant Cell. 2006, 18: 831-851. 10.1105/tpc.105.039255
De Cooman L, Everaert ESW, Faché P, Vande Casteele K, Van Sumere CF: Flavonoid biosynthesis in petals of Rhododendron simsii. Phytochem. 1993, 33: 1419-1426. 10.1016/0031-9422(93)85102-W
De Schepper S, Debergh P, Van Bockstaele E, De Loose M, Gerats A, Depicker A: Genetic and epigenetic aspects of somaclonal variation: flower colour bud sports in azalea, a case study. South African J Bot. 2003, 69: 117-128.
Heursel J, Horn W: A hypothesis on the inheritance of flower colours and flavonoids in Rhododendron simsii Planch. Zeitschrift für Pflanzenzüchtung. 1977, 79: 238-249.
Sasaki N, Nishizaki Y, Uchida Y, Wakamatsu E, Umemoto N, Momose M, Okamura M, Yoshida H, Yamaguchi M, Nakayama M, Ozeki Y, Itoh Y: Identification of the glutathione S-transferase gene responsible for flower color intensity in carnations. Plant Biotechnol. 2012, 29: 223-227. 10.5511/plantbiotechnology.12.0120a
De Keyser E, Lootens P, Van Bockstaele E, De Riek J: Image analysis for QTL mapping of flower colour and leaf characteristics in pot azalea (Rhododendron simsii hybrids). Euphytica. 2013, 189: 445-460. 10.1007/s10681-012-0809-7
Nakamura N, Fukuchi-Mizutani M, Fukui Y, Ishiguro K, Suzuki K, Suzuki H, Okazaki K, Shibata D, Tanaka Y: Generation of pink flower varieties from blue Torenia hybrida by redirecting the flavonoid biosynthetic pathway from delphinidin to pelargonidin. Plant Biotechnol. 2010, 27: 375-383. 10.5511/plantbiotechnology.10.0610a
Boase MR, Lewis DH, Davies KM, Marshall GB, Patel D, Schwinn KE, Deroles SC: Isolation and antisense suppression of flavonoid 3′, 5′-hydroxylase modifies flower pigments and colour in cyclamen. BMC Plant Biol. 2010, 10: 107- 10.1186/1471-2229-10-107
Nishihara M, Nakatsuka T: Genetic engineering of flavonoid pigments to modify flower color in floricultural plants. Biotechnol Lett. 2011, 33: 433-441. 10.1007/s10529-010-0461-z
Chen W-H, Hsu C-Y, Cheng H-Y, Chang H, Chen H-H, Ger M-J: Downregulation of putative UDP-glucose: flavonoid 3-O-glucosyltransferase gene alters flower coloring in Phalaenopsis. Plant Cell Rep. 2011, 30: 1007-1017. 10.1007/s00299-011-1006-1
Kamiishi Y, Otani M, Takagi H, Han D-S, Mori S, Tatsuzawa F, Okuhara H, Kobayashi H, Nakano M: Flower color alteration in the liliaceous ornamental Tricyrtis sp. By RNA interference-mediated suppression of the chalcone synthase gene. Mol Breeding. 2012, 30: 671-680. 10.1007/s11032-011-9653-z
Nakatsuka T, Mishiba K, Kubota A, Abe Y, Yamamura S, Nakamura N, Tanaka Y, Nishihara M: Genetic engineering of novel flower colour by suppression of anthocyanin modification genes in gentian. J Plant Physiol. 2010, 167: 231-237. 10.1016/j.jplph.2009.08.007
Akita Y, Kitamura S, Hase Y, Narumi I, Ishizaka H, Kondo E, Kameari N, Nakayama M, Tanikawa N, Morita Y, Tanaka A: Isolation and characterization of the fragrant cyclamen O-methyltransferase involved in flower coloration. Planta. 2011, 234: 1127-1136. 10.1007/s00425-011-1466-0
Yamamizo C, Noda N, Ohmiya A: Anthocyanin and carotenoid pigmentation in flowers of section Mina, subgenus Quamoclit, genus Ipomoea. Euphytica. 2012, 184: 429-440. 10.1007/s10681-011-0618-4
Sui X, Gao X, Ao M, Wang Q, Yang D, Wang M, Fu Y, Wang L: cDNA cloning and characterization of UDP-glucose: anthocyanidin 3-O-glucosyltransferase in Freesia hybrida. Plant Cell Rep. 2011, 30: 1209-1218. 10.1007/s00299-011-1029-7
Mizuta D, Nakatsuka A, Miyajima I, Ban T, Kobayashi N: Pigment composition patterns and expression analysis of flavonoid biosynthesis genes in the petals of evergreen azalea ‘Oomurasaki’ and its red flower sport. Plant Breeding. 2010, 129: 558-562.
De Schepper S, Debergh P, Van Bockstaele E, De Loose M: Molecular characterisation of flower colour genes in azalea sports (Rhododendron simsii hybrids). Acta Hort. 2001, 552: 143-150.
Liu X-J, Chuang Y-N, Chiou C-Y, Chin D-C, Shen F-Q, Yeh K-W: Methylation effect on chalcone synthase gene expression determines anthocyanin pigmentation in floral tissues of two Oncidium orchid cultivars. Planta. 2012, 236: 401-409. 10.1007/s00425-012-1616-z
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001, 25: 402-408. 10.1006/meth.2001.1262
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT: The MIQE guidelines: M inimum I nformation for publication of Q uantitative real-time PCR E xperiments. Clin Chemistry. 2009, 55: 611-622. 10.1373/clinchem.2008.112797
Guttierrez L, Mauriat M, Guénin S, Pelloux J, Lefebvre J, Louvet R, Rusterrucci C, Moritz T, Guerineau F, Bellini C, Van Wuytswinkel O: The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription – polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnol J. 2008, 6: 609-618. 10.1111/j.1467-7652.2008.00346.x
Guttierrez L, Mauriat M, Pelloux J, Bellini C, Van Wuytswinkel O: Towards a systematic validation of references in real-time RT-PCR. Plant Cell. 2008, 20: 1734-1735. 10.1105/tpc.108.059774
Guénin S, Mauriat M, Pelloux J, Van Wuytswinkel O, Bellini C, Guttierrez L: Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. J Exp Bot. 2009, 60: 487-493. 10.1093/jxb/ern305
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F: Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3: 1-11.
Thellin O, Elmoualij B, Heinen E, Zorzi W: A decade of improvements in quantification of gene expression and internal standard selection. Biotechnol Adv. 2009, 4: 323-333.
Nolan T, Hands RE, Bustin SA: Quantification of mRNA using real-time RT-PCR. Nat Protoc. 2006, 1: 1559-1582. 10.1038/nprot.2006.236
Die JV, Roman B: RNA quality assessment: a view from plant qPCR studies. J Exp Bot. 2012, 63: 6069-6077. 10.1093/jxb/ers276
Nakatsuka A, Mizuta D, Kii Y, Myajima I, Kobayashi N: Isolation and expression analysis of flavonoid biosynthesis genes in evergreen azalea. Sci Hort. 2008, 118: 314-320. 10.1016/j.scienta.2008.06.016
Baelde HJ, Cleton-Jansen AM, Van Beerendonck H, Namba K: High quality RNA isolation from tumors with low cellularity and high extra-cellular matrix component for cDNA microarrays. Application to chondrosarcoma. J Clin Pathol. 2001, 54: 778-782. 10.1136/jcp.54.10.778
Shultz DJ, Craig R, Cox-Foster DL, Mumma RO, Medford JI: RNA isolation from recalcitrant plant tissue. Plant Mol Biol Rep. 1994, 12: 310-316. 10.1007/BF02669273
Mueller O, Hahnenberger K, Dittmann M, Yee H, Dubrow R, Nagle R, Ilsley D: A microfluidic system for high-speed reproducible DNA sizing and quantitation. Electrophoresis. 2000, 21: 128-134. 10.1002/(SICI)1522-2683(20000101)21:1<128::AID-ELPS128>3.0.CO;2-M
Nolan T, Hands RE, Ogunkolade W, Bustin SA: SPUD: A quantitative PCR assay for the detection of inhibitors in nucleic acid preparations. Anal Biochem. 2006, 351: 308-310. 10.1016/j.ab.2006.01.051
Vermeulen J, De Preter K, Lefever S, Nuytens J, De Vloed F, Derveaux S, Hellemans J, Speleman F, Vandesompele J: Measurable impact of RNA quality on gene expression results from quantitative PCR. Nucl Acids Res. 2011, 39: 1-12. 10.1093/nar/gkq742
Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T: The RIN: and RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 2006, 7: 3- 10.1186/1471-2199-7-3
Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J: qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8: 10.1186/gb-2007-8-2-r19.
De Keyser E, De Riek J, Van Bockstaele E: Discovery of species-wide EST-derived markers in Rhododendron by intron-flanking primer design. Mol Breeding. 2009, 23: 171-178. 10.1007/s11032-008-9212-4
Ovstebo R, Bente Foss Haug K, Lande K, Kierulf P: PCR-based calibration curves for studies of quantitative gene expression in human monocytes: development and evaluation. Clinical Biochem. 2003, 49: 425-432.
Wang QT, Xiao W, Mindrinos M, Davis RW: Yeast tRNA as carrier in the isolation of microscale RNA for global amplification and expression profiling. Biotechniques. 2002, 33: 788-796.
Quattrocchio F, Wing JF, van der Woude K, Souer E, de Vetten N, Mol J, Koes R: Molecular analysis of the anthocyanin2 gene of petunia and its role in the evolution of flower color. Plant Cell. 1999, 11: 1433-1444.
Streisfeld MA, Rausher MD: Altered trans-regulatory control of gene expression in multiple anthocyanin genes contributes to adaptive flower color evolution in Mimulus aurantiacus. Mol Biol Evol. 2009, 26: 433-444. 10.1093/molbev/msn268
Bustin SA, Nolan T: Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J Biomol Tech. 2004, 15: 155-166.
Fleige S, Walf V, Huch S, Prgomet C, Sehm J, Pfaffl M: Comparison of relative mRNA quantification models and the impact of RNA integrity in quantitative real-time RT-PCR. Biotechnol letters. 2006, 28: 1601-1613. 10.1007/s10529-006-9127-2
D’haene B, Hellemans J: The importance of quality control during qPCR data analysis. Int Drug Disc. 2010, 18-24.
Imbeaud S, Graudens E, Boulanger V, Barlet X, Zaborski P, Eveno E, Mueller O, Schroeder A, Auffray C: Towards standardization of RNA quality assessment using user-independent classifiers of microcapillary electrophoresis traces. Nucl Acids Res. 2005, 33: e56- 10.1093/nar/gni054
Pfaffl MW, Fleige S, Riedmaier I: Validation of lab-on-chip capillary electrophoresis systems for total RNA quality and quantity control. Biotechnol Biotechnol Equip. 2008, 22: 839-843.
Denisov V, Strong W, Walder M, Gringich J, Wintz H: Development and validation of RQI: an RNA quality indicator for the ExperionTM automated gel electrophoresis system. Tech Note Bio-Rad. 2008, 5761:
Krupp G: Agilent Technologies application note. Stringent RNA quality control using the Agilent 2100 bioanalyzer. 2005,
Pico de Coana Y, Parody N, Fernandez-Caldas E, Alonso C: A modified protocol for RNA isolation from high polysaccharide containing Cupressus arizonica pollen. Applications for RT-PCR and phage display library construction. Mol Biotechnol. 2009, 10.1007/s12033-009-9219-z.
Philips MA, D’auria JC, Luck K, Gershenzon J: Evaluation of candidate reference genes for real-time quantitative PCR of plant samples using purified cDNA as template. Plant Mol Biol Rep. 2009, 27: 407-416. 10.1007/s11105-008-0072-1
Onate-Sanchez L, Vicente-Carbajosa J: DNA-free RNA isolation protocols for Arabidopsis thaliana, including seeds and siliques. BMC Reseach Notes. 2008, 1: 93-10.1186/1756-0500-1-93. 10.1186/1756-0500-1-93
De Santis C, Smith-Keune C, Jerry DR: Normalizing RT-qPCR data: are we getting the right answers? An appraisal of normalization approaches and internal reference genes from a case-study in the Finfish Lates Calcarifer. Mar Biotechnol. 2011, 13: 170-180. 10.1007/s10126-010-9277-z
Grunwald U, Guo W, Fischer K, Isayenkov S, Ludwig-Muller J, Hause B, Yan X, Kuster H, Franken P: Overlapping expression patterns and differential transcript levels of phosphate transporter genes in arbuscular mycorrhizal, Pi-fertilised and phytohormone treated Medicago truncatula roots. Planta. 2009, 229: 1023-1034. 10.1007/s00425-008-0877-z
Laurell H, Iacovoni JS, Abot A, Svec D, Maoret J-J, Arnal J-F, Kubista M: Correction of RT-qPCR data for genomic DNA-derived signals with ValidPrime. Nucl Acids Res. 2012, 40: e51- 10.1093/nar/gkr1259
Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR: Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005, 139: 5-17. 10.1104/pp.105.063743
Popovici V, Goldstein DR, Antonov J, Jaggi R, Delorenzi M, Wirapati P: Selecting control genes for RT-qPCR using public microarray data. BMC Bioinforma. 2009, 10: 1186/1471-2105-10-42.
Coker JS, Davies E: Selection of candidate housekeeping controls in tomato plants using EST data. Biotechniques. 2003, 35: 740-748.
Cruz F, Kalaoun S, Nobile P, Colombo C, Almeida J, Barros LMG, Romano E, Grossi-de-Sa MF, Vaslin M, Alves-Ferreira M: Evaluation of coffee reference genes for relative expression studies by quantitative real-time RT-PCR. Mol Breeding. 2009, 23: 607-616. 10.1007/s11032-009-9259-x
Klie M, Debener T: Identification of superior reference genes for data normalisation of expression studies via quantitative PCR in hybrid roses (Rosa hybrida). BMC Reserach Notes. 2011, 4: 518-10.1186/1756-0500-4-518. 10.1186/1756-0500-4-518
Mallona I, Lischewski S, Weiss J, Hause B, Egea-Cortines M: Validation of reference genes for quantitative real-time PCR during leaf and flower development in Petunia hybrida. BMC Plant Biol. 2010, 10: 10.1186/1471-2229-10-4.
Regier N, Frey B: Experimental comparison of relative RT-qPCR quantification approaches for gene expression studies in poplar. BMC Mol Biol. 2010, 11: 57- 10.1186/1471-2199-11-57
Ramakers C, Ruijter JM, Lekanne-Deprez RH, Moorman AFM: Assumption-free analysis of quantitative real-time PCR data. Neuroscience Lett. 2002, 339: 62-66.
Liu W, Saint DA: A new quantitative method of real time reverse transcription polymerase chain reaction assay based on simulation of polymerase chain reaction kinetics. Anal Biochem. 2002, 302: 52-59. 10.1006/abio.2001.5530
Liu W, Saint DA: Validation of a quantitative method for real time PCR kinetics. Biochem Biophys Res Comm. 2002, 294: 347-353. 10.1016/S0006-291X(02)00478-3
Lalam N: Estimation of the reaction efficiency in polymerase chain reaction. J Theor Biol. 2006, 242: 947-953. 10.1016/j.jtbi.2006.06.001
Rieu I, Powers SJ: Real-time quantitative RT-PCR: design, calculations and statistics. Plant Cell. 2009, 21: 1031-1033. 10.1105/tpc.109.066001
Nordgard O, Kvaloy JT, Farmen RK, Heikkila R: Error propagation in relative real-time reverse transcription polymerase chain reaction quantification models: the balance between accuracy and precision. Anal Biochem. 2006, 356: 182-193. 10.1016/j.ab.2006.06.020
Marino JH, Cook P, Miller KS: Accurate and statistically verified quantification of relative mRNA abundances using SYBR Green I and real-time RT-PCR. J Immunol Meth. 2003, 283: 291-306. 10.1016/S0022-1759(03)00103-0
Cook P, Fu C, Hickey M, Han E-S, Miller K: SAS programs for real-time RT-PCR having multiple independent samples. Bioinformatics. 2004, 37: 990-995.
Derveaux S, Vandesompele J, Hellemans J: How to do successful gene expression analysis using real-time PCR. Methods. 2010, 50: 227-230. 10.1016/j.ymeth.2009.11.001
Kunert R, Gach JS, Vorauer-Uhl K, Engel E, Katinger H: Validated method for quantification of genetically modified organisms in samples of maize flour. J Agricul Food Chem. 2006, 54: 678-681. 10.1021/jf052257s
Taverniers I, Van Bockstaele E, De Loose M: Cloned plasmid DNA fragments as calibrators for controlling GMOs: diffeent real-time duplex quantitative PCR methods. Anal Bioanal Chem. 2004, 378: 1198-1207. 10.1007/s00216-003-2372-5
Jansen RC, Nap JP: Genetical genomics: the added value from segregation. Trends Genet. 2001, 17: 388-391. 10.1016/S0168-9525(01)02310-1
Joosen RVL, Ligterink W, Hilhorst HWM, Keurentjes JJB: Advances in genetical genomics of plants. Curr Genomics. 2009, 10: 540-549. 10.2174/138920209789503914
Motulsky H: Choosing an appropriate sample size. Intuitive Biostatistics. 1995, 195-204. New York: Oxford University Press,
Shi C, Uzarowska A, Ouzunova M, Landbeck M, Wenzel G, Lübberstedt T: Identification of candidate genes associated with cell wall digestibility and eQTL (expression quantitative trait loci) analysis in a Flint x Flint maize recombinant inbred line population. BMC Genomics. 2007, 8: 22- 10.1186/1471-2164-8-22
Schwinn KE, Davies KM: Plant Pigments and their manipulation. Edited by: Davies K. 2004, 92-149. Flavonoids, Oxford: Blackwell Publishing Ltd, Annual Plant Reviews, Volume 14.
Larsen ES, Alfenito MR, Briggs WR, Walbot V: A carnation anthocyanin mutant is complemented by the glutathione S-transferases encoded by maize Bz2 and petunia An9. Plant Cell Rep. 2003, 21: 900-904.
De Jong WS, Eannetta NT, De Jong DM, Bodis M: Candidate gene analysis of anthocyanin pigmentation loci in the Solanaceae. Theor Appl Genet. 2004, 108: 423-432. 10.1007/s00122-003-1455-1
Jung CS, Griffiths HM, De Jong DM, Cheng S, Bodis M, Kim TS, De Jong WS: The potato developer (D) locus encodes an R2R3 MYB transcription factor that regulates expression of multiple anthocyanin structural genes in tuber skin. Theor Appl Genet. 2009, 120: 45-57. 10.1007/s00122-009-1158-3
Kliebenstein DJ, West MA, van Leeuwen H, Loudet O: Dioerge RW and St Clair DA: Identification of QTLs controlling gene expression networks defined a priori. BMC Bioinforma. 2006, 7: 308-10.1186/1471-2105-7-308. 10.1186/1471-2105-7-308
De Keyser E, Shu QI, Van Bockstaele E, De Riek J: Multipoint-likelihood maximization mapping on 4 segregating populations to achieve an integrated framework map for QTL analysis in pot azalea (Rhododendron simsii hybrids). BMC Mol Biol. 2010, 11: 1- 10.1186/1471-2199-11-1
Moore D, Dowhan D: Manipulation of DNA. Current Protocols Mol Biol. 2002, 59: 2.1.1-2.1.10.
Dhanasekaran S, Doherty TM, Kenneth J: Comparison of different standards for real-time PCR-based absolute quantification. J Immunol Methods. 2010, 354: 34-39. 10.1016/j.jim.2010.01.004
Luu-The V, Paquet N, Calvo E, Cumps J: Improved real-time RT-PCR method for high-throughput measurements using second derivative calculation and double correction. Biotechniques. 2005, 38: 287-293. 10.2144/05382RR05
Van Ooijen JW: MapQTL®5, software for the mapping of quantitative trait loci in experimental populations. 2004, Wageningen, The Netherlands: Kyazma BV,
Mantel N: The detection of disease clustering and a generalized regression approach. Cancer Res. 1976, 27: 209-220.
Liedloff AC: Mantel Nonparametric Test Calculator. Version 2.0. 1999, Australia: School of Natural Resource Sciences, Queensland University of Technology,
Acknowledgements
The authors are grateful to Romain Uytterhaegen for his outstanding technical assistance in plant cultivation. The whole staff of the biotech lab of ILVO-Plant Sciences Unit was very much appreciated for their support. We want to express our gratitude to Bio-Rad laboratories for giving us the opportunity to test the Experion on our plant material. This research was funded by the Ministry of Agriculture of Flanders.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EDK was responsible for design of the study, data analysis and statistics and drafted the manuscript; LD was involved in the acquisition of data and assisted in data analysis; EVB participated in the study’s design and contributed to editing the manuscript; JDR conceived the study, participated in data analysis and helped to draft the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12867_2012_656_MOESM1_ESM.pdf
Additional file 1:RNA concentration and purity. Description: RNA quantity and purity was measured of each biological replicate per sample using a NanoDrop spectrophotometer. For each sample, the assay is indicated in which the sample was analysed. Flower colour is indicated as well (0 = white, 1 = red, 2 = carmine red, 3 = pink). Samples used for analysis in the SPUD-assay and the Experion are indicated with an *. (PDF 138 KB)
12867_2012_656_MOESM2_ESM.pdf
Additional file 2:Evaluation of the optimal number of reference genes for normalization. Description: A cut-off value of 0.15 is proposed (top panel). Average expression stability (M) of the reference genes tested in azalea. M is calculated at each step during stepwise exclusion of the least stable reference gene. Genes are ranked from the least (left) to the most stable (right). Only genes with an M-value < 0.5 are valid in homogeneous samples (lower panel). Both graphs are generated in GeNorm [30]. (PDF 80 KB)
12867_2012_656_MOESM3_ESM.pdf
Additional file 3:PCR efficiencies of the standard curves. Description: Summary of slopes and derived PCR efficiencies (E) of the standard curves of dilution series analysed on different plates in 3 independent assays. E and the standard deviation on E (SD(E)) were calculated according to the formulas described in Hellemans et al. [41]. (PDF 163 KB)
12867_2012_656_MOESM4_ESM.pdf
Additional file 4:Description: Gene expression results. In the left part of the table, gene expression values were calculated on samples of a single assay (assay 1, 2 or 3). On the right, results are presented per assay but calculations occurred on the entire dataset of 72 samples. For each sample the geometric mean of the biological replicates is presented and (C)NRQ values have been log-transformed. (PDF 289 KB)
12867_2012_656_MOESM5_ESM.pdf
Additional file 5:Description: Fold differences between 2 CNRQ values of biological replicates. Samples are grouped according to flower colour (0 = white, 1 = red, 2 = carmine red, 3 = pink). Empty cells indicate one of the biological replicates was discarded after noRT analysis. (PDF 251 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
De Keyser, E., Desmet, L., Van Bockstaele, E. et al. How to perform RT-qPCR accurately in plant species? A case study on flower colour gene expression in an azalea (Rhododendron simsii hybrids) mapping population. BMC Molecular Biol 14, 13 (2013). https://doi.org/10.1186/1471-2199-14-13
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2199-14-13