
Abstract
Background
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are a diverse group of biologically active bacterial molecules. Due to the conserved genomic arrangement of many of the genes involved in their synthesis, these secondary metabolite biosynthetic pathways can be predicted from genome sequence data. To date, however, despite the myriad of sequenced genomes covering many branches of the bacterial phylogenetic tree, such an analysis for a broader group of bacteria like anaerobes has not been attempted.
Results
We investigated a collection of 211 complete and published genomes, focusing on anaerobic bacteria, whose potential to encode RiPPs is relatively unknown. We showed that the presence of RiPP-genes is widespread among anaerobic representatives of the phyla Actinobacteria, Proteobacteria and Firmicutes and that, collectively, anaerobes possess the ability to synthesize a broad variety of different RiPP classes. More than 25% of anaerobes are capable of producing RiPPs either alone or in conjunction with other secondary metabolites, such as polyketides or non-ribosomal peptides.
Conclusion
Amongst the analyzed genomes, several gene clusters encode uncharacterized RiPPs, whilst others show similarity with known RiPPs. These include a number of potential class II lanthipeptides; head-to-tail cyclized peptides and lactococcin 972-like RiPP. This study presents further evidence in support of anaerobic bacteria as an untapped natural products reservoir.
Similar content being viewed by others


Background
The increasing number of multiresistant bacteria pose a constant challenge for medicine and dictate the necessity of developing new antimicrobial compounds to treat life-threatening infections. Ribosomally synthesized and post-translationally modified peptides (RiPPs) are a promising addition to antibiotics biosynthesized via polyketide or non-ribosomal pathways. As antimicrobial agents this group of compounds often possess a narrow activity spectrum, most often targeting near relatives of the producing organism, although some broader spectrum RiPPs have been identified [1–3]. Their limited range of activity makes RiPPs potential targets for clinical applications as they can avoid the off-target effects seen with broad spectrum antibiotic agents, which can disturb the normal flora and open the door to undesired secondary infections by resistant organisms [3]. Although their target organisms may be highly specific, RiPPs have been shown to interrupt a variety of cellular processes, including the disruption of DNA, RNA or protein biosynthesis, although they commonly form pores in cell membranes by either targeting lipid II, a cell wall building block, or by direct pore formation via insertion into the cell wall [3]. As the targets of these compounds are conserved amongst many bacteria and are not subject to heavy modification, the potential for the development of resistance against RiPPs is significantly diminished [3, 4].
Despite the fact that RiPPs cover a diverse range of structural classes, they all follow a simple biosynthetic logic: a precursor peptide consisting of an N-terminal leader sequence and a C-terminal core sequence, encoded by a single gene is translated, the leader sequence is removed by a series of transporters, peptidases or a combination of both, and the remaining active peptide moiety is further processed by other enzymes, often encoded by genes within close proximity to the precursor gene [1, 2, 5].
The genetic basis for the production of many RiPP classes is well understood, and in most cases, gene content and structure is conserved amongst different arms of the bacterial phylogenetic tree. As such, comparison of well characterized biosynthetic genes or gene clusters against new genome sequences can identify putative RiPPs and in some cases, even the structure of the target metabolite can be predicted [6]. This "genome mining" approach allows for the discovery of potentially novel natural products in a completely culture independent fashion, with the potential to reduce the rediscovery rate of known molecules. Furthermore, genome mining has expanded the definition of exactly what constitutes a secondary metabolite producer and has revealed that the biosynthetic potential of many microorganisms has been widely underestimated [7–9]. Amongst these newly identified producers lie the anaerobic bacteria, a group that were believed to be incapable of producing secondary metabolites, as life without oxygen was presumed to not provide the required energy for the complex biosynthesis of antibiotics [10]. These "neglected" bacteria include those that are known to produce highly toxic peptides (botulinum toxin, tetanus toxin), and more recently several species have been identified as the source of novel natural products [8, 11–13]. An extensive investigation of 211 genomes of anaerobic bacteria for the presence of polyketide synthase (PKS) or non-ribosomal peptide synthetase (NRPS) encoding genes revealed a much larger potential than previously suspected and many of these PKS or NRPS loci appeared to be novel, with limited homology to previously characterized gene clusters [8]. Furthermore, it showed that certain genera have a predisposition towards increased secondary metabolite potential (such as members of the phyla Proteobacteria and Firmicutes) and that the natural habitat of the organisms seems to play an important role – isolates from environmental strains (soil, mud) often contain up to three times more genes for secondary metabolite biosynthesis than all other habitats combined. In particular, the clostridia were shown to be a potential treasure trove of novel secondary metabolites, which the isolation of the novel antibiotics closthioamide and clostrubin have recently confirmed [13, 14].
Despite the recent investigation of anaerobes for their potential to produce polyketide or non-ribosomal peptide metabolites [8], little is known about their ability to produce RiPPs. As anaerobes have been shown to possess a wealth of novel biosynthetic gene clusters, this suggests that there is also the potential to identify novel RiPP genetic loci amongst these organisms. This may, in turn, lead to the discovery of novel antimicrobial compounds to treat multi-drug resistant infections. Here we present an in-depth investigation of RiPP-encoding genes within the genomes of 211 anaerobic bacteria. As the nomenclature for RiPPs was redefined in 2013, with the support of many in the natural products community, we have chosen to follow these recommendations here, and have placed a size limit of 10 kDa for inclusion as a potential RiPP [1]. We have used a variety of bioinformatic tools in our analysis, including antiSMASH [15, 16], Bagel and bactibase database screening [17, 18], and BLAST searches to identify and predict the presence of RiPP gene clusters. Here we have shown that anaerobes have the potential to produce a variety of different RiPPs and that there is tendency towards the presence of RiPP biosynthetic gene clusters within those that already possess genetic loci for other secondary metabolites.
Results and discussion
General features of anaerobe genomes with respect to RiPPs
To survey the diversity of RiPPs we have undertaken a bioinformatic investigation of 211 complete and published anaerobe genomes for the presence of RiPP genes and gene clusters. Of note is the fact that anaerobes are a potential source of RiPPs, with >25% of currently sequenced anaerobe genomes encoding at least one or more RiPP classes (Table 1). It appears as though the RiPP biosynthetic gene clusters are more likely to be found in strains that possess other secondary metabolite biosynthetic gene loci, with only 10.4% of analyzed genomes containing only RiPP-encoding genes. However, these trends may only be predictable for the phyla Firmicutes, Actinobacteria, Bacteriodetes, Proteobacteria and Spirochaetes, which comprise a sufficient number of genomes for a representative analysis (Table 1, Figure 1). To what extent the present results also represent a general trend for the other phyla is difficult to estimate and more genomes of these phyla are required. The combination of PKS/NRPS and RiPPs appears to be limited to the phyla Actinobacteria, Proteobacteria and Firmicutes, confirming previous reports in aerobic organisms [1]. Notably, RiPP biosynthetic gene clusters were not identified in any anaerobes from the phylum Bacteriodetes, although aerobes from this phylum have been shown to possess lanthipeptide gene clusters [1]. In contrast to the situation with PKS/NRPS gene clusters, which are absent in Spirochaetes genomes, a small number of these organisms appear capable of producing RiPPs (Table 1, Figure 1). As is the case with PKS/NRPS biosynthetic gene clusters, of the sequenced genomes in our analysis, the Firmicutes appear to contain the highest percentage of RiPP producers, with approximately 75% of the Clostridium species analyzed being capable of producing PKS/NRPS or RiPPs.

Potential of anaerobic bacteria for PKS/NRPS/RiPP production and distribution among different phyla. A Distribution of genes for secondary metabolite production; percentage of strains containing: no PKS/NRPS/RiPP genes (green); both PKS/NRPS and RiPP (blue); only PKS/NRPS (yellow); only RiPP (red) B Distribution of secondary metabolite containing strains according to phyla and ability for secondary metabolite production (no PKS/NRPS/RiPP genes (green); both PKS/NRPS and RiPP genes (blue); only PKS/NRPS genes (yellow); only RiPP genes (red)). Firmicutes are additionally divided into Clostridia and others.
When it comes to the kind of RiPPs which are produced by the respective strains lanthipeptides, sactipeptides and linear azol(in)e-containing peptides (LAP) are the most common types (each group of RiPPs is explained in further detail below). LAP- and lactococcin-like genes are present predominantly in human pathogenic strains, whilst strains from extreme environments tend to contain DNA encoding head-to-tail cyclized peptides, as well as lanthipeptides and sactipeptides (Table 2, Figure 2). Lasso peptide biosynthetic loci appear to be mainly contained within non-pathogen genomes, and the lanthipeptides also appear to follow a similar distribution. Proteobacteria predominantly contain lasso peptide gene clusters and these are also more common in non-clostridia Firmicutes as well as head-to-tail cyclized peptides (Table 2, Figure 2).

Distribution of different RiPP biosynthetic gene clusters by habitat and pathogenicity. Animal (yellow); human (blue), environment (soil/mud) (red); extreme (green); other (purple); pathogenic (orange); non- pathogenic (black).
Although the focus here is on RiPP classes, several other peptides with potential antimicrobial activity, such as holins, linocins or peptidases were also identified. However, as their predicted size is much bigger than for the RiPPs, they were excluded from the following analyses.
Lanthipeptides
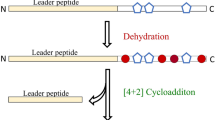
The lanthipeptides are defined by the presence of the non-proteinogenic amino acids lanthionine and 3-methyllanthionine, which are crosslinked via a thioether linkage at their β-carbon atoms [1, 2, 5, 19, 20]. The best-known and characterized lanthipeptide is nisin, which was first reported in 1928, although its structure was only finally elucidated in the 1970s [9, 10]. The biosynthetic genes for nisin had to wait until the late 1980s to be uncovered, and since this time many lan biosynthetic loci have been identified. The synthesis of the unusual lanthionine and 3-methyllanthionine residues occurs by dehydration of serine and threonine to dehydroalanine (dha) and dehydrobutyrine (dhb), respectively, via phosphorylated intermediates, which subsequently undergo a Michael-type addition (cyclization) with a cysteine residue [1, 5, 19] (Figure 3).

Lichenicidin-like lanthipeptides. A Lichenicidin biosynthetic gene cluster (lic) of B. licheniformis in comparison to putative lichenicidin gene clusters of C. botulinum H04402 065 and C. cellulovorans 743B; Numbers represent the locus tag for each gene within the genome sequence of each organism. B Comparison of lichenicidin peptide precursors (LicA1 and LicA2) and the putative precursor peptides of C. botulinum H04402 065 (H04402_00614 and H04402_00615) and C. cellulovorans 743B (Clocel_4229 and Clocel_4226); Glycine-Glycine motif indicates the cleavage site of leader sequence and core peptide (bold) C Formation of lanthionine (lan) and methyl-lanthionine (me-lan) moieties by dehydration of serine/threonine residues to dehydroalanine (dha) / dehydrobutyrine (dhb) and subsequent cyclization with a cysteine residue catalyzed by LanM D Amino acid structure of lichenicidin α-subunit (Bliα).
Based on the enzymes that are responsible for the post-translational modifications of the precursor peptides, lanthipetides are divided into four different groups. Class I lanthipeptides possess two distinct enzymes that carry out the dehydration (LanB) and cyclization (LanC). Class II-IV lanthipeptide are modified by multifunctional enzymes. Class II lanthipeptides possess a bifunctional enzyme with a N-terminal dehydratation (LanM- similar to LanB), and a C-terminal cyclisation domain (similar LanC). In the case of class III and IV lanthipeptides the modifications are carried out by a specific tri functional enzymes LanKC and LanL, respectively, which consist of an N-terminal phosphoserine- or phosphothreoninelyase, a central kinase and a C-terminal cyclisation domain [1, 2, 5, 19]. Class III lanthipeptides may also contain another non-proteinogenic amino acid – labionin [21–23]. Labionin follows a similar biosynthetic route to lanthionine, with cyclodehydration of a cysteine and two serine residues, which then react in a second Michael addition with another dehydroalanine [21–23]. Due to the strong conservation of these modifying enzymes they can be used for genome mining approaches [24, 25]. To date, characterized labionin-containing lanthipeptides are the labyrinthopeptide [21], erythreapeptin [26], avermipeptin [26], griseopeptin [26], catenulipeptin [27] and NAI112 [23]. The activity spectrum of lanthipeptides are mostly limited to Gram-positive bacteria and the mode of action is often associated with the disturbance of cell wall biosynthesis and pore formation [19], however, biosurfactant lanthipeptides have also been identified [28, 29].
In total 15 putative lanthipeptide biosynthetic gene clusters were detected in the analysed bacterial genomes, with class II lanthipeptides being the most common (Table 3, Figures 3 and 4). The lanthipeptides were once thought to be restricted to the Actinobacteria and Firmicutes phyla and in terms of anaerobes, this also appears to be the case [1]. Whilst some predicted lanthipeptide biosynthetic gene clusters exhibit similarity to previously characterized 2-component lanthipeptides, such as those for lichenicidin VK21 (Bacillus licheniformis VK21) [30] or lichenicidin (Bacillus licheniformis DSM 13 (ATCC 14580)), the remaining lanthipeptide gene clusters appear to be unique. The lichenicidins consist of two single peptides which gain their full activity only in combination [30] and they are encoded by two different precursor peptides as well as modified by two separate LanM enzymes. The leader sequence is removed during the transport out of the cell by the bifunctional enzyme LanT (transporter with N-terminal protease) [31].

Detected putative lanthipeptide gene clusters sorted by similar biosynthetic origin. Numbers represent the locus tag for each gene within the genome sequence of each organism.
Similar to the lichenicidin gene cluster, two precursor peptide encoding genes (A1, A2) and two LanM (M1, M2) encoding genes were detected in the genomes of Clostridium botulinum H04402 065 and Clostridium cellulovorans 743B (Figure 3). The arrangement of the genes is different in the respective clusters, but all the necessary core proteins appear to be encoded. The lichenicidin gene cluster, however, possesses a number of genes for immunity, which were not detected in the orthologous gene clusters in the clostridia. However, the heterologous expression of the B. licheniformis lichenicidin gene cluster in E. coli has shown that the immunity genes are not necessary for production of the lantibiotic, suggesting that the clostridial lichenicidin-like gene clusters may also be capable of producing an active lantibiotic [33]. It is also possible that the immunity gene(s) are located elsewhere in the genome. BLAST analysis of the putative precursor peptides of Caldicellulosiruptor bescii Z-1320 also showed similarities to lichenicidin, but only one precursor peptide and one modifying LanM protein are encoded in this cluster (Figure 4).
Sactipeptides
Sactipeptides or sactibiotics (sulphur to alpha-carbon antibiotic) are peptides in which a sulfur bridge is post-translationally formed between a cysteine residue and the α-carbon of another residue (Figure 5B & C), in contrast to lanthipeptides where the sulfur bridge is installed via the β-carbon [1, 34]. The sulfur linkage is introduced via a special radical SAM enzyme whose gene is co-localized in all sactipetide gene clusters and can be used for genome mining approaches [1, 35–37]. Several sactipeptides have so far been elucidated, all from Bacillus species, and include subtilosin A (B. subtilis, hemolytic) [38, 39], thuricin CD with its components Trn-α and Trn-β (B. thuringiensis, anticlostridial) [40], thurincin H (B. thuringiensis) [41] and the sporulation killing factor (SKF) (B. subtilis) [42]. Approximately 0.5% of the total protein content of anaerobic bacteria is represented by highly diverse radical SAM enzymes [43], and using putative radical SAM enzymes as a means of identifying sactipeptide loci returned a large number of enzymes putatively involved in RiPP formation. A similar approach was previously taken by Murphy et al., using the radical SAM enzyme of the thuricin CD gene cluster as BLAST template, which identified several thuricin CD-like biosynthetic gene clusters, including several in anaerobic bacteria [37].

Detected putative sactipeptides. A Thuricidin CD gene cluster (tm) of B. thuringiensis DPC 6431 and subtilosin A gene cluster (alb) of B. subtilis 168 in comparison to detected putative sactipeptide gene clusters; Numbers represent the locus tag for each gene within the genome sequence of each organism. B Amino acid structure of thuricin CD α-subunit (Trnα) C Characteristic sulfur bridge between a cysteine residue and the α-carbon of another residue in sactipeptides.
In this study many putative sactipeptide like gene clusters were obtained by using BAGEL database in a similar fashion to those reported previously [37]. Screening of the genes surrounding the encoded radical SAM proteins for sactipeptide like accessory genes (such as transporters and other proteins related to peptide maturation or secretion) led to the exclusion of many putative gene clusters, with those remaining listed in Table 4. Several of the gene clusters showed similarities to thuricin CD (Figure 5A) as mentioned above, however, the gene organization and number of precursor peptides differ between strains. It appears that the number of radical SAM enzymes encoded within a gene cluster correlates with the number of putative precursor peptides, except in case of Clostridium cellulolyticum H10 where only one radical SAM per two precursor peptides and Clostridium difficile 630 where two radical SAM enzymes per precursor peptide are encoded (Figure 5A).
Linear azol(in)e- containing peptides (LAP)
Many RiPPs are characterized by the presence of heterocyclic functional groups, such as oxazoles and thiazoles. One such group are the linear azol(in)e-containing peptides (LAP), whose heterocycles are derived from the cysteine, serine and threonine of a small precursor peptide [1]. LAP comprise of four essential components: a precursor peptide (known as ‘A’), and a heterotrimeric enzyme complex consisting of a dehydrogenase (‘B’) and cyclodehydratase (‘C’ and ‘D’). Biosynthetically, the first step towards a LAP is the formation of an azoline-heterocycle by the ‘C/D’ complex from serine or threonine and a cysteine residue, followed by dehydrogenation by ‘B’ leading to the corresponding azole (Figure 6C).

Detected putative LAP gene cluster. A Gene cluster of plantazolicin (pzn) (B. amyloliquefeaciens FZB42), streptolysin S (sag) (S. pyrogenes) and clostridiolysin S (clos) (C. botulinum ATCC 3502) in comparison to putative LAP gene clusters of B. intermedia, B. hyodysenteriae and T. mathranii mathranii A3; Numbers represent the locus tag for each gene within the genome sequence of each organism. B Comparison of precursor peptides of plantazolicin (PlnA), streptolysin S (SagA), clostridiolysin S (ClosA) with putative precursor peptides of B. intermedia, B. hyodysenteriae, and T. mathranii mathranii A3; Cleavage site of leader and core peptide in bold. C Introduction of heterocycles in plantazolicin by cyclodehydrogenase (PznC) and dehydrogenase (PznB) enzyme complex, X = S,O. D Chemical structure of plantazolicin.
Known LAP include streptolysin S (Streptococcus pyogenes) [44], microcin B17 (Escherichia coli) [45], plantazolicin (Bacillus amyloliquefaciens FBZ42) [46, 47] (Figure 6D), goadsporin (Streptomyces sp. TP- A0584) [48, 49] and clostridiolysin S (Clostridium botulinum) [50]. Despite the fact that the ‘BCD’ enzyme complex exhibits rather low amino acid identity between LAP loci, several studies have shown that ‘BCD’ genes from one LAP biosynthetic gene cluster can complement different LAP synthesis pathways, with the precursor peptide being converted into the active RiPP [47, 51]. As a result, these genes can be used for genome mining approaches [24].
The detected LAP gene clusters are found exclusively in the phyla of Firmicutes and Spirochaetes (Table 5). The gene cluster for clostridiolysin S is conserved in almost all Clostridium botulinum strains [50], except the strains BKT015925 and E3 str. Alaska E43, where it is absent. Like other LAP, the complete structure of clostridiolysin S has not yet been solved, owing to the difficulty inherent in the structure elucidation of heterocycles [50]. Several strains within the genus Brachyspira (B. pilosicoli 95/1000, B. intermedia PWS/A, B. murdochii 56–150 and B. hyodysenteriae WA1) also share an identical gene cluster, with only the precursor peptide of B. hyodysenteriae WA1 having a slightly different amino acid sequence (Figure 6A & B). The LAP gene cluster contained with the genome of Thermoanaerobacter mathranii mathranii A3 has a different gene organization.
Thiopeptides
Thiopeptides are characterized by a highly modified peptide macrocycle including several thiozole rings, a six-membered nitrogenous ring (either present as piperidine, dehydropiperidine or pyridine) and a side chain containing multiple dehydrated amino acid residues [1, 52, 53]. The introduction of a second macrocycle increases the complexity of these peptides and tryptophan-derived quinaldic acid or indolic acid residues are incorporated into the peptide scaffold. As for LAP biosynthesis, the thiozole rings are formed by dehydrogenation and cyclodehydratation of serine and cysteine residues [1, 52, 53]. The central nitrogen heterocycle is installed by a cycloaddition of two dehydroalanines catalyzed by similar proteins found in lanthipeptide biosynthesis (Figure 7C). Depending on the oxidation state and substitution pattern of the central nitrogen heterocycle, thiopetides are classified into different series (A-E) [1, 52, 53]. Thiomuracin A, isolated from a Nonomuraea species with strong activity against S. aureus[54] (Figure 7D), represents a series D thiopeptide with a tri-substituted pyridine ring as the central nitrogen heterocycle (Figure 7C). Besides the strong activity of many thiopeptides against Gram-positive bacteria by interfering with protein synthesis, some show additional antimalarial or anticancer activities (thiostrepton A) [1, 52, 53].

Detected putative thiopeptides. A Gene cluster of thiomuracin (tpd) (Nonomuraea str. Bp3714-39) in comparison to putative thiopeptide gene clusters of C. cellulovorans 743B and P. acnes KPA171202; Numbers represent the locus tag for each gene within the genome sequence of each organism. B Comparison of precursor peptides of thiomuracin (TpdA) and putative precursor peptides of C. cellulovorans 753B and P. acnes KPA171202; Cleavage site of leader and core peptide in bold. C Introduction of the central nitrogen heterocycle (red) in series d thiopeptides. D Chemical structure of thiomuracin A.
Two putative thiopeptide gene clusters have been detected in C. cellulovorans 743B and P. acnes KPA171202, both most likely encoding a series D thiopeptide (Table 6). The C. cellulovorans gene cluster (Figure 7A) encodes a LAP-like portion, with genes encoding the dehydrogenase and docking protein of a potential LAP but missing the cyclodehydratase protein. Furthermore lanthionine modifying proteins and three putative precursor peptides are located within the cluster, differing slightly in their protein sequence and showing greater similarity to LAP than to lanthipeptide precursors (Figure 7B).
Nitrile hydratase-related leader peptides (NHLP)
An intersection between LAP and lanthipeptides is formed by the class of NHLP (nitrile hydratase-related leader peptides) and Niff11 (nitrogen-fixing) related RiPPs [55]. On the one hand representatives of these RiPPs can contain the cyclodehydratase and dehydrogenase enzyme complex of LAP (introduction of heterocycles), and on the other hand the LanM enzymes involved in lanthipeptide biosynthesis (see above) [55]. A characteristic feature of these RiPPs are their precursor peptides, where NHLP precursors show sequence similarity with the α-subunit of nitrile hydratases (NHase), but without the active site motif [55]. The so-called Niff11 precursor peptides resemble an uncharacterized protein, which can be frequently found in nitrogen-fixing bacteria (including cyanobacteria) [55]. Compared to LAP, whose typical leader peptide sequences are about 24 amino acids in length, NHLP/Niff11 precursor peptides have much longer leader sequences, typically in the range of 70–83 amino acids [55]. The leader sequence is often terminated by a glycine-glycine motif. In contrast to their N-terminal sequences, the C-terminal ends of NHLP/Niff11 precursors vary considerably between different gene clusters and are rich in cysteine, serine and threonine, which are required for the posttranslational modifications [55].
The putative anaerobic NHLP/Niff11 clusters are located exclusively in the Actinobacteria, δ-Proteobacteria and Firmicutes phyla and all putative precursor peptides are annotated as NHLP or Niff11-superfamily proteins. The leader sequences (taken as the amino acid sequence before the GG motif) have a range between 66–85 amino acids, whilst the core sequences, taken as the amino acid sequence following the conserved VAGG or VSGG motif, are quite variable in length (14–59 amino acids) (Table 7, Figure 8B). The number of putative precursor peptides also differs from one to three depending on the individual gene cluster (Figure 8A). It is striking that cyclodehydratase and dehydrogenase related genes were only observed in the gene clusters present in Syntrophomonas wolfei subsp. wolfei str. Goettingen and Pelotomaculum thermopropionicum (Figure 8A). In the other cases a transporter with an N-terminal peptidase was identified, as well as several radical SAM proteins, which may be responsible for the modification steps of the NHLP/Niff11 precursors. Furthermore, proteins important for secretion are also located within several of the gene clusters (Figure 8A).

Detected putative NHLP/ Niff. A Structure of putative NHLP/Niff related gene clusters of D. baarsii 2st14, E. lenta VPI 0255, D. hafniense DCP-2, D. acetoxidans DSM 771, S. wolfei subsp. wolfei str. Goettingen, P. thermopropionicum SI; Numbers represent the locus tag for each gene within the genome sequence of each organism. B Comparison of the putative precursor peptides with VAGG-motif separating the leader and core peptide in bold.
Lasso peptides
Lasso peptides are among the most extraordinary RiPPs, and their rigid structure gives them enormous stability against heat, chemical attack and proteases [1, 56, 57]. So named because of their particular knotted structure, the lasso peptides are usually 16–23 amino acids in length and contain an 8–9 membered macrolactam ring, which is formed between the N-terminal amino group and the carboxylate of a conserved aspartate or glutamate residue at position 8 or 9, by a putative asparagine synthase like enzyme, resulting in a C-terminal loop and tail formation [1, 56, 57] (Figure 9B & C). Three subgroups of the lasso peptides have been characterized. The prototypical members of the group I lasso peptides include siamycin I [58], siamycin II [58] and RP71955 [59], all of which possess two disulfide bonds and an N-terminal cysteine [1, 56, 57]. In contrast, group II lasso peptides contain no disulfide bonds, and the N-terminal amino acid is glycine [1, 56, 57], with examples in the form of microcin J25 [60, 61], lariatin [62] and capistruin [63, 64]. Lasso peptide BI-32169 [65, 66] is the only member of group III, having one disulfide bridge and glycine as the N-terminal amino acid [1, 56, 57].
Studies on the biosynthesis of microcin J25 from E. coli AY25 [67, 68] and capistruin from Burkholderia thailandensis[63, 64] have shown that four genes (‘A-D’) are necessary for lasso peptide formation. In each case, the leader sequence is cleaved by an ATP-dependent protease (‘B’) from the precursor peptide (‘A’), with the simultaneous activation of the aspartate or glutamate residues [1, 56, 57]. Isopeptide bond formation is catalyzed by an ATP-dependent enzyme (‘C’), which has similarities to asparagine synthetase B, and the resulting product is transported out of the cell through ‘D’, which also ensures immunity of the producer to the mature RiPP [1, 56, 57]. Only the first eight N-terminal amino acids and the second last threonine of the leader sequence are required for its recognition by the modifying enzymes [69]. Due to conservation of the ‘B’ and ‘C’ enzymes, as well as conserved motifs in the precursor sequences, these can all be used as the basis for genome mining [24, 56, 67, 70–72].
Previous attempts at genome mining for lasso peptides identified putative gene clusters within the following anaerobe genomes: Spirochaeta smaragdinae DSM 11293, Syntrophomonas wolfei subsp. wolfei str. Goettingen, Treponema pallidum, Treponema cuniculi paraluiscuniculi A, Pelobacter propionicus DSM 2379, Desulfobacca acetoxidans DSM 111069 and Geobacter uraniireducens[71, 72]. However, upon closer investigation, several of these gene clusters were either undetected in the present study, or lacked the necessary genes encoding the characteristic lasso peptide modifying enzymes and as such they were not included in the current analysis. In the case of Desulfobacca acetoxidans both studies identified identical gene clusters for putative lasso peptides, with the only difference being the prediction of the precursor peptide (Figure 9A (* = precursor peptide identified in this study, # = precursor peptide identified by [71])).

Detected putative lasso peptides. A Microcin J25 (mcj) (E. coli) and Lariatin (lar) (R. jostii K01-B0171) gene clusters in comparision to putative lasso peptide gene clusters of G. uraniireducens Rf4, P. propionicus DSM 2379, D. acetoxidans DSM 11069 (* = precursor peptide identified in this study, # = precursor peptide identified by [71]), B. proteoclasticum B313, D. acetoxidans DSM 771, S. glycolicus DSM 8271 and C. perfringens str. 13; Annotation of the putative precursor peptide was not conclusively possible in most cases; Numbers represent the locus tag for each gene within the genome sequence of each organism. B Cleavage of the lariatin precursor peptide by a putative protease (LarD); Isopeptide bond (green) formation by LarB between the N-terminal amino acid glycine (red) and a glutamate (red) leads to the formation of a 8- membered macrolactame ring in lariatin. C Lasso peptide structure of lariatin (isopeptide bond (green)).
The biosynthetic gene clusters for microcin J25 and lariatin are shown in Figure 9A [73, 74]. Unlike microcin J25 and other lasso peptides, lariatins A and B, produced by Rhodococcus jostii, are formed by a five-gene cluster, larABCDE. Similar to other lasso peptides, LarA is the precursor peptide which is processed by LarB, LarC and LarD and then exported by the transporter LarF [73]. Whilst LarB and LarD appear to have similar functions, the role of LarC remains unclear, although it appears that larC is specific for Gram-positive bacteria [73]. Indeed, this appeared to be the case, as all anaerobic strains in which lasso peptide gene clusters were identified (Table 8) were Gram-positive and contained larC orthologues (Figure 9A). Interestingly, additional enzymes, such as a HPr kinase and a sulfotransferase, were also identified in some gene clusters. The role that these proteins play in the modification of the precursor peptides is currently unclear, although they may be involved in previously unidentified lasso peptide modifications [55].
Lactococcin
Like many other RiPPs, lactococcins possess an N-terminal leader sequence, which terminates in a glycine-glycine motif. This motif is an important signal unit for the respective transporters which secrete the substance and simultaneously cleave off the leader sequence [75]. Lactococcin 972 is homodimeric RiPP, which is only encoded by one structural gene [76]. This gene encodes a 91 amino acid precursor peptide of which 25 amino acids comprise the leader sequence and the remainder, the core sequence [76, 77]. In addition to the precursor peptide named LclA the gene cluster encodes a transporter (LclB) and an additional protein that is important for immunity (Figure 10A). Lactococcin 972 blocks the incorporation of lipid II, an essential cell wall building block [77–79].

Detected putative lactococcins like RiPPs. A Lactococcin 972 gene cluster (lcl) of L. lactis subsp. lactis in comparison to detected putative lactococcin 972 like gene clusters in D. hafniense Y51, B. longuminfantis JCM 1222 and P. acnes KPA171202; Numbers represent the locus tag for each gene within the genome sequence of each organism. B Lactococcin A gene cluster (lcn) of L. lactis subsp. cremoris and detected putative lactococcin A-like gene cluster in C. perfringens SM 101; (T = transposase); Numbers represent the locus tag for each gene within the genome sequence of each organism.
In almost all propionibacteria a lactococcin 972 like precursor peptide is present, with the exception of Propionibacterium acnes ATCC 11828, where it is absent (Figure 10A). The N-terminal leader sequence of P. acnes 266 includes an additional 23 amino acids in comparison to the other P. acnes strains. The gene organization in the P. acnes strains is different to Desulfitobacterium hafniese Y51, Bifidobacterium longum infantis and in comparison to the characterized lactococcin 972 gene cluster of Lactococcus lactis (Figure 10A).
Lactococcin like genes were detected in 11 genomes, in particular genes encoding lactococcin 972 and lactococcin A like proteins (Table 9). Unlike lactococcin 972, lactococcin A (Lactococcus lactis subsp. cremoris, Lactococcus lactis subsp. lactis biovar diacetylactis WM4) is a linear RiPP with a 75 amino acid precursor, containing a 21 amino acid leader sequence [80, 81]. Both lactococcins have a limited spectrum of activity against various Lactococcus strains and the antimicrobial effect of these peptides is based on the binding of the peptide core to a mannose-phosphotransferase that is localized in the cell wall of the target organism, resulting in increased cell wall permeability [77, 80, 82]. In lactococcin A biosynthesis, the precursor peptide LcnA is processed by LcnC, a transporter with N-terminal peptidase, and secreted by LcnD [81] (Figure 10B). A co-localized self-resistance gene guarantees immunity to lactococcin A. A homologous gene cluster to lactococcin A could be detected in C. perfringens SM 101, where the cluster is flanked by transposases (Figure 10B).
Head-to-tail cyclized peptides
As the name head-to-tail (HtT) suggests, these peptides are cyclized between their N- and C-terminus and range in size between 30–70 amino acids [1]. As with other cyclized peptides, they show a higher stability to heat, pH changes and proteases. These peptides tend to be hydrophobic and exert their effects by the formation of pores in the cell membrane of target organisms [1, 83]. The most famous and best known representative of this group is enterocin AS-48 (Enterococcus sp.) [84]. Other HtT-like cyclized peptides are the cyanobactins, amatoxins and cyclotides but they differ in their size and their biosynthetic origin [1, 83]. For example, HtT-cyclized peptides have no additional amino acids at the C-terminal end, which contribute to the cyclization and ring formation [1, 83]. It is still not completely clear how the C-terminal carboxyl group is activated, however, a protein containing a conserved domain of unknown function is present in most of the identified gene clusters [1, 83]. ATP-binding proteins are also present in the majority of the known gene clusters, which may also be involved in the activation of the carboxyl group [1, 83]. Because of their presence in many HtT-cyclized protein gene clusters these genes can be used for genome mining approaches.
Relatively few HtT-cyclized peptides were identified amongst the genomes analyzed here. Those that were identified were found in the phyla Firmicutes and Chloroflexi (Table 10), with several exhibiting homology to circularin A (Figure 11A), a previously characterized peptide of Clostridium beijerinckii ATCC 25752 [85, 86] (Figure 11C). The gene order in the Caldicellulosiruptor gene clusters is identical to each other and the precursor sequences differ by only a few amino acids (Figure 11B). The putative circularin A gene cluster of C. perfringens SM 101 is quite different, however, and it has limited conservation with the circularin A gene cluster in C. beijerinckii ATCC 25752 and is flanked by numerous transposases (Figure 11A).

Detected circularin A-like RiPPs. A Circularin A gene cluster (cir) of C. beijerinckii ATCC 25752 in comparison to putative circularin A like gene cluster of C. bescii Z-1320 and C. perfringens SM 101; Numbers represent the locus tag for each gene within the genome sequence of each organism. B Alignment of circularin A precursor sequence (CirA) and circularin A-like precursor sequences of C. bescii Z-1320 (Athe_2617), C. saccharolyticus DSM 8903 (Csac_0526) and C. perfringens SM 101 (CPR_0761) C Amino acid structure of circularin A.
Conclusion
Here we have surveyed the genomes of 211 anaerobic bacteria for the presence of RiPP biosynthetic gene clusters. As such, we have identified >25% of anaerobes are capable of producing RiPPs either alone or in conjunction with other secondary metabolites, such as polyketides or non-ribosomal peptides. As with the possession of NRPS and PKS gene clusters, the most likely RiPP producer organisms lie within the phyla Proteobacteria and Firmicutes. However, in contrast to their NRPS and PKS biosynthetic potential, which was minimal, anaerobic Actinobacteria appear to have a greater propensity for RiPP production. Interestingly, we found that the phylum Spirochaetes also contains a number of potential RiPP producing organisms, something that has not previously been found. In general, it also appears that non-pathogenic organisms have a greater potential for RiPP production, which aligns well with what is known about NRPS/PKS potential in anaerobes. Remarkably, anaerobes were found to have the potential to produce a variety of different RiPP classes, with the LAPs and lactococcins appearing to be favored by pathogenic anaerobes, whilst the other classes are more prominent in non-pathogenic isolates. Surprisingly, isolates from extreme environments contain a wide range of different RiPPs, in particular head-to-tail cyclized peptides and lanthipeptides. Despite the fact that their environmental niche is already restricted, it appears as though it must still be necessary for these organisms to have some way to defend themselves against competitors. In total we identified 81 putative RiPP clusters of which 43 had not been previously described and appear to be unique among known RiPP biosyntetic gene clusters. Furthermore, we were able to identify 23 gene clusters with similarities to known RiPP biosynthetic gene clusters, but that have not been previously identified in anaerobes and we were able to confirm a further 15 previously identified RiPP gene clusters.
Amongst the analyzed genomes, several gene clusters with good correlation to known RiPPs were identified. These include a number of potential class II lanthipeptides from the phyla Firmicutes and Actinobacteria, with similarity to the lichenicidin gene cluster from Bacillus licheniforme; sactipeptides identified in the phylum Firmicutes with similarities to the thuricin CD gene cluster of B. thuringiensis; head-to-tail cyclized peptides within the phyla Chloroflexi and Firmicutes with homology to the circularin A biosynthetic gene cluster from C. beijerinckii ATCC 25752; and lactococcin 972-like RiPPs from the phylum of Actinobacteria. The distribution of similar gene clusters amongst diverse organisms suggests that horizontal gene transfer has been active in the distribution of RiPP gene clusters amongst organisms that share similar environments.
Despite the fact that several identified gene clusters and precursor peptides show similarities to previously characterized RiPPs, in many instances the prediction of the final products remains difficult. Differences in the precursor peptide sequence between similar RiPP products may have an impact on the final modified structure of the peptide, meaning that prediction of RiPP homology between species where a similar gene cluster exists is also difficult.
In consideration of the increasing number of multiresistant strains, RiPPs are a promising alternative to classical antibiotic treatment. This investigation is the first report of the potential of anaerobic bacteria for the production of RiPPs and the detected putative RiPPs may represent future lead compounds in the fight against multirestistant pathogens. Nevertheless, the identification of all these potential metabolites remains a challenge for the future and more methods are needed to connect the detected genotypes to chemotypes [87].
Methods
Genome sequences
Complete and published genome sequences of 211 anaerobic bacteria (Additional file 1: Table S1) were obtained from the NCBI Refseq and draft genome repository.
Analysis of anaerobe genomes
Genomes were analyzed for the presence of RiPP encoding gene clusters by using the web-based bioinformatic tools antiSMASH [15, 16], Bagel and bactibase [17, 18]. Predicted gene clusters from each of the database outputs were inspected manually and compared using BLAST searches. Putative gene clusters were classified according to Arnison et al. [1] (antiSMASH data collected in April/ May 2012; Bagel database data collected in January 2014).
References
Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, Cotter PD, Craik DJ, Dawson M, Dittmann E, Donadio S, Dorrestein PC, Entian KD, Fischbach MA, Garavelli JS, Göransson U, Gruber CW, Haft DH, Hemscheidt TK, Hertweck C, Hill C, Horswill AR, Jaspars M, Kelly WL, Klinman JP, Kuipers OP, et al: Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat Prod Rep. 2013, 30: 108-160. 10.1039/c2np20085f.
McIntosh JA, Donia MS, Schmidt EW: Ribosomal peptide natural products: bridging the ribosomal and nonribosomal worlds. Nat Prod Rep. 2009, 26: 537-559. 10.1039/b714132g.
Cotter PD, Ross RP, Hill C: Bacteriocins - a viable alternative to antibiotics?. Nat Rev Microbiol. 2013, 11: 95-105.
Sang Y, Blecha F: Antimicrobial peptides and bacteriocins: alternatives to traditional antibiotics. Anim Health Res Rev. 2008, 9: 227-235. 10.1017/S1466252308001497.
Willey JM, van der Donk WA: Lantibiotics: peptides of diverse structure and function. Annu Rev Microbiol. 2007, 61: 477-501. 10.1146/annurev.micro.61.080706.093501.
Winter JM, Behnken S, Hertweck C: Genomics-inspired discovery of natural products. Curr Opin Chem Biol. 2011, 15: 22-31. 10.1016/j.cbpa.2010.10.020.
Donadio S, Sosio M, Stegmann E, Weber T, Wohlleben W: Comparative analysis and insights into the evolution of gene clusters for glycopeptide antibiotic biosynthesis. Mol Genet Genomics. 2005, 274: 40-50. 10.1007/s00438-005-1156-3.
Letzel A-C, Pidot SJ, Hertweck C: A genomic approach to the cryptic secondary metabolome of the anaerobic world. Nat Prod Rep. 2013, 30: 392-428. 10.1039/c2np20103h.
Udwary DW, Gontang EA, Jones AC, Jones CS, Schultz AW, Winter JM, Yang JY, Beauchemin N, Capson TL, Clark BR, Esquenazi E, Eustáquio AS, Freel K, Gerwick L, Gerwick WH, Gonzalez D, Liu WT, Malloy KL, Maloney KN, Nett M, Nunnery JK, Penn K, Prieto-Davo A, Simmons TL, Weitz S, Wilson MC, Tisa LS, Dorrestein PC, Moore BS: Significant natural product biosynthetic potential of actinorhizal symbionts of the genus frankia, as revealed by comparative genomic and proteomic analyses. Appl Environ Microbiol. 2011, 77: 3617-3625. 10.1128/AEM.00038-11.
Seedorf H, Fricke WF, Veith B, Bruggemann H, Liesegang H, Strittmatter A, Miethke M, Buckel W, Hinderberger J, Li F, Hagemeier C, Thauer RK, Gottschalk G: The genome of Clostridium kluyveri, a strict anaerobe with unique metabolic features. Proc Natl Acad Sci U S A. 2008, 105: 2128-2133. 10.1073/pnas.0711093105.
Behnken S, Hertweck C: Cryptic polyketide synthase genes in non-pathogenic Clostridium spp. PLoS One. 2012, 7: e29609-10.1371/journal.pone.0029609.
Behnken S, Hertweck C: Anaerobic bacteria as producers of antibiotics. Appl Microbiol Biotechnol. 2012, 96: 61-67. 10.1007/s00253-012-4285-8.
Pidot S, Ishida K, Cyrulies M, Hertweck C: Discovery of clostrubin, an exceptional polyphenolic polyketide antibiotic from a strictly anaerobic bacterium. Angew Chem Int Ed. 2014, 53: 7856-7859. 10.1002/anie.201402632.
Lincke T, Behnken S, Ishida K, Roth M, Hertweck C: Closthioamide: an unprecedented polythioamide antibiotic from the strictly anaerobic bacterium Clostridium cellulolyticum. Angew Chem Int Ed. 2010, 49: 2011-2013. 10.1002/anie.200906114.
Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R: antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39: W339-W346. 10.1093/nar/gkr466.
Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T: antiSMASH 2.0–a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013, 41: W204-W212. 10.1093/nar/gkt449.
van Heel AJ, de Jong A, Montalban-Lopez M, Kok J, Kuipers OP: BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013, 41: W448-W453. 10.1093/nar/gkt391.
Hammami R, Zouhir A, Le Lay C, Ben Hamida J, Fliss I: BACTIBASE second release: a database and tool platform for bacteriocin characterization. BMC Microbiol. 2010, 10: 22-10.1186/1471-2180-10-22.
Bierbaum G, Sahl HG: Lantibiotics: mode of action, biosynthesis and bioengineering. Curr Pharm Biotechnol. 2009, 10: 2-18. 10.2174/138920109787048616.
Chatterjee C, Paul M, Xie L, van der Donk WA: Biosynthesis and mode of action of lantibiotics. Chem Rev. 2005, 105: 633-684. 10.1021/cr030105v.
Meindl K, Schmiederer T, Schneider K, Reicke A, Butz D, Keller S, Guhring H, Vertesy L, Wink J, Hoffmann H, Brönstrup M, Sheldrick GM, Süssmuth RD: Labyrinthopeptins: a new class of carbacyclic lantibiotics. Angew Chem Int Ed. 2010, 49: 1151-1154. 10.1002/anie.200905773.
Pesic A, Henkel M, Sussmuth RD: Identification of the amino acid labionin and its desulfurised derivative in the type-III lantibiotic LabA2 by means of GC/MS. Chem Comm. 2011, 47: 7401-7403. 10.1039/c1cc11573a.
Iorio M, Sasso O, Maffioli SI, Bertorelli R, Monciardini P, Sosio M, Bonezzi F, Summa M, Brunati C, Bordoni R, Corti G, Tarozzo G, Piomelli D, Reggiani A: A glycosylated, labionin-containing lanthipeptide with marked antinociceptive activity. ACS Chem Biol. 2014, 9: 398-404. 10.1021/cb400692w.
Velasquez JE, van der Donk WA: Genome mining for ribosomally synthesized natural products. Curr Opin Chem Biol. 2011, 15: 11-21. 10.1016/j.cbpa.2010.10.027.
Begley M, Cotter PD, Hill C, Ross RP: Identification of a novel two-peptide lantibiotic, lichenicidin, following rational genome mining for LanM proteins. Appl Environ Microbiol. 2009, 75: 5451-5460. 10.1128/AEM.00730-09.
Voller GH, Krawczyk JM, Pesic A, Krawczyk B, Nachtigall J, Sussmuth RD: Characterization of new class III lantibiotics–erythreapeptin, avermipeptin and griseopeptin from Saccharopolyspora erythraea, Streptomyces avermitilis and Streptomyces griseus demonstrates stepwise N-terminal leader processing. Chembiochem. 2012, 13: 1174-1183. 10.1002/cbic.201200118.
Wang H, van der Donk WA: Biosynthesis of the class III lantipeptide catenulipeptin. ACS Chem Biol. 2012, 7: 1529-1535. 10.1021/cb3002446.
Kodani S, Hudson ME, Durrant MC, Buttner MJ, Nodwell JR, Willey JM: The SapB morphogen is a lantibiotic-like peptide derived from the product of the developmental gene ramS in Streptomyces coelicolor. Proc Natl Acad Sci U S A. 2004, 101: 11448-11453. 10.1073/pnas.0404220101.
Willey JM, Willems A, Kodani S, Nodwell JR: Morphogenetic surfactants and their role in the formation of aerial hyphae in Streptomyces coelicolor. Mol Microbiol. 2006, 59: 731-742. 10.1111/j.1365-2958.2005.05018.x.
Shenkarev ZO, Finkina EI, Nurmukhamedova EK, Balandin SV, Mineev KS, Nadezhdin KD, Yakimenko ZA, Tagaev AA, Temirov YV, Arseniev AS, Ovchinnikova TV: Isolation, structure elucidation, and synergistic antibacterial activity of a novel two-component lantibiotic lichenicidin from Bacillus licheniformis VK21. Biochemistry. 2010, 49: 6462-6472. 10.1021/bi100871b.
Caetano T, Krawczyk JM, Mosker E, Sussmuth RD, Mendo S: Heterologous expression, biosynthesis, and mutagenesis of type II lantibiotics from Bacillus licheniformis in Escherichia coli. Chem Biol. 2011, 18: 90-100. 10.1016/j.chembiol.2010.11.010.
Singh M, Sareen D: Novel LanT associated lantibiotic clusters identified by genome database mining. PLoS One. 2014, 9: e91352-10.1371/journal.pone.0091352.
Caetano T, Krawczyk JM, Mosker E, Sussmuth RD, Mendo S: Lichenicidin biosynthesis in Escherichia coli: licFGEHI immunity genes are not essential for lantibiotic production or self-protection. Appl Environ Microbiol. 2011, 77: 5023-5026. 10.1128/AEM.00270-11.
Fluhe L, Marahiel MA: Radical S-adenosylmethionine enzyme catalyzed thioether bond formation in sactipeptide biosynthesis. Curr Opin Chem Biol. 2013, 17: 605-612. 10.1016/j.cbpa.2013.06.031.
Fluhe L, Knappe TA, Gattner MJ, Schafer A, Burghaus O, Linne U, Marahiel MA: The radical SAM enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat Chem Biol. 2012, 8: 350-357. 10.1038/nchembio.798.
Fluhe L, Burghaus O, Wieckowski BM, Giessen TW, Linne U, Marahiel MA: Two [4Fe-4S] clusters containing radical SAM enzyme SkfB catalyze thioether bond formation during the maturation of the sporulation killing factor. J Am Chem Soc. 2013, 135: 959-962. 10.1021/ja310542g.
Murphy K, O'Sullivan O, Rea MC, Cotter PD, Ross RP, Hill C: Genome mining for radical SAM protein determinants reveals multiple sactibiotic-like gene clusters. PLoS One. 2011, 6: e20852-10.1371/journal.pone.0020852.
Kawulka K, Sprules T, McKay RT, Mercier P, Diaper CM, Zuber P, Vederas JC: Structure of subtilosin A, an antimicrobial peptide from Bacillus subtilis with unusual posttranslational modifications linking cysteine sulfurs to alpha-carbons of phenylalanine and threonine. J Am Chem Soc. 2003, 125: 4726-4727. 10.1021/ja029654t.
Huang T, Geng H, Miyyapuram VR, Sit CS, Vederas JC, Nakano MM: Isolation of a variant of subtilosin A with hemolytic activity. J Bacteriol. 2009, 191: 5690-5696. 10.1128/JB.00541-09.
Rea MC, Sit CS, Clayton E, O'Connor PM, Whittal RM, Zheng J, Vederas JC, Ross RP, Hill C: Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc Natl Acad Sci U S A. 2010, 107: 9352-9357. 10.1073/pnas.0913554107.
Sit CS, van Belkum MJ, McKay RT, Worobo RW, Vederas JC: The 3D solution structure of thurincin H, a bacteriocin with four sulfur to alpha-carbon crosslinks. Angew Chem Int Ed. 2011, 50: 8718-8721. 10.1002/anie.201102527.
Liu WT, Yang YL, Xu Y, Lamsa A, Haste NM, Yang JY, Ng J, Gonzalez D, Ellermeier CD, Straight PD, Pevzner PA, Pogliano J, Nizet V, Pogliano K, Dorrestein PC: Imaging mass spectrometry of intraspecies metabolic exchange revealed the cannibalistic factors of Bacillus subtilis. Proc Natl Acad Sci U S A. 2010, 107: 16286-16290. 10.1073/pnas.1008368107.
Haft DH, Basu MK: Biological systems discovery in silico: radical S-adenosylmethionine protein families and their target peptides for posttranslational modification. J Bacteriol. 2011, 193: 2745-2755. 10.1128/JB.00040-11.
Nizet V, Beall B, Bast DJ, Datta V, Kilburn L, Low DE, De Azavedo JC: Genetic locus for streptolysin S production by group A streptococcus. Infect Immun. 2000, 68: 4245-4254. 10.1128/IAI.68.7.4245-4254.2000.
San Millan JL, Hernandez-Chico C, Pereda P, Moreno F: Cloning and mapping of the genetic determinants for microcin B17 production and immunity. J Bacteriol. 1985, 163: 275-281.
Scholz R, Molohon KJ, Nachtigall J, Vater J, Markley AL, Sussmuth RD, Mitchell DA, Borriss R: Plantazolicin, a novel microcin B17/streptolysin S-like natural product from Bacillus amyloliquefaciens FZB42. J Bacteriol. 2011, 193: 215-224. 10.1128/JB.00784-10.
Molohon KJ, Melby JO, Lee J, Evans BS, Dunbar KL, Bumpus SB, Kelleher NL, Mitchell DA: Structure determination and interception of biosynthetic intermediates for the plantazolicin class of highly discriminating antibiotics. ACS Chem Biol. 2011, 6: 1307-1313. 10.1021/cb200339d.
Onaka H, Tabata H, Igarashi Y, Sato Y, Furumai T: Goadsporin, a chemical substance which promotes secondary metabolism and morphogenesis in streptomycetes. I. Purification and characterization. J Antibiot. 2001, 54: 1036-1044. 10.7164/antibiotics.54.1036.
Igarashi Y, Kan Y, Fujii K, Fujita T, Harada K, Naoki H, Tabata H, Onaka H, Furumai T: Goadsporin, a chemical substance which promotes secondary metabolism and Morphogenesis in streptomycetes. II Structure determination. J Antibiot. 2001, 54: 1045-1053. 10.7164/antibiotics.54.1045.
Gonzalez DJ, Lee SW, Hensler ME, Markley AL, Dahesh S, Mitchell DA, Bandeira N, Nizet V, Dixon JE, Dorrestein PC: Clostridiolysin S, a post-translationally modified biotoxin from Clostridium botulinum. J Biol Chem. 2010, 285: 28220-28228. 10.1074/jbc.M110.118554.
Lee SW, Mitchell DA, Markley AL, Hensler ME, Gonzalez D, Wohlrab A, Dorrestein PC, Nizet V, Dixon JE: Discovery of a widely distributed toxin biosynthetic gene cluster. Proc Natl Acad Sci U S A. 2008, 105: 5879-5884. 10.1073/pnas.0801338105.
Bagley MC, Dale JW, Merritt EA, Xiong X: Thiopeptide antibiotics. Chem Rev. 2005, 105: 685-714. 10.1021/cr0300441.
Li C, Kelly WL: Recent advances in thiopeptide antibiotic biosynthesis. Nat Prod Rep. 2010, 27: 153-164. 10.1039/b922434c.
Morris RP, Leeds JA, Naegeli HU, Oberer L, Memmert K, Weber E, LaMarche MJ, Parker CN, Burrer N, Esterow S, Hein AE, Schmitt EK, Krastel P: Ribosomally synthesized thiopeptide antibiotics targeting elongation factor Tu. J Am Chem Soc. 2009, 131: 5946-5955. 10.1021/ja900488a.
Haft DH, Basu MK, Mitchell DA: Expansion of ribosomally produced natural products: a nitrile hydratase- and Nif11-related precursor family. BMC Biol. 2010, 8: 70-10.1186/1741-7007-8-70.
Maksimov MO, Link AJ: Prospecting genomes for lasso peptides. J Ind Microbiol Biotechnol. 2014, 41: 333-344. 10.1007/s10295-013-1357-4.
Maksimov MO, Pan SJ, James Link A: Lasso peptides: structure, function, biosynthesis, and engineering. Nat Prod Rep. 2012, 29: 996-1006. 10.1039/c2np20070h.
Tsunakawa M, Hu SL, Hoshino Y, Detlefson DJ, Hill SE, Furumai T, White RJ, Nishio M, Kawano K, Yamamoto S: Siamycins I and II, new anti-HIV peptides: I. Fermentation, isolation, biological activity and initial characterization. J Antibiot. 1995, 48: 433-434. 10.7164/antibiotics.48.433.
Helynck G, Dubertret C, Mayaux JF, Leboul J: Isolation of RP 71955, a new anti-HIV-1 peptide secondary metabolite. J Antibiot. 1993, 46: 1756-1757. 10.7164/antibiotics.46.1756.
Wilson KA, Kalkum M, Ottesen J, Yuzenkova J, Chait BT, Landick R, Muir T, Severinov K, Darst SA: Structure of microcin J25, a peptide inhibitor of bacterial RNA polymerase, is a lassoed tail. J Am Chem Soc. 2003, 125: 12475-12483. 10.1021/ja036756q.
Rosengren KJ, Clark RJ, Daly NL, Goransson U, Jones A, Craik DJ: Microcin J25 has a threaded sidechain-to-backbone ring structure and not a head-to-tail cyclized backbone. J Am Chem Soc. 2003, 125: 12464-12474. 10.1021/ja0367703.
Iwatsuki M, Tomoda H, Uchida R, Gouda H, Hirono S, Omura S: Lariatins, antimycobacterial peptides produced by Rhodococcus sp. K01-B0171, have a lasso structure. J Am Chem Soc. 2006, 128: 7486-7491. 10.1021/ja056780z.
Knappe TA, Linne U, Zirah S, Rebuffat S, Xie X, Marahiel MA: Isolation and structural characterization of capistruin, a lasso peptide predicted from the genome sequence of Burkholderia thailandensis E264. J Am Chem Soc. 2008, 130: 11446-11454. 10.1021/ja802966g.
Knappe TA, Linne U, Robbel L, Marahiel MA: Insights into the biosynthesis and stability of the lasso peptide capistruin. Chem Biol. 2009, 16: 1290-1298. 10.1016/j.chembiol.2009.11.009.
Knappe TA, Linne U, Xie X, Marahiel MA: The glucagon receptor antagonist BI-32169 constitutes a new class of lasso peptides. FEBS Lett. 2010, 584: 785-789. 10.1016/j.febslet.2009.12.046.
Potterat O, Wagner K, Gemmecker G, Mack J, Puder C, Vettermann R, Streicher R: BI-32169, a bicyclic 19-peptide with strong glucagon receptor antagonist activity from Streptomyces sp. J Nat Prod. 2004, 67: 1528-1531. 10.1021/np040093o.
Duquesne S, Destoumieux-Garzon D, Zirah S, Goulard C, Peduzzi J, Rebuffat S: Two enzymes catalyze the maturation of a lasso peptide in Escherichia coli. Chem Biol. 2007, 14: 793-803. 10.1016/j.chembiol.2007.06.004.
Clarke DJ, Campopiano DJ: Maturation of McjA precursor peptide into active microcin MccJ25. Org Biomol Chem. 2007, 5: 2564-2566. 10.1039/b708478a.
Pan SJ, Rajniak J, Maksimov MO, Link AJ: The role of a conserved threonine residue in the leader peptide of lasso peptide precursors. Chem Comm. 2012, 48: 1880-1882. 10.1039/c2cc17211a.
Severinov K, Semenova E, Kazakov A, Kazakov T, Gelfand MS: Low-molecular-weight post-translationally modified microcins. Mol Microbiol. 2007, 65: 1380-1394. 10.1111/j.1365-2958.2007.05874.x.
Maksimov MO, Pelczer I, Link AJ: Precursor-centric genome-mining approach for lasso peptide discovery. Proc Natl Acad Sci U S A. 2012, 109: 15223-15228. 10.1073/pnas.1208978109.
Hegemann JD, Zimmermann M, Zhu S, Klug D, Marahiel MA: Lasso peptides from proteobacteria: genome mining employing heterologous expression and mass spectrometry. Biopolymers. 2013, 100: 527-542. 10.1002/bip.22326.
Inokoshi J, Matsuhama M, Miyake M, Ikeda H, Tomoda H: Molecular cloning of the gene cluster for lariatin biosynthesis of Rhodococcus jostii K01-B0171. Appl Microbiol Biotechnol. 2012, 95: 451-460. 10.1007/s00253-012-3973-8.
Solbiati JO, Ciaccio M, Farias RN, Gonzalez-Pastor JE, Moreno F, Salomon RA: Sequence analysis of the four plasmid genes required to produce the circular peptide antibiotic microcin J25. J Bacteriol. 1999, 181: 2659-2662.
Nissen-Meyer J, Rogne P, Oppegard C, Haugen HS, Kristiansen PE: Structure-function relationships of the non-lanthionine-containing peptide (class II) bacteriocins produced by gram-positive bacteria. Curr Pharm Biotechnol. 2009, 10: 19-37. 10.2174/138920109787048661.
Martinez B, Fernandez M, Suarez JE, Rodriguez A: Synthesis of lactococcin 972, a bacteriocin produced by Lactococcus lactis IPLA 972, depends on the expression of a plasmid-encoded bicistronic operon. Microbiology. 1999, 145 (Pt 11): 3155-3161.
Martinez B, Suarez JE, Rodriguez A: Lactococcin 972: a homodimeric lactococcal bacteriocin whose primary target is not the plasma membrane. Microbiology. 1996, 142 (Pt 9): 2393-2398.
Martinez B, Rodriguez A, Suarez JE: Lactococcin 972, a bacteriocin that inhibits septum formation in lactococci. Microbiology. 2000, 146 (Pt 4): 949-955.
Martinez B, Bottiger T, Schneider T, Rodriguez A, Sahl HG, Wiedemann I: Specific interaction of the unmodified bacteriocin Lactococcin 972 with the cell wall precursor lipid II. Appl Environ Microbiol. 2008, 74: 4666-4670. 10.1128/AEM.00092-08.
Holo H, Nilssen O, Nes IF: Lactococcin A, a new bacteriocin from Lactococcus lactis subsp. cremoris: isolation and characterization of the protein and its gene. J Bacteriol. 1991, 173: 3879-3887.
Stoddard GW, Petzel JP, van Belkum MJ, Kok J, McKay LL: Molecular analyses of the lactococcin A gene cluster from Lactococcus lactis subsp. lactis biovar diacetylactis WM4. Appl Environ Microbiol. 1992, 58: 1952-1961.
van Belkum MJ, Kok J, Venema G, Holo H, Nes IF, Konings WN, Abee T: The bacteriocin lactococcin A specifically increases permeability of lactococcal cytoplasmic membranes in a voltage-independent, protein-mediated manner. J Bacteriol. 1991, 173: 7934-7941.
van Belkum MJ, Martin-Visscher LA, Vederas JC: Structure and genetics of circular bacteriocins. Trends Microbiol. 2011, 19: 411-418. 10.1016/j.tim.2011.04.004.
Sanchez-Hidalgo M, Montalban-Lopez M, Cebrian R, Valdivia E, Martinez-Bueno M, Maqueda M: AS-48 bacteriocin: close to perfection. Cell Mol Life Sci. 2011, 68: 2845-2857. 10.1007/s00018-011-0724-4.
Kemperman R, Kuipers A, Karsens H, Nauta A, Kuipers O, Kok J: Identification and characterization of two novel clostridial bacteriocins, circularin A and closticin 574. Appl Environ Microbiol. 2003, 69: 1589-1597. 10.1128/AEM.69.3.1589-1597.2003.
Kemperman R, Jonker M, Nauta A, Kuipers OP, Kok J: Functional analysis of the gene cluster involved in production of the bacteriocin circularin A by Clostridium beijerinckii ATCC 25752. Appl Environ Microbiol. 2003, 69: 5839-5848. 10.1128/AEM.69.10.5839-5848.2003.
Mohimani H, Kersten RD, Liu WT, Wang M, Purvine SO, Wu S, Brewer HM, Pasa-Tolic L, Bandeira N, Moore BS, Pevzner PA, Dorrestein PC: Automated Genome Mining of Ribosomal Peptide Natural Products. ACS Chem Biol. 2014, 9: 1545-1551. 10.1021/cb500199h.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ACL and SJP collected and analyzed the genome mining data. ACL, SJP and CH wrote the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2014_6858_MOESM1_ESM.docx
Additional file 1: Table S1: Genomes (finished and published) of anaerobic bacteria analyzed in this study. (DOCX 23 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Letzel, AC., Pidot, S.J. & Hertweck, C. Genome mining for ribosomally synthesized and post-translationally modified peptides (RiPPs) in anaerobic bacteria. BMC Genomics 15, 983 (2014). https://doi.org/10.1186/1471-2164-15-983
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-983