
Abstract
In recent years, genome mining has become a powerful strategy for the discovery of new specialized metabolites from microorganisms. However, the discovery of new groups of ribosomally synthesized and post-translationally modified peptides (RiPPs) by employing the currently available genome mining tools has proven challenging due to their inherent biases towards previously known RiPP families. In this chapter we provide detailed guidelines on using RiPPER, a recently developed RiPP-oriented genome mining tool conceived for the exploration of genomic database diversity in a flexible manner, thus allowing the discovery of truly new RiPP chemistry. In addition, using TfuA proteins of Alphaproteobacteria as an example, we present a complete workflow which integrates the functionalities of RiPPER with existing bioinformatic tools into a complete genome mining strategy. This includes some key updates to RiPPER (updated to version 1.1), which substantially simplify implementing this workflow.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
- Biosynthetic gene cluster
- RiPP
- Genome mining
- Bioinformatics
- Antibiotics
- Natural products
- Specialized metabolites
- Peptides
1 Introduction
Microbial specialized metabolites (also known as secondary metabolites or natural products) constitute an essential source of bioactive molecules for both pharmaceutical and agrochemical industries. In the last 15 years, traditional bioactivity-based discovery methods have found an invaluable sidekick in “genome mining” approaches that leverage affordable next-generation genome sequencing [1, 2]. These strategies are focused on the bioinformatic exploration of growing genomic databases for the identification of biosynthetic gene clusters (BGCs) responsible for making novel bioactive compounds, thus avoiding re-discovery of already known molecules.
Standard genome mining approaches have proven to be very effective for the identification of BGCs containing large modular biosynthetic systems, as in the case of polyketide synthases (PKSs) or non-ribosomal peptide synthetases (NRPSs ), as well as smaller BGCs containing characteristic enzymes (e.g., terpene synthases or cyclodipeptide synthases) [3,4,5]. However, it is much more challenging to use standard genome mining methods to comprehensively identify BGCs for ribosomally synthetized and post-translationally modified peptides (RiPPs) [6], one of the major classes of specialized metabolites. This is a consequence of: (a) the unrivalled chemical diversity of their biosynthetic pathways, and (b) a lack of unifying features that could be employed as general rules for their detection.
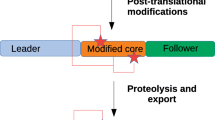
The biosynthesis of RiPPs is characterized by the use of a genetically encoded precursor peptide as a substrate for a series of chemical modifications catalyzed by clustered tailoring enzymes (Fig. 1). These are made to a core peptide region of the precursor peptide, which is usually at its C-terminus. The unmodified leader peptide region is then cleaved as a late-stage step in the pathway. Due to the high variability of their sequences (specific for each pathway) and their short length (usually <120 amino acids, AAs), the genes encoding these precursor peptides are sometimes not annotated in databases, so are not ideal starting points for the discovery of new RiPP BGCs. In addition, the accompanying tailoring enzymes are also specific for different RiPP subclasses or families.

Overview of RiPP biosynthesis
Different approaches have been recently developed for the discovery of new RiPP BGCs by genome mining in a user-friendly manner [3, 4, 7,8,9,10,11,12]. However, most are designed for the identification of BGCs belonging to specific RiPP families, searching for defined tailoring enzymes and considering certain pre-established sequence features for the correct identification of the precursor peptides. Although such specialized genome mining tools allow the reliable identification of RiPP BGCs, their inherent biases constrain the user’s freedom to explore truly new RiPP metabolic landscapes, thus limiting the likelihood of finding strikingly new RiPP chemistry.
We therefore developed RiPPER (RiPP Precursor Peptide Enhanced Recognition) as a RiPP genome mining tool to help overcome these limitations. This was achieved by providing the user with a highly customizable tool to explore genomic databases in a strongly bias-reduced manner [13]. RiPPER functions as a command line tool that employs its own RiPP-specific functionality alongside a series of pre-existing tools to retrieve and annotate precursor peptide candidates encoded within the same genomic locus as a user-defined tailoring enzyme.
RiPPER first uses RODEO2 [7] to retrieve a genomic region associated with a given tailoring enzyme as a GenBank file. RODEO2 was itself designed for the identification of RiPP BGCs, but it is used here to retrieve accessions and to generate an html output for the genomic locus. RiPPER then re-annotates open reading frames (ORFs) encoding short peptides (20–120 AAs) in the putative BGC using Prodigal-short, a modified version of the established gene finding tool, Prodigal [14] (Fig. 2). This is much more effective at identifying true coding regions compared to a crude 6-frame translation approach. RiPPER specifically searches for short ORFs in intergenic regions and re-annotates short peptides (<120 AAs) already annotated in the retrieved GenBank file. This yields a modified GenBank file that can be viewed in Artemis [15] (Fig. 2). Here, the gene encoding the “bait” tailoring protein is colored green, and the short peptides are color coded on a white-red scale for their Prodigal-short score, where Prodigal-short assigns a score to these ORFs based on numerous genetic factors. The top three scoring peptides are retained for downstream analysis, as are any additional ORFs that are over a defined score threshold (default = 7.5). This RiPPER output includes tables of numerous peptide parameters, such as distance and orientation of gene in relation to the bait gene, as well as a conserved domain search against both the Pfam database [16] and a NCBI database with conserved domains specifically for RiPP precursor peptides [17] (Fig. 2).

Overview of RiPPER 1.1 outputs using an input of accessions of cytochrome P450s with homology to the P450 in the tryptorubin BGC [18]. Dark grey arrows indicate automated steps in the RiPPER tool. The GenBank files are visualized in Artemis [15], and the tryptorubin-like precursor peptide is highlighted in red in the table and starred in the alignment
In this chapter, we will describe a workflow that combines RiPPER with a series of pre-existing bioinformatic functionalities, allowing identification of phylogenetic correlations between networks of homologous precursor peptide candidates and groups of homologous tailoring enzymes (Fig. 3). The RiPPER workflow requires more commitment from the user than web server-based genome mining tools, but its advantages are substantial. RiPPER facilitates the exploration of the RiPP diversity landscapes in a very flexible way as it allows the user to focus on any tailoring enzyme (well-characterized or suspected) as starting bait. We believe this is critical for the discovery of truly new RiPP chemistry. Therefore, instead of looking for predefined RiPP classes across a given genome, the RiPPER workflow described here allows exploration of the whole diversity of RiPP BGCs containing a given tailoring enzyme.

The biological logic of the RiPPER 1.1 mining workflow described in this chapter
We have now updated RiPPER in numerous ways to generate RiPPER 1.1. This includes the integration of sequence similarity networking by the EGN (Evolutionary gene and genome network) tool [19], which allows the user to rapidly identify groups of related precursor peptides. In the workflow we describe, the output is then assessed for the co-occurrence of tailoring enzymes with co-evolving candidate precursor peptides. Further data processing is then used to dissect this diversity into subgroups by comparing BGC architecture. We have found this approach invaluable for the identification of large families of novel RiPP BGCs, where the existence of conserved precursor peptides that belong to evolutionarily conserved gene clusters highlights the likelihood of new RiPP families.
2 Materials
-
1.
A PC, Macintosh or Linux computer with administrator privileges and an internet connection. The example reported here was carried out using a Mac with macOS Catalina (10.15), a 1.8 GHz (two cores) processor and 8 GB RAM. if a Windows PC is used, this requires Windows 10 Pro, Education or Enterprise for the installation of Docker. This requires a 64-bit processor and a minimum of 4 GB RAM. Hyper-V and Windows Container Features must be enabled. Approximately 14 GB hard drive is required for the storage of Docker images.
-
2.
Installed versions of Docker (https://www.docker.com/products/docker-desktop), Cytoscape (https://cytoscape.org), Microsoft Excel, and a text editor capable of displaying both Unix and DOS line endings correctly (we recommend Sublime Text or jEdit, which is free to download). The software versions we use in the steps below are Docker Desktop 2.2.0.3, Cytoscape v3.7.2, Excel 16.16.17, and Sublime Text 3.0. On a Windows PC the user must specify using Linux containers rather than using Windows containers upon installation of Docker. This is done by not clicking the box that says “use Windows containers instead of Linux containers”.
-
3.
Tools to generate and visualize phylogenetic trees. In the steps below, we use iToL [20] (https://itol.embl.de) and CIPRES Science Gateway [21] (https://www.phylo.org), which both require free online accounts.
3 Methods
The RiPPER-based genome mining strategy we will describe here involves several data processing steps (Fig. 3). The initial input consists of the accession of a single protein that is suspected or known to be involved in RiPP biosynthesis. In the method presented here, we use AAB17515.1, which is a TfuA domain protein from Rhizobium leguminosarum bv. trifolii. TfuA domain proteins function with YcaO domain proteins to catalyze thioamidation of peptide backbones [13, 22]. This was believed to be a very rare modification, but we had previously shown that TfuA domain proteins are widespread in novel RiPP BGCs in Actinobacteria [13]. In the steps below, as an example of how to operate with the RiPPER-based genome mining workflow, we provide a detailed summary of how to search for potential RiPP BGCs in Alphaproteobacteria using a TfuA domain protein.
This input accession can be used in a number of ways to retrieve related proteins, such as BLAST analysis to identify related proteins. Here, we will instead use the Conserved Domain Architecture Retrieval Tool (CDART) [23] to download all proteins with the same conserved domain. CDART can additionally filter by taxonomic classification if required (for example, if the user wants to focus on a particular area of biodiversity). As the number of retrieved proteins can potentially be enormous and difficult to handle, a trimming process is normally performed to reduce the redundancy of protein set. Here, the Enzyme Function Initiative-Enzyme Similarity (EFI-EST) tool is used [24]. A list of accession numbers corresponding to the proteins “baits” is now ready to be used with the RiPPER tool.
RiPPER uses this accession list to retrieve the genomic context of each of those proteins in a flexible manner, and annotates and scores the potential precursor peptides existing within a given genomic window around the bait homologue following a series of customisable parameters. Following RiPPER analysis, we will have a list of baits associated with three or more top-scoring co-occurring short peptides, as well as a GenBank files relating to each putative BGC.
The ultimate aim of the RiPPER-based strategy described here is to identify groups of related precursor peptides that have undergone co-evolution alongside groups of homologous bait proteins. The identification of conserved short peptides associated with conserved genomic loci suggests that potential precursor peptides are genuine, and potentially acted on by the bait proteins. It is possible that short peptides with other functions also associate with conserved BGCs, such as transcription factors and chaperones like PqqD [25]. Here, the conserved domain search for the retrieved short peptides assists the user. We have also recently updated RiPPER to automatically carry out sequence similarity networking analysis of the resulting short peptides by incorporating the EGN tool with predefined settings for short peptide networking. This provides a series of peptide networks, which should each contain closely related peptides, and this can all be visualized in Cytoscape [26]. In the workflow described below, a phylogenetic tree of the bait proteins is created. The association of networks of precursor peptides alongside defined branches of the phylogenetic tree is a strong indication of the evolution of a family of related RiPP BGCs and suggests that the precursor peptide and tailoring enzyme are co-evolving. We also describe how the architecture of the candidate BGCs can be explored using MultiGeneBlast [27], providing additional insight into their conservation and diversity. The software and bioinformatic tools associated with this workflow are summarized in Fig. 4.

Suggested workflow for identification of RiPP gene clusters and precursor peptides from genomes using bait proteins in RiPPER 1.1. Tools are shown in colored boxes, with arrows indicating the order of steps described in this chapter. Some steps are facilitated by the use of Excel formulas and/or usage of the command line; the formulas/code we use are presented in the text. Note, the presented workflow is suggested but is not the only possible means of conducting the analysis
3.1 Obtain an Input List of Protein Accessions
3.1.1 CDART
-
1.
Begin by selecting a desired bait protein, which you suspect may be involved in RiPP biosynthesis. In the example here, we use a TfuA domain protein, AAB17515.1, from the alphaproteobacterium Rhizobium leguminosarum bv. trifolii. Similar proteins can be found in other organisms using CDART from NCBI (https://www.ncbi.nlm.nih.gov/Structure/lexington/lexington.cgi).
-
2.
Within the CDART interface, use the “Filter your results” option (NCBI Taxonomy Tree setting) to limit the search to your organism(s) of interest and then select “Lookup sequences in Entrez”. Here, we have chosen to focus on proteins from Alphaproteobacteria.
-
3.
Download (“Send to” option) as an Accession List, and you can simply proceed to Section 3.2 if desired. However, we recommend the use of EFI-EST to reduce the dataset size by reducing bait protein redundancy. This is useful as it reduces the likelihood of a set of very closely related proteins from a highly sequenced species dominating the RiPPER output (see Note 1).
3.1.2 EFI-EST
-
1.
Use the sequence similarity networking tools at EFI-EST (https://efi.igb.illinois.edu/efi-est/) to reduce the size of the dataset, where the accession list is inputted via the Accession IDs input option. We generally use a 95% identity reduced dataset, where any sequences more than 95% identical are collapsed to a single representative sequence and accession. In this example, we used the default settings for the initial submission. EFI-EST will then require you to finalize the Sequence Similarity Networks (SSNs) by selecting size and alignment score cutoffs.
-
2.
It is advised within EFI-EST to select an alignment score corresponding to 35% similarity. In this example, we selected 120 AAs as the lower end cutoff, with no upper limit, and an alignment score threshold of 22.
-
3.
Once the SSN is finalized (see Note 2), download the 95% identity network and open it using Cytoscape.
-
4.
Export the associated attribute table as a .csv file, and open this using Excel. Find the column containing the list of accession IDs. In our experience, these are in a column called “Query IDs” or “Description” and include multiple IDs if the node in the network represents multiple proteins that are >95% identical.
-
5.
To obtain a list that includes one accession from each node, we do the following: copy this column to column A of a new Excel sheet, and extract the first accession from each row using the following Excel command in a different column of that new sheet, where “|” represents the symbol that follows the first accession. In some examples, this is instead a space (“ ”, see Note 3):
= IF(ISERR(FIND("∣",A2)),A2,LEFT(A2,FIND("|",A2)-1))
-
6.
Apply this to all rows, thereby generating a new column with only the one accession per row. Now copy this column and paste it into a new .txt file, generating a shortened accession list in which entries share less than 95% identity to use as input for RiPPER and phylogenetic analysis. Note that Section 3.4 (phylogenetic analysis of bait proteins) can take significant time to compute and only requires this input accession list, so can be started prior to Sections 3.2 and 3.3 if the user prefers.
3.2 Short Peptide Searching and Networking Using RiPPER 1.1
3.2.1 BGC Retrieval and Short Peptide Searching
-
1.
Online details on how to use RiPPER 1.1 are found at https://github.com/streptomyces/ripper. We highly recommend that it is run from a Docker container, which includes all dependencies and can be run from Linux/Mac/Windows systems. Docker must be installed and running before RiPPER 1.1 can be used (https://www.docker.com/get-started). To install RiPPER 1.1, open the command line interface (terminal in Mac, command prompt in Windows) with Docker running and use the command:
docker pull streptomyces/ripdock
-
2.
Following installation, run the container using the following command, where your input accession list is stored in “filepath”. Do not change /home/mnt as this refers to a directory in the container and is required for the scripts to run:
# Example usage on Linux / Mac
docker run -it -v /filepath:/home/mnt streptomyces/ripdock
# Example usage on MS Windows.
docker run -it -v C:/filepath:/home/mnt streptomyces/ripdock
-
3.
Now you can run RiPPER 1.1 on your accession list, to search for short peptides co-occurring with your bait protein and obtain all RiPPER output files (see summary below). RiPPER 1.1 will retrieve short peptides based on a set of parameters that have sensible defaults for RiPP analysis (Fig. 2): using Prodigal-short, peptides between 20 AA (minPPlen) and 120 AA (maxPPlen) in length are searched for within a 17.5-kb window (flanklen) either side of the bait protein, where a score boost of 5 (sameStrandReward) is included if the peptide is encoded on the same strand as the bait protein, which is a common feature in RiPP BGCs. Peptides within a window of 8 kb (maxDistFromTE) either side of the bait protein are considered for the output peptide list, and three are retrieved (fastaOutputLimit) based on the Prodigal-short score ranking. Additional peptides are retrieved if they have a score above 7.5 (prodigalScoreThresh). We use these defaults for the analysis described here, but these parameters can be modified (see Note 4). Assuming your list of bait proteins is stored in a file called “Accession_IDs.txt”, use the following command to run RiPPER:
./ripper_run.sh /home/mnt/Accession_IDs.txt
-
4.
RiPPER 1.1 may take several hours to run, due to the time required to download GenBank files from NCBI sequentially based on the accession ID list, which requires a stable internet connection (see Note 5).
-
5.
A number of output files and folders will be generated:
orgnamegbk: A folder containing GenBank files for all retrieved gene clusters. These files are named by the host organism and the input accession ID.
out.txt: This is a tab-delimited file that reports peptide data for all peptides retrieved by RiPPER (as defined by the analysis parameters above) for all gene clusters. This includes various associated data for the peptides, including sequence, species, Prodigal-short score, Pfam domain, and distance from the bait protein.
distant.txt: This is a tab-delimited file that reports peptide data for all peptides retrieved by RiPPER via a precursor peptide Hidden Markov Model (HMM) search across the full size of the retrieved gene cluster (i.e., ignores maxDistFromTE), and only reports peptides that were not identified in the original RiPPER search. Peptide identifiers are unique from out.txt, so these text files can be combined for downstream analysis. This is useful for retrieving peptides from large RiPP BGCs (e.g., thiopeptides).
out.faa: A fasta file for peptides reported in out.txt that is formatted as an input file for similarity networking using EGN. An equivalent distant.faa file is also provided.
rodeohtml: A folder containing RODEO [8] html output files for all retrieved gene clusters. Additional RODEO data is provided in a separate rodout folder. The data stored in rodout can be further analyzed via approaches described on the RODEO website (http://ripp.rodeo/advanced.html).
pna: A folder containing the output of EGN-based sequence similarity networking, which includes files for visualization in Cytoscape (see below).
Networks: A folder containing subfolders (e.g., “Network1”, “Network2”) that contain GenBank files associated with each peptide network identified by EGN networking (see below).
-
6.
Following the RiPPER run, exit the Docker container by typing “exit”.
3.2.2 Short Peptide Networking
-
1.
It is possible to utilize the output of RiPPER directly, for example, by looking at the .gbk files stored in orgnamegbk, but in earlier studies [13] we found it very helpful to generate sequence similarity networks (SSNs) of the output short peptides using EGN [19]. This step identifies families of related short peptide sequences, which can highlight the existence of conserved RiPP-like BGCs [13]. Other SSN software may be used, but we find that the customisable nature of EGN works best with the short sequences generated in this analysis.
-
2.
To significantly simplify the workflow, we have now improved RiPPER to incorporate an automated EGN-based networking analysis. The default parameters we have defined within RiPPER are that peptides are networked if they have over 40% identity and there is over 40% sequence coverage, with a minimum alignment region of 14 AA. These are relatively permissive networking settings, but our testing indicates they are highly effective at grouping families of related precursor peptides. An advanced user may wish to use EGN themselves to assess alternative settings [19]. The EGN output is provided in folder “pna.” Within this folder the network file is found in /GENENET_10.40.40.0.0/CYTOSCAPE as a file titled “cc_1.to.X.txt”, which in our example is “cc_1.to.78.txt”.
-
3.
This should be imported into Cytoscape to visualize the series of networks generated with the short peptides found by RiPPER (Fig. 5). Open Cytoscape and import the file “cc_1.to.X.txt” by dragging it into the network panel on the left.
-
4.
Following this, the Cytoscape attributes file outputted by RiPPER (“cytoattrib.txt” in “pna” folder) can be imported into Cytoscape by dragging into the table panel along the bottom. Mapping attributes to the network means that the nodes are associated with the information provided in the RiPPER output, such as peptide sequence, Prodigal-short score, and conserved domain. This is therefore a suitable interface for assessing the RiPPER output.
-
5.
The network can now be colored by using the “Style” pane. Under the “Fill Color” option, select “Column: Color” and “Mapping type: Passthrough Mapping”. The networks will now be color-coded the same as in a phylogenetic tree that is color-coded in Section 3.5 (Fig. 5). Node formatting can also be used to highlight numerous other peptide features (see Note 6).
-
6.
Save a graphic of the networks using “Export”—“Network to Image” (.png) and export the attributes table as a .csv file for use in Sections 3.3 and 3.4.

The short peptide network output when using alphaproteobacterial TfuA proteins as the input is described in this chapter. The network file is found in the /GENENET_10.40.40.0.0/CYTOSCAPE/ folder and then recolored using the “cytoattrib.txt” file
3.3 Assessing Conservation of BGC Architecture
The retrieved GenBank files, peptide sequences, and networks constitute a rich dataset for downstream BGC analysis. In this step, conservation within putative BGCs will be assessed, as will sequence conservation among networked peptides. We use MultiGeneBlast (MGB) [27] to assess for conservation of genes within the retrieved BGCs. MGB uses multiple proteins in a GenBank file as the query sequences to performs BLAST analysis against multiple other GenBank files. This can help determine whether retrieved genomic loci represent conserved BGCs, and also help identify the boundaries of the cluster (Fig. 6). Other options are available that can perform similar analyses, such as CORASON [28] and BiG-SCAPE [28], but we prefer the output provided by MGB.

MultiGeneBlast (MGB) output for genes surrounding a selected network of putative precursor peptides (Network 2 of our analysis). Analysis of surrounding genes using MGB can give clues as to whether the identified putative precursor peptides and associated bait proteins are part of a genuine RiPP BGC or not. Conservation of a set of genes for many or all members of a peptide network is suggestive of a real BGC. This analysis can also help identify the boundaries of the BGC
3.3.1 MultiGeneBlast (MGB)
-
1.
MGB can be very useful to use on genomic loci associated with each network of short peptides. To achieve this, the GenBank files outputted by RiPPER are divided into different networks identified in Section 3.2. RiPPER now does this automatically for GenBank files associated with the top 30 networks, which are saved in folders for each network in /Networks/NetworkX (e.g., “Network1”, “Network2”).
-
2.
MGB for Windows is currently only available as 32-bit software, which is incompatible with modern Windows operating systems. We have therefore generated a Docker image for MGB which contains the 64-bit Linux version of MGB, which we recommend is used by both PC and Mac users. This can be used as follows. Start Docker desktop, open a new terminal window, and type the command:
docker pull streptomyces/multigeneblast
-
3.
Start the Docker container as below, where “filepath” relates to the path to the “Networks” folder (or equivalent if the user moves the “NetworkX” folders):
docker run -it -v /filepath:/home/work streptomyces/multigeneblast
-
4.
MGB requires a custom database to be searched against. Therefore, make a database for each relevant network with the following command (DBnetwork_X = database name, Network_folder = folder with desired GenBank files):
makedb DBnetwork_X Network_folder
Example:
makedb DBnetwork_2 Network2
Where the files for creating the “DBnetwork_2” database are GenBank format files from the RiPPER output, stored in the directory /Networks/Network2.
-
5.
You can then run MGB using the following commands (“DBnetwork_X” = database previously generated; Network_folder/example.gbk = query file; “mgbout” = destination folder name for MGB results). This assumes the query file is in a “NetworkX” folder, so specify a different path if this is not the case:
multigeneblast -db DBnetwork_X -in Network_folder/example.gbk -from 0 -to 35000 -out mgbout
Example:
multigeneblast -db DBnetwork_2 -in Network2/WP_003184680.1.gbk -from 0 -to 35000 -out Network2_ WP_003184680_MGB
-
6.
The folder “Network2_ WP_003184680_MGB” will be created, which contains .svg files that visualize each GenBank file compared to your query GenBank file, and a combined .svg file with all gene clusters compared to the query (Fig. 6). .xhtml files are also outputted that can be viewed in a web browser. This provides an interactive color-coded visualization of all gene clusters compared to the query file. The user must ensure the output folder name is changed in the command when running another search, or the folder will be overwritten.
-
7.
Following MGB analysis, exit the Docker container by typing “exit”.
3.3.2 Peptide Alignment
-
1.
It can be informative to directly assess the sequence conservation of peptides that belong to a given network (Fig. 6). This step can help identify potential outliers within a Network. Network-specific FASTA files are generated automatically by EGN running within RiPPER (saved in /pna/GENENET_10.40.40.0.0/FASTA/) and are named “p_ccX.faa”, where X is the network number.
-
2.
These can therefore be used directly as the input for multiple sequence alignment using MUSCLE (https://www.ebi.ac.uk/Tools/msa/muscle/).
3.4 Building Phylogenetic Tree of Bait Proteins
Building a phylogenetic tree of your selected bait proteins (TfuA domain proteins in Alphaproteobacteria in this example) can help illustrate the diversity of these proteins in your dataset, and aids downstream identification of evolutionarily associated BGCs.
3.4.1 Batch Entrez
-
1.
The first step is to retrieve sequences for all the proteins in your accession list. We do this using the Batch Entrez tool from NCBI (https://www.ncbi.nlm.nih.gov/sites/batchentrez). Simply upload the 95% identity filtered list that was generated in Section 3.1, and click “retrieve”, making sure to select the “protein” option from the dropdown menu. This results in a list of these proteins on the NCBI website. Simply click the “Send to” option, select “File”, and change format to “FASTA”, then “Create file.” This will download the full list in FASTA format with amino acid sequences for each accession ID in the list. We will use this as an input to create the phylogenetic tree.
3.4.2 CIPRES
-
1.
We use the CIPRES Science Gateway (https://www.phylo.org) to create a phylogenetic tree of our sequences. Users will need to create a free account in order to use the service.
-
2.
Create a new project and upload your FASTA file from the above step in the “Data” subfolder. In the “Tasks” subfolder, create a new task with your FASTA file as the input, and use the “Muscle” tool to perform an alignment. Ensure that sufficient time is allocated via the maximum run time setting, then save and run the analysis. We use default alignment parameters here.
-
3.
Once the alignment has run, download the “output.fasta” file, and upload this in the “Data” subfolder as before.
-
4.
Then create a new task to build the phylogenetic tree, selecting the “output.fasta” file as input. We find the “RAxML-HPC2 on XSEDE” tool works well, which we use with default parameters in this example, ensuring that sufficient time is allocated via the maximum run time setting and that “Protein” is set for Data Type.
-
5.
Once this has run, download the “RAxML_bestTree” output file. This can then be imported directly into Interactive tree of Life (iTOL, https://itol.embl.de) [20].
3.5 Visualize the Integrated Output
3.5.1 Mapping Short Peptide Networks to Protein Clades
-
1.
In order to map short peptide networks to protein clades in iTOL, it is necessary to make use of iTOL definition files. A template is available at https://itol.embl.de/help.cgi as a file “colors_styles_template.txt”, which should be downloaded. To convert the data into the right format to import into iTOL requires processing using Microsoft Excel and a text editor, such as Sublime Text.
Start by opening the network attribute table .csv file from Section 3.2. Cluster 1 peptides (cc1 in “Network” column) occur in rows 2 to X (2–67 in our example), all cluster 2 peptides (cc2) in rows X + 1 to Y (68–91 in our example), and so on. Save as an Excel Workbook (e.g., “iTOL_definitions.xlsx”) and create a new sheet with the following columns defined: “Accession” (column A), “Full Accession” (B), “Color type” (C), “Color” (D), “Style” (E), “Network” (F). Copy and paste columns “Accession”, “Color”, and “Network” from the original sheet into the relevant columns. The “Full Accession” column is required to generate an accession code format that matches the format in the tree. The following Excel command is therefore used in cell B2 and then applied to all rows in the column:
= A2&".1"
“Color type” has multiple options, but we find “branch” works well; simply type “branch” in the top row for the “Color type” column, and use the fill down option to apply this to all rows. For “Style”, simply type “normal” into the top row and use the fill down option to apply it to all rows.
-
2.
Once this is done, columns B–E for the relevant networks can be pasted into the bottom of the iTOL-supplied “colors_styles_template.txt” file below “DATA”. It will also be necessary to change the separator value in the template file to TAB; do this by typing “#” before the “Space” and “Comma” options, and remove the “#” before “Tab”; failing to do so will render iTOL unable to locate the nodes in the tree to assign colors to.
3.5.2 Final Output
-
1.
The above steps provide a large amount of data that simply originates from a single input bait protein: (i) 35 kb GenBank files centered on putative BGCs with short peptides scored and visualized; (ii) tabulated data for short peptides and their associated attributes, including conserved domains; (iii) Peptide networks that highlight conserved sets of peptides across multiple BGCs; (iv) MultiGeneBlast comparison of BGCs for specified networks; (v) A phylogenetic tree of non-redundant homologues of the original bait protein. This data can now be integrated and visualized.
-
2.
Firstly, to recolor the tree based on peptide networks, the iTOL definition file(s) generated above are simply dragged into a web browser while the phylogenetic tree is open in iTOL. The branches of the bait proteins that have nearby short peptides belonging to a given network will be automatically recolored (Fig. 7). Some networks may map to monophyletic groups, while others may not. The recolored tree can be exported as a .png file.
-
3.
It can be useful to investigate the MultiGeneBlast output to view the surrounding genes to help explain this. Graphics generated in Section 3.2 (Cytoscape-generated network images) and Section 3.3 (MultiGeneBlast outputs and peptide sequence alignments) can then be visualized alongside the colored tree using software such as Microsoft PowerPoint or Adobe Photoshop (Fig. 7).

Final RiPPER workflow output combining bait protein phylogenetics, precursor peptide identification, and genetic architecture investigation. The final output shows a phylogenetic tree of supplied bait proteins across a user-defined phylogenetic group, onto which are mapped networks of similar putative RiPP precursor peptides identified by RiPPER. MGB can then inform the user whether the surrounding gene architecture is conserved, potentially supporting the hypothesis that these are real RiPP biosynthetic gene clusters
Where closely related bait proteins are associated with similar short peptides, and the surrounding gene architecture is highly conserved, it highlights the possibility that this represents a genuine RiPP biosynthetic gene cluster that is conserved across multiple species (Fig. 7).
4 Notes
-
1.
Bait proteins from heavily sequenced species, such as widely studied clinical pathogens, can potentially distort the output, as proteins with almost identical sequences are very likely to retrieve the same precursor peptide and BGC via RiPPER, and will also be over-represented in phylogenetic analyses. This can lead to an overestimation of the natural prevalence of the resulting RiPP BGC family. These problems are solved by using an identity cutoff to reduce redundancy. We use 95% identity to filter any proteins that are almost identical, but lower cutoffs can be used, especially if the input protein list is very large. EFI-EST provides the option to filter based on numerous percentage identity cutoffs. The user is warned that the use of a lower cutoff (e.g., 70% identity) will likely mean that RiPP diversity is reduced in the output.
-
2.
In our experience, there can sometimes be a disparity between the number of NCBI accessions and the number of proteins that are used for network analysis in EFI-EST. This is due to issues with matching NCBI accessions with UniProt IDs. This can be overcome by using the FASTA input option in EFI-EST instead. See Section 3.4.1 for instructions on how to generate a FASTA file from an accession list via Batch Entrez.
-
3.
Excel formulas provided are tailored towards the example presented. Most will work as presented with data from the RefSeq database. A simple modification to the presented formula = IF(ISERR(FIND(“|”,A2)),A2,LEFT(A2,FIND(“|”,A2)-1)) would be to change the “|” character to whichever character is used to separate the accession IDs in the Cytoscape output “Query IDs” column.
-
4.
The RiPPER parameter file is called “local.conf”. If you would like to modify this, once the Docker container is running, copy it into the working directory (i.e., “filepath”) on your computer using the following command:
cp local.conf /home/mnt
Modify and save this locally stored file using a text editor and then use the following command to copy it back into the working directory for RiPPER:
cp /home/mnt/local.conf /home/work
Following this step, we recommend testing a small subset of input accessions to ensure parameters have been properly modified.
-
5.
Occasionally, RiPPER may fail to retrieve a sequence for a given accession, so an error will be listed on the command line, and RiPPER will then proceed to the next accession. This can occur if the sequence is corrupted, if there is an error with the accession ID (for example, a nucleotide accession is used, an accession ID has changed, or there is no nucleotide accession associated with the protein), if a lapse in internet connection occurs or if there is a problem with the NCBI server. At present, RiPPER does not attempt to re-run these at the end of the analysis, and they will be absent from your dataset. However, this will be addressed in a future version of RiPPER.
-
6.
In our experience, Cytoscape visualization is a powerful tool to assess the properties of RiPPER-identified peptides. For example, as an alternative to coloring by network, color-coding and/or node size can be used to assess Prodigal score (using Continuous Mapping to visualize low to high scores), conserved domains (“hdesc” using Discrete Mapping), or genus (Discrete Mapping).
Change history
29 September 2021
In the original version of this book, chapter 14 was published non-open access. It has now been changed to open access under a CC BY 4.0 license and the copyright holder has been updated to “The Author(s).” This book has also been updated with these changes.
References
Katz L, Baltz RH (2016) Natural product discovery: past, present, and future. J Ind Microbiol Biotechnol 43:155–176
Genilloud O (2019) Natural products discovery and potential for new antibiotics. Curr Opin Microbiol 51:81–87
Medema MH, Blin K, Cimermancic P et al (2011) antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39:W339–W346
Blin K, Shaw S, Steinke K et al (2019) antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res 47:W81–W87
Skinnider MA, Merwin NJ, Johnston CW et al (2017) PRISM 3: expanded prediction of natural product chemical structures from microbial genomes. Nucleic Acids Res 45:W49–W54
Montalbán-López M, Scott TA, Ramesh S et al (2021) New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep. 38: 130–239 https://doi.org/10.1039/d0np00027b
Tietz JI, Schwalen CJ, Patel PS et al (2017) A new genome-mining tool redefines the lasso peptide biosynthetic landscape. Nat Chem Biol 13:470–478
Schwalen CJ, Hudson GA, Kille B et al (2018) Bioinformatic expansion and discovery of thiopeptide antibiotics. J Am Chem Soc 140:9494–9501
Li J, Qu X, He X et al (2012) ThioFinder: a web-based tool for the identification of thiopeptide gene clusters in DNA sequences. PLoS One 7:e45878
Agrawal P, Khater S, Gupta M et al (2017) RiPPMiner: a bioinformatics resource for deciphering chemical structures of RiPPs based on prediction of cleavage and cross-links. Nucleic Acids Res 45:W80–W88
Skinnider MA, Johnston CW, Edgar RE et al (2016) Genomic charting of ribosomally synthesized natural product chemical space facilitates targeted mining. Proc Natl Acad Sci U S A 113:E6343–E6351
Mohimani H, Kersten RD, Liu W-T et al (2014) Automated genome mining of ribosomal peptide natural products. ACS Chem Biol 9:1545–1551
Santos-Aberturas J, Chandra G, Frattaruolo L et al (2019) Uncovering the unexplored diversity of thioamidated ribosomal peptides in Actinobacteria using the RiPPER genome mining tool. Nucleic Acids Res 47:4624–4637
Hyatt D, Chen G-L, LoCascio PF et al (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119
Carver T, Harris SR, Berriman M et al (2012) Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28:464–469
Finn RD, Coggill P, Ry E et al (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279–D285
Haft DH, DiCuccio M, Badretdin A et al (2018) RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res 46:D851–D860
Reisberg SH, Gao Y, Walker AS et al (2020) Total synthesis reveals atypical atropisomerism in a small-molecule natural product, tryptorubin A. Science 367:458–463
Halary S, McInerney JO, Lopez P et al (2013) EGN: a wizard for construction of gene and genome similarity networks. BMC Evol Biol 13:146
Letunic I, Bork P (2019) Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47:W256–W259
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES science gateway for inference of large phylogenetic trees. In: 2010 gateway computing environments workshop (GCE). IEEE, New Orleans, LA, pp 1–8
Mahanta N, Liu A, Dong S et al (2018) Enzymatic reconstitution of ribosomal peptide backbone thioamidation. Proc Natl Acad Sci U S A 115:3030–3035
Geer LY, Domrachev M, Lipman DJ et al (2002) CDART: protein homology by domain architecture. Genome Res 12:1619–1623
Gerlt JA, Bouvier JT, Davidson DB et al (2015) Enzyme function initiative-enzyme similarity tool (EFI-EST): a web tool for generating protein sequence similarity networks. Biochim Biophys Acta 1854:1019–1037
Latham JA, Iavarone AT, Barr I et al (2015) PqqD is a novel peptide chaperone that forms a ternary complex with the radical S-adenosylmethionine protein PqqE in the pyrroloquinoline quinone biosynthetic pathway. J Biol Chem 290:12908–12918
Shannon P, Markiel A, Ozier O et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Medema MH, Takano E, Breitling R (2013) Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol Biol Evol 30:1218–1223
Navarro-Muñoz JC, Selem-Mojica N, Mullowney MW et al (2020) A computational framework to explore large-scale biosynthetic diversity. Nat Chem Biol 16:60–68
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this protocol

Cite this protocol
Moffat, A.D., Santos-Aberturas, J., Chandra, G., Truman, A.W. (2021). A User Guide for the Identification of New RiPP Biosynthetic Gene Clusters Using a RiPPER-Based Workflow. In: Barreiro, C., Barredo, JL. (eds) Antimicrobial Therapies. Methods in Molecular Biology, vol 2296. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-1358-0_14
Download citation
DOI: https://doi.org/10.1007/978-1-0716-1358-0_14
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-1357-3
Online ISBN: 978-1-0716-1358-0
eBook Packages: Springer Protocols