
Abstract
Background
Amur ide (Leuciscus waleckii) is an economically and ecologically important cyprinid species in Northern Asia. The Dali Nor population living in the soda lake Dali Nor can adapt the extremely high alkalinity, providing us a valuable material to understand the adaptation mechanism against extreme environmental stress in teleost.
Results
In this study, we generated high-throughput RNA-Seq data from three tissues gill, liver and kidney of L. waleckii living in the soda lake Dali Nor and the fresh water lake Ganggeng Nor, then performed parallel comparisons of three tissues. Our results showed that out of assembled 64,603 transcript contigs, 28,391 contigs had been assigned with a known function, corresponding to 20,371 unique protein accessions. We found 477, 2,761 and 3,376 differentially expressed genes (DEGs) in the gill, kidney, and liver, respectively, of Dali Nor population compared to Ganggeng Nor population with FDR ≤ 0.01and fold-change ≥ 2. Further analysis revealed that well-known functional categories of genes and signaling pathway, which are associated with stress response and extreme environment adaptation, have been significantly enriched, including the functional categories of “response to stimulus”, “transferase activity”, “transporter activity” and “oxidoreductase activity”, and signaling pathways of “mTOR signaling”, “EIF2 signaling”, “superpathway of cholesterol biosynthesis”. We also identified significantly DEGs encoding important modulators on stress adaptation and tolerance, including carbonic anhydrases, heat shock proteins, superoxide dismutase, glutathione S-transferases, aminopeptidase N, and aminotransferases.
Conclusions
Overall, this study demonstrated that transcriptome changes in L. waleckii played a role in adaptation to complicated environmental stress in the highly alkalized Dali Nor lake. The results set a foundation for further analyses on alkaline-responsive candidate genes, which help us understand teleost adaptation under extreme environmental stress and ultimately benefit future breeding for alkaline-tolerant fish strains.
Similar content being viewed by others


Background
Amur ide (Leuciscus waleckii) belongs to the family of cyprinid, inhabiting the Heilongjiang (Amur) River basin in Russia, Mongolia, China and Korea. Although L. waleckii inhabits fresh water in rivers, streams and lakes, it also has great tolerance on high salinity and alkalinity (http://www.fishbase.org). As an extreme instance, L. waleckii inhabiting Dali Nor lake, Inner Mongolia (E116o25′-116o45′,N43o13′-43o23′) can survive in water of ultra-high alkalinity up to pH 9.6. Dali Nor lake is a typical saline-alkaline lake with high concentrations of carbonate salts. It locates in an endorheic basin on eastern Inner Mongolia Plateau. The evaporation is greater than precipitation and inflows, making the lake shrink consistently from 1600 to less 200 km2 since early Holocene (11,500–7,600 cal yr BP). The alkalinity and salinity are increasing steadily [1]. Currently the pH value ranged from 8.25 to 9.6, with the alkaline content (ALK) over 50 mg/L and the salinity around 6 ‰. Combining geological and biological evidence, it’s commonly believed that L. waleckii population in Dali Nor lake were used to be fresh water fish that evolved fast in the past several thousand years and developed great tolerance on high alkalinity [2, 3].
L. waleckii is economically important to local Mongolian who live around the Dali Nor lake, and ecologically important to wild birds on their migration journeys from Siberia to the south which feed on L. waleckii as major food source [4]. In spite of economic and ecological importance, the mechanism of its high tolerance on alkalinity is still a puzzle. Very limited physiological and genetic studies had been performed, and rare genetic resources had been developed. So far, only a few genetic markers had been developed for population genetics evaluation and phylogeny analysis [3, 5]. Mitochondrial genome had been completely sequenced and annotated, providing basic molecular tools for ecological and genetic study [6]. Scientists are paying more attention to L. waleckii with gradually recognized importance. Recently, high throughput transcriptome sequencing was performed on Illumina platform and analyzed, providing the genomic basis for further investigation of the mechanism of its alkaline tolerance [7]. L. waleckii has been recently developed as potential aquaculture species in the widely distributed saline and alkaline water in northern China. The breeding program also eagerly desires better understanding of its physiological and genetic basis of the tolerance adaptation and stress resistant on alkaline environment. Besides, scientists are also interested on the mechanism of microevolution on L. waleckii which evolved fast to adapt paleoenvironmental changes since early Holocene.
Comparative study between organisms inhabiting distinct environments could provide insight into the mechanism that responding to the environmental difference. In some cases, scientists apply artificial treatments to create the difference in the experiments, and facilitate the comparison [8, 9]. To better understand the physiological and genetic changes and mechanism of alkaline tolerance and adaptation in L. waleckii, comparative analysis between the fish living in alkaline water and fresh water is the efficient method. Fortunately, there is a sister lake of Dali Nor called Ganggeng Nor, which is fresh water lake and connected to Dali Nor through the short Shali river. L. waleckii also inhabits the fresh water of Ganggeng Nor lake. There is frequent genetic communication between the population in Dali Nor lake (alkaline water type, AW) and those in Ganggeng Nor lake (fresh water type, FW) through anadromous spawning migration annually. Both types of L. waleckii are derived from same ancestors and have consistent genetic background, which provide us unique natural samples to explore gene expression changes in response to high alkaline environment.
Transcriptome profiling and differential gene expression analyses traditionally use microarray technology, which requires cDNA library, Expressed Sequence Tags (EST) dataset and array hybridization. With the emerging of the next generation sequencing, RNA sequencing (RNA-Seq) is relatively new technology for transcriptomic study across the whole genome. Comparing to traditional cDNA microarray, RNA-Seq provides deep sequencing data for direct quantification of transcripts, which is more sensitive to detect all expressed genes without the hassles of EST collection, probe synthesis, microarray design and hybridization [10, 11]. In the past several years, RNA-Seq has been widely used in many teleost for differential gene expression analysis in various organisms. For instance, RNA-Seq were used to unveil gene expression differences in response to various pathogenic challenge in Lateolabrax japonicas[12], catfish (Ictalurus punctatus) [13, 14], Grouper (Epinephelus spp.) [15], European sea bass (Dicentrarchus labrax) [16] and Asian sea bass (Lates calcarifer) [8]. It was even used to quantify the gene expression changes in Fundulus grandis in the Gulf of Mexico to evaluate the impact of oil contamination after the disaster of Deepwater Horizon drilling platform [17]. Gene expression changes responding to abiotic stress are generally very significant comparing to those control counterparts. Thus, RNA-Seq was also used to profile DEGs and pathways under certain environmental stress. For instance, drought-responsive genes were identified and analyzed using RNA-Seq to compare drought-treated and well-watered fertilized ovary and basal leaf meristem tissue [18]. Gene expression changes in response to extreme dehydration on Belgica Antarctica were characterized using RNA-Seq, unveiling the tolerance mechanisms on dehydration in Antarctic insect [19]. RNA-Seq results also revealed gene expression changes in various metabolic pathways in response to osmotic stress and exogenous abscisic acid challenge, providing global gene expression overview of drought stress sorghum [20].
In this study, we use RNA-Seq to investigate the genome-wide gene expression differences in L. waleckii population inhabiting soda water of Dali Nor lake and their sister population inhabiting fresh water of Ganggeng Nor lake. Gene expression changes are identified from whole transcriptome background. Our study highlights those reactive pathways in response to high alkaline stress by using gene ontology and pathway analysis. This study provides us useful information to explain mechanism of alkaline stress tolerance in teleost.
Results and discussion
RNA-Seq data processing, reference assembly and alignment
To provide comprehensive understanding of the expression difference between L. waleckii inhabiting AW and FW, we collected and deeply sequenced the RNA samples from liver, kidney and gill. A total of 187,430,252 paired-end reads were generated from six samples with 101-bp read length. The number of sequences from each sample ranged from 28.7 to 35.7 million. After removal of ambiguous nucleotides, low-quality sequences (Phred quality scores < 20), contaminated microbial sequences, ribosomal RNA sequences, a total of 154,265,700 cleaned reads (82.3%) were harvested for further analysis. The cleaned sequences of each sample ranged from 23.7 to 29.1 million reads, showing the stability and consistence on sampling, library preparation and sequencing. Cleaned RNA-Seq reads of six samples were mapped to assembled transcriptome reference by using the ultrafast short read aligner Bowtie (version 0.12.3) [21]. The mapping ratio ranged from 80.8% to 88.1% with an average of 84.1%. All RNA-Seq data in this study have been deposited in the NCBI SRA database (Accession number SRR949612) (Table 1).
The cleaned reads of six samples were pooled and assembled by using Trinity assembler [22] to generate the transcriptome reference. As shown in Table 2, the trancriptome were assembled into 64,603 contigs, ranging from 201 to 16,177 bp in length. The average length is 879 bp, N50 length is 1,776 bp and median length is 404 bp. The contig length distribution was shown in Figure 1. We then annotated assembled contigs to provide expression background and facilitate the functional analysis of DEGs. We compared our assembly with three protein databases, including NCBI non-redundant (nr) protein database, uniprot database, and zebrafish reference protein database, by using BLASTx with e-value cutoff of 1e-10. A total of 28,391 contigs have significant hit at least in one database, corresponding to 20,371 unique protein accessions (Table 2). Gene ontology (GO) analysis was conducted to assign GO term to each of those 20,371 unique proteins. A total of 14,326 unique proteins were assigned at least one GO term for describing biological processes, molecular functions and cellular components, corresponding to 22,460 assembled contigs (Table 3).

Length distribution of assembled transcriptome contigs of L. waleckii .
Identification of differentially expressed genes
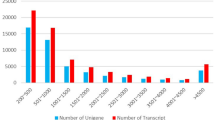
We found 477, 2,761 and 3,376 DEGs in the gill, kidney, and liver, respectively, of AW population compared to FW population with FDR ≤ 0.01 and fold-change ≥ 2 (Figure 2). M-A plots were drafted using “eps” format files as shown in Figure 3. Of these differentially expressed genes, 154, 1,087, 1,949 genes showed higher expression in gill, kidney, and liver of the AW population, respectively; and 323, 1,674, 1,427 genes showed higher expression in gill, kidney, and liver of the FW population, respectively. Of these, 127, 64, 314 genes were exclusively expressed in gill, kidney, and liver of the AW population, and 85, 335, 125 genes were exclusively expressed in in gill, kidney, and liver of the FW population (Additional file 1: Table S1). Venn diagram of the DEGs illustrated that majority of these genes were not shared in three tissues, suggesting that the mechanism and pathways in response to alkaline stress are significant different in gill, kidney, and liver (Figure 4).

DEGs in three tissues between AW and FW for L. waleckii .

M-A plots showing gene expression in three tissues. (A) M-A plot showing gene expression in liver; (B) M-A plot showing gene expression in kidney; (C) M-A plot showing gene expression in gill.

Venn diagram of DEGs among three tissues.
To validate RNA-Seq results, 35 genes with high level of significance or important stress-responding functions were selected for qRT-PCR analysis with beta-actin as reference gene. Primers for all genes are listed in Additional file 2: Table S2. Overall, the expression patterns of 30 genes were in agreement across the RNA-Seq and qRT-PCR analyses with minor differences in the expression level (Figure 5). There were only 5 genes that not showed the consistency of expression in the two assays. Thus, these genes showed similar patterns of mRNA abundance in RNA-Seq analysis and qRT-PCR, validated the genome-wide expressed profiling in gill, kidney, and liver in response to AW stress.

Differentially expressed genes validated by qRT-PCR. Comparison between RNA-Seq results and qRT-PCR validation results. X-axis shows genes in three tissues validated in this study; Y-axis shows Log2Ratio of expression of AW (alkaline water) versus FW (fresh water).
Functional analysis on differential expressed genes in gill
In response to AW stress, we observed significant gene enrichment of several Gene Ontology (GO) terms in gill that related to stress response. These GO terms include “transcription regulator activity (GO:0030528)”, “metabolic process (GO:0008152)” and “cell communication (GO:0007154)” in up-regulated genes, and “response to stimulus (GO:0050896)” in the down-regulated genes. Notably, there are a total of 26 DEGs in the category of “response to stimulus”. Detailed analysis revealed that 12 genes are related to “response to stress (GO:0006950)”, 12 genes are related to “response to chemical stimulus (GO:0042221)”, 7 genes are related to “response to external stimulus (GO:0009605)” and 6 genes are related to “cellular response to stimulus (GO:0051716)”.
We further investigated those highly DEGs in gill, and observed that 7 heat shock protein genes, 4 Cathepsin genes and 3 proteasome subunit genes are highly up-regulated in gill of AW (Additional file 1: Table S1). Heat shock proteins target damaged proteins to the proteasome to prevent accumulation of dysfunctional proteins and to recycle peptides and amino acids [23]. This result suggested that the high level of autophagy occurred in gill under AW stress.
In the IPA analysis, we observed significant Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment of the differential expressed genes in gill in response to AW environment stress. “Eukaryotic Initiation Factor 2 (EIF2) Signaling”, “Regulation of eIF4 (Eukaryotic Initiation Factor 4) and p70S6K (P70S6 kinase) Signaling” and “mTOR (Mammalian Target of Rapamycin) Signaling” were listed in the top enriched pathways, and all of them had been reported that played essential roles on stress response and tolerance. Eukaryotic Initiation Factor 2 (EIF2) is a GTP (Guanosine Triphosphate)-binding protein that escorts the initiation-specific form of Met-tRNA (Met-tRNAi) onto the ribosome, it also plays a role in identifying the translational start site. EIF2 signaling is the protein synthesis pathway in eukaryotic organisms. Because protein synthesis is energetically costly, stressed cells usually inhibit this process to devote resources to stress responses. In many cases EIF2α phosphorylation is a biological response that facilitates cells to cope with stressful environments by down-regulation of protein synthesis [24, 25]. mTOR is a serine/threonine kinase distributed within two protein complexes (mTORC1 and mTORC2) in the cell [26], which plays important roles in response to stress, including activation of the autophagy [27] and modulation of protein synthesis [28]. These responses can conserve energy and promote survival during prolonged periods of stress [19]. “Regulation of eIF4 and p70S6K Signaling” pathway has similar functions on protein synthesis regulation. p70s6k has been considered an mTOR activation mirror and a marker of increased protein synthesis induced by stress and stimulation [29].
According to above evidences from GO and KEGG pathway analysis, we hypothesized that high alkaline stress suppressed protein synthesis and increased the level of autophagy in gill of L. waleckii in Dali Nor, which could conserve energy and provide sufficient amino acids and macromolecules for surviving in high alkaline environment.
Functional analysis on differential expressed genes in liver
In response to AW stress, we observed significant gene enrichment under several GO terms in liver, including “oxidoreductase activity (GO:0016491) (52 genes)” and “transporter activity (GO:0005215) (139 genes)” in up-regulated genes, and “transferase activity (GO:0016740) (201 genes)”, “electron carrier activity (GO:0009055)” and “oxidoreductase activity (GO:0016491) (132 genes)” in the down-regulated genes. The genes in the molecular function category of “oxidoreductase activity” and “electron carrier activity” are widely studied and recognized to associate with oxidative stress and adaptation on environmental stimuli in coupling with mitochondrial functions. For instance, Mitchell et al. performed genome-wide gene expression profiling on two model microorganisms, Escherichia coli and Saccharomyces cerevisiae, in response to environmental stimuli, showed significant functional enrichment of oxidative stress categories, including oxidoreductase activity [30]. The expression profiling studies on teleost species also showed similar response that a cluster of genes of oxidoreductase activity differential expressed in response to environmental stress, such as temperature stress and confinement stress [31, 32]. Here we identified 6 oxidoreductase genes differentially expressed in AW and FW environment. Genes in the category of “transporter activity” are in charge of the movement of substances, such as macromolecules, small molecules and ions, etc. They were significantly enriched in the sub-terms of “substrate-specific transmembrane transporter activity” and “ion transmembrane transporter activity” in this study, suggesting their important roles in regulating homeostasis of various substrates in response to environmental stress, which were consistent with those previous reports on stress adaptation and resistance of many organisms. For instance, amino acid and ion transmembrane transporters were reported to be essential factors to salt and osmotic stress response in many plants [33, 34], as well as in many aquatic animals including mollusks [35, 36] and teleosts [37, 38] etc. We further inspected those highly DEGs in liver and confirmed that 128 accessions encoding various transporter proteins or solute carrier (SLC) family members, suggesting they were important regulators in response to alkaline stress in liver of L. waleckii. IPA pathway enrichment analysis on those DEGs in liver showed significant pathway enrichment on “Superpathway of cholesterol biosynthesis”, suggesting the cholesterol synthesis had been significantly induced under the severe environmental stress in the liver of L. waleckii.
Functional analysis on differential expressed genes in kidney
Kidney is the essential organ which serves homeostatic functions such as the regulation of electrolytes, maintenance of acid–base balance, and salt and water balance in the body. From those 2,761 DEGs in kidney, we identified significant enrichment on several GO terms, including “oxidoreductase activity (GO:0016491) (150 genes)”, “transferase activity (GO:0016740) (165 genes)”, “transporter activity (GO:0005215) (77 genes)”, “electron carrier activity (GO:0009055) (32 genes)”, “enzyme regulator activity (GO:0030234) (56 genes)”, “response to stimulus (GO:0050896) (91 genes)” in the up-regulated genes, and “oxidoreductase activity (GO:0016491) (41 genes)”, “transporter activity (GO:0005215) (124 genes)”, “enzyme regulator activity (GO:0030234) (93 genes)”, “transcription factor activity (GO:0003700) (75 genes)” in the down-regulated genes. The enrichment profile is similar to those DEGs in liver on “oxidoreductase activity”, suggesting that both tissues were facing oxidative stress caused by environmental stimuli, and the genes with oxidoreductase activity changed their expression to adapt the changes. The genes of “transporter activity” were enriched in both up- and down-regulated genes. A significant portion of these transporters are substrate-specific transmembrane transporters (GO:0022891). The active transmembrane transporters are much more than those passive transmembrane transporters, which indicate that active transporters play the essential roles to transport specific substrates such as ion and organic acid across membranes under severe osmotic and alkaline stress with great energy consumption than those in FW environment. The increased energy requirement leads to active proteolysis in the kidney, which can be demonstrated by observed up-regulation of genes encoding various aminotransferases, including tyrosine aminotransferase, aspartate aminotransferase, ornithine aminotransferase, and alanine aminotransferase, etc. Other than aminotransferases, abundant genes under the term of “transferase activity” are significantly enriched in up-regulated genes, which are mainly comprised of “transferring one-carbon groups”, “transferring acyl groups”, “transferring glycosyl groups”, “transferring phosphorus-containing groups”, suggesting their important roles in response to environmental stress in AW. One of well-studied transferase families is glutathione S-transferase (GST) gene family, which have been confirmed their essential functions in protection against oxidative stress caused by various stress from toxic heavy metal ions [39–41], osmotic imbalance [42], salinity [43] and pH change [44]. GSTs have been even used as biomarkers for environmental pollution and toxins monitoring recently [45, 46]. In the biological process, we identified 47 DEGs that belong to the subcategory of “response to chemical stimulus” in those 91 up-regulated genes of “response to stimulus” in kidney, corresponding to the essential roles of kidney in response to external and endogenous chemical stresses in AW environment.
Expression of the genes under positive selection
Previous dN/dS analysis on transcriptome of L. waleckii from Dali Nor Lake revealed that there were 61 genes experienced strong positive selection under severe environmental stress [7]. We investigated the genes under strong positive selection and found that significant portions of these genes were also expressed differentially under AW and FW environment (Table 4). For instance, we identified 5 carbonic anhydrase genes, 2 superoxide dismutase genes, 5 glutathione S-transferase genes, 3 aminopeptidase N genes, and 2 perforin-1 genes from the DEG list of liver, and identified 4 carbonic anhydrase genes, 2 superoxide dismutase genes, 8 glutathione S-transferase genes, 3 aminopeptidase N genes, and 2 Perforin-1 genes from the DEG list of kidney. Obviously, a number of genes that retain specific nucleotide changes under strong positive selection also change their expression profiles under severe alkaline stress, as well as osmotic, salt and heavy metal stress in the Dali Nor lake.
Conclusion
We performed comparative transcriptome profiling study on L. waleckii inhibiting in alkaline water of Dali Nor lake and in fresh water of Ganggeng Nor lake, and identified a relatively large number of genes that displayed distinct differences on their expression in gill, liver and kidney. Further analysis revealed that several well-known functional categories of genes and signaling pathway, which are associated with stress response and extreme environment adaptation, had been significantly enriched, including the functional categories of “response to stimulus”, “transferase activity”, “transporter activity” and “oxidoreductase activity”, etc., and signaling pathways of “mTOR signaling”, “EIF2 signaling”, “superpathway of cholesterol biosynthesis”, etc. We also identified significantly DEGs in three tissues, encoding important modulators on stress adaptation and tolerance, including carbonic anhydrases, heat shock proteins, superoxide dismutase, glutathione S-transferases, aminopeptidase N, and aminotransferases. Overall, this study demonstrated that transcriptome changes in L. waleckii played a role in adaptation to complicated environmental stress in the highly alkalized Dali Nor lake. The results set a foundation for further analyses on alkaline-responsive candidate genes, which would help us understand teleost adaptation under extreme environmental stress and ultimately benefit future breeding for alkaline-tolerant fish strains.
Methods
Ethics statement
This study was approved by the Animal Care and Use committee of Centre for Applied Aquatic Genomics at Chinese Academy of Fishery Sciences.
Fish sampling
Ten individuals (five males, five females) of L. waleckii inhabiting alkaline water were sampled at north shore of Dali Nor lake, Inner Mongolia, China (43o22′43"N, 116o39'24"E). Nine individuals (five males, four females) inhabiting fresh water were sampled at west shore of Ganggeng Nor lake (43o17'48"N, 116o53'27"E). Both groups weighted ranging from 130 grams to 150 grams. Liver, kidney and gill were dissected and collected, due to previous report that these organs play important role in hyperosmotic and hypersaline conditions [47]. Tissue samples were stored in RNAlater (Qiagen, Hilden, Germany) and transported to laboratory in Beijing at room temperature, then stored at −20°C prior to RNA extraction.
RNA extraction and quality control
Total RNA was extracted from each tissue using TRIZOL Kit (Invitrogen, Carlsbad, CA, USA) with manufacturer’s instructions. RNA samples were then digested by DNase I to eliminate potential genomic DNA. Integrity and size distribution were checked on Bioanalyzer 2100 with RNA 6000 Nano Labchips (Agilent technologies, Santa Clara, CA, USA). Equal amounts of the high quality RNA samples from each tissue were then pooled for RNA-Seq.
cDNA library construction and sequencing
RNA-Seq library preparation and sequencing was carried out by HudsonAlpha Genomic Services Lab (Huntsville, AL, USA) as previously described [48]. cDNA libraries were prepared with ∼2.5 μg of starting total RNA following the protocols of the Illumina TruSeq RNA Sample Preparation Kit (Illumina). The final library had an average fragment size of 270 bp and final yields of 400 ng. After KAPA quantitation and dilution, the library was sequenced on an Illumina HiSeq 2000 with 101 bp paired-end reads.
Sequence data processing and de novo assembly
Adaptor sequences were trimmed and low quality reads were removed. Then read length less than 10 were removed. TRINITY was used to assemble all cleaned reads with default parameters [22] and generate reference sequences for comparative transcriptome study.
Functional annotation of assembled contigs
The assembled transcriptome contigs were subjected to similarity search against NCBI non-redundant (nr) protein database using BLASTx with e-value cutoff of 1E-10. Gene name and description was assigned to each contig based on the top BLASTx hit with the highest score. Gene ontology (GO) analysis was conducted on assembled transcriptome using InterProScan (http://www.ebi.ac.uk/Tools/pfa/iprscan/) and integrated protein databases with default parameters. The GO terms associated with transcriptome contigs were then obtained for describing their biological processes, molecular functions and cellular components.
Read mapping and differential gene expression analysis
All the cleaned reads were mapped to the assembled reference transcriptome by Bowtie [21], and about 84.1% of the reads can be mapped to the reference for each sample (Table 1). RSEM was then used to estimate and quantify the gene and isoform abundances according to the trinity assembled transcriptome. Finally, we used edgeR to normalize the expression levels in each of these samples and obtain the differentially expressed transcripts by pairwise comparisons [49].
Quantitative reverse transcription-PCR (qRT-PCR)
qRT-PCR was used to validate the RNA-Seq results on randomly selected 30 gene accessions. The beta-actin gene was used as an internal reference, and primers were designed as below, forward primer: 5′- TGCAAAGCCGGATTCGCTGG -3′; reverse primer: 5′- AGTTGGTGACAATACCGTGC -3′. Briefly, qRT-PCR was performed in the optical 96-well plates with an ABI PRISM 7500 Real-time Detection System (Life Technology). The amplification was performed in a total volume of 15 μl, containing 7.5 μl 2X SYBR Green Master Mix reagent (Life Technology), 1 μl of cDNA (100 ng/μl), and 0.3 μl of 10 μM of each gene-specific primer. The PCR cycle was 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min. All reactions were set up in triplicate including the negative controls with no template. To assess PCR efficiency, five 10-fold serial dilutions of a randomly selected cDNA sample were used on both the target genes and the reference gene to assess the PCR efficiency. After the PCR, data were analyzed with ABI 7500 SDS software. The comparative CT method (2-∆∆CT method) was used to analyze the expression of the target genes. All data were given at levels relative to the expression of the beta-actin gene.
IPA analysis
The genes differentially expressed in 3 tissues were further analyzed using the Ingenuity Pathway Analysis program (IPA; https://analysis.ingenuity.com) in order to identify the biochemical pathways affected. The IPA software contains the biological function, interaction, and information of a curated gene set and many biochemical pathways, identifying global canonical pathways, dynamical biological networks and functions from a given list of genes. Basically, the accession numbers of DEGs were uploaded into the IPA and compared with the genes included in each canonical pathway using the whole gene set as the background. All the pathways with one or more genes overlapping the candidate genes were extracted. During IPA analyses, each of the pathways was assigned a P value from Fisher’s exact test, denoting the probability of overlap between the pathway and the input genes.
Availability of supporting data
All supporting files have been deposited to LabArchives entitled ‘amur ide DGE’ with a DOI of 10.6070/H4PZ56RJ (https://mynotebook.labarchives.com/share/xujian/Mi42fDIxOTY2LzIvVHJlZU5vZGUvMzc5NDY5ODU2MHw2LjY=).
References
Xiao J, Si B, Zhai D, Itoh S, Lomtatidze Z: Hydrology of Dali Lake in central-eastern Inner Mongolia and Holocene East Asian monsoon variability. J Paleolimnol. 2008, 40 (1): 519-528. 10.1007/s10933-007-9179-x.
Geng K, Zhang Z: Geomorphologic features and evolution of the Holocene lakes in Dali Nor Area, the Inner Mongolia (in Chinese). 4, 94–100. J Beijing Normal University. 1988, 4: 94-100.
Chi B, Chang Y, Yan X, Cao D, Li Y, Gao Y, Liu Y, Liang L: Genetic variability and genetic structure of Leuciscus waleckii Dybowski in Wusuli River and Dali Lake. J Fishery Sci China. 2010, 17 (2): 228-235.
Zhang Y, Liang L, Jiang P, Li D, Lu C, Sun X: Genome evolution trend of common carp (Cyprinus carpio L.) as revealed by the analysis of microsatellite loci in a gynogentic family. J Genet Genomics. 2008, 35 (2): 97-103. 10.1016/S1673-8527(08)60015-6.
Liu J, Chang Y, Xu L, Liu C, Liang L, Han G, Gao Y, Liu Y: Isolation and characterization of microsatellite from genome of Leuciscus waleckii Dybowski. Acta Agriculturae Boreali-Sinic. 2011, 26 (1): 87-93.
Wang B, Ji P, Xu J, Sun J, Yang J, Xu P, Sun X: Complete mitochondrial genome of Leuciscus waleckii (Cypriniformes: Cyprinidae: Leuciscus). Mitochondrial DNA. 2012, 24 (2): 126-128.
Xu J, Ji P, Wang B, Zhao L, Wang J, Zhao Z, Zhang Y, Li J, Xu P, Sun X: Transcriptome sequencing and analysis of wild amur ide (Leuciscus waleckii) inhabiting an extreme alkaline-saline lake reveals insights into stress adaptation. PLoS ONE. 2013, 8 (4): e59703-10.1371/journal.pone.0059703.
Xia JH, Liu P, Liu F, Lin G, Sun F, Tu R, Yue GH: Analysis of stress-responsive transcriptome in the intestine of Asian seabass (Lates calcarifer) using RNA-Seq. DNA Res. 2013, Epub of ahead
Smith S, Bernatchez L, Beheregaray LB: RNA-seq analysis reveals extensive transcriptional plasticity to temperature stress in a freshwater fish species. BMC Genomics. 2013, 14: 375-10.1186/1471-2164-14-375.
Hale MC, Xu P, Scardina J, Wheeler PA, Thorgaard GH, Nichols KM: Differential gene expression in male and female rainbow trout embryos prior to the onset of gross morphological differentiation of the gonads. BMC Genomics. 2011, 12: 404-10.1186/1471-2164-12-404.
Xu P, McIntyre LM, Scardina J, Wheeler PA, Thorgaard GH, Nichols KM: Transcriptome profiling of embryonic development rate in rainbow trout advanced backcross introgression lines. Mar Biotechnol (NY). 2011, 13 (2): 215-231. 10.1007/s10126-010-9283-1.
Xiang L-x, He D, Dong W-r, Zhang Y-w, Shao J-z: Deep sequencing-based transcriptome profiling analysis of bacteria-challenged Lateolabrax japonicus reveals insight into the immune-relevant genes in marine fish. BMC Genomics. 2010, 11 (1): 472-10.1186/1471-2164-11-472.
Ji P, Liu G, Xu J, Wang X, Li J, Zhao Z, Zhang X, Zhang Y, Xu P, Sun X: Characterization of common carp transcriptome: sequencing, de novo assembly, annotation and comparative genomics. PLoS ONE. 2012, 7 (4): e35152-10.1371/journal.pone.0035152.
Sun F, Peatman E, Li C, Liu S, Jiang Y, Zhou Z, Liu Z: Transcriptomic signatures of attachment, NF-κB suppression and IFN stimulation in the catfish gill following columnaris bacterial infection. Dev Comp Immunol. 2012, 38 (1): 169-180. 10.1016/j.dci.2012.05.006.
Lu M-W, Ngou F-H, Chao Y-M, Lai Y-S, Chen N-Y, Lee F-Y, Chiou P: Transcriptome characterization and gene expression of Epinephelus spp in endoplasmic reticulum stress-related pathway during betanodavirus infection in vitro. BMC Genomics. 2012, 13 (1): 651-10.1186/1471-2164-13-651.
Sarropoulou E, Galindo-Villegas J, García-Alcázar A, Kasapidis P, Mulero V: Characterization of European sea bass transcripts by RNA-Seq after oral vaccine against V. anguillarum. Mar Biotechnol. 2012, 14 (5): 634-642. 10.1007/s10126-012-9466-z.
Garcia T, Shen Y, Crawford D, Oleksiak M, Whitehead A, Walter R: RNA-Seq reveals complex genetic response to deepwater horizon oil release in Fundulus grandis. BMC Genomics. 2012, 13 (1): 474-10.1186/1471-2164-13-474.
Kakumanu A, Ambavaram MM, Klumas C, Krishnan A, Batlang U, Myers E, Grene R, Pereira A: Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed by RNA-Seq. Plant Physiol. 2012, 160 (2): 846-867. 10.1104/pp.112.200444.
Teets NM, Peyton JT, Colinet H, Renault D, Kelley JL, Kawarasaki Y, Lee RE, Denlinger DL: Gene expression changes governing extreme dehydration tolerance in an Antarctic insect. Proc Natl Acad Sci U S A. 2012, 109 (50): 20744-20749. 10.1073/pnas.1218661109.
Dugas DV, Monaco MK, Olsen A, Klein RR, Kumari S, Ware D, Klein PE: Functional annotation of the transcriptome of Sorghum bicolor in response to osmotic stress and abscisic acid. BMC Genomics. 2011, 12: 514-10.1186/1471-2164-12-514.
Langmead B, Trapnell C, Pop M, Salzberg SL: Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10 (3): R25-10.1186/gb-2009-10-3-r25.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q: Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011, 29 (7): 644-652. 10.1038/nbt.1883.
Goldberg AL: Protein degradation and protection against misfolded or damaged proteins. Nature. 2003, 426 (6968): 895-899. 10.1038/nature02263.
Muaddi H, Majumder M, Peidis P, Papadakis AI, Holcik M, Scheuner D, Kaufman RJ, Hatzoglou M, Koromilas AE: Phosphorylation of eIF2alpha at serine 51 is an important determinant of cell survival and adaptation to glucose deficiency. Mol Biol Cell. 2010, 21 (18): 3220-3231. 10.1091/mbc.E10-01-0023.
Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG: Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002, 22 (21): 7405-7416. 10.1128/MCB.22.21.7405-7416.2002.
Bhaskar PT, Hay N: The two TORCs and AKT. Dev Cell. 2007, 12 (4): 487-502. 10.1016/j.devcel.2007.03.020.
Jung CH, Ro SH, Cao J, Otto NM, Kim DH: mTOR regulation of autophagy. FEBS letters. 2010, 584 (7): 1287-1295. 10.1016/j.febslet.2010.01.017.
Sengupta S, Peterson TR, Sabatini DM: Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010, 40 (2): 310-322. 10.1016/j.molcel.2010.09.026.
Zanchi NE, Lancha AH: Mechanical stimuli of skeletal muscle: implications on mTOR/p70s6k and protein synthesis. Eur J Appl Physiol. 2008, 102 (3): 253-263.
Mitchell A, Romano GH, Groisman B, Yona A, Dekel E, Kupiec M, Dahan O, Pilpel Y: Adaptive prediction of environmental changes by microorganisms. Nature. 2009, 460 (7252): 220-224. 10.1038/nature08112.
Calduch-Giner JA, Davey G, Saera-Vila A, Houeix B, Talbot A, Prunet P, Cairns MT, Perez-Sanchez J: Use of microarray technology to assess the time course of liver stress response after confinement exposure in gilthead sea bream (Sparus aurata L.). BMC Genomics. 2010, 11: 193-10.1186/1471-2164-11-193.
Thorne MA, Burns G, Fraser KP, Hillyard G, Clark MS: Transcription profiling of acute temperature stress in the Antarctic plunderfish Harpagifer antarcticus. Mar Genomics. 2010, 3 (1): 35-44. 10.1016/j.margen.2010.02.002.
Uozumi N, Schroeder JI: Ion channels and plant stress: past, present, and future. 2010, In: Ion Channels and Plant Stress Responses. Springer, 1-22.
Zhu JK: Regulation of ion homeostasis under salt stress. Curr Opin Plant Biol. 2003, 6 (5): 441-445. 10.1016/S1369-5266(03)00085-2.
Zhao X, Yu H, Kong L, Li Q: Transcriptomic responses to salinity stress in the pacific oyster Crassostrea gigas. PLoS ONE. 2012, 7 (9): e46244-10.1371/journal.pone.0046244.
Lockwood BL, Somero GN: Transcriptomic responses to salinity stress in invasive and native blue mussels (genus Mytilus). Mol Ecol. 2011, 20 (3): 517-529. 10.1111/j.1365-294X.2010.04973.x.
Kiilerich P, Tipsmark CK, Borski RJ, Madsen SS: Differential effects of cortisol and 11-deoxycorticosterone on ion transport protein mRNA levels in gills of two euryhaline teleosts, Mozambique tilapia (Oreochromis mossambicus) and striped bass (Morone saxatilis). J Endocrinol. 2011, 209 (1): 115-126. 10.1530/JOE-10-0326.
Buhariwalla H, Osmond E, Barnes K, Cozzi R, Robertson G, Marshall W: Control of ion transport by mitochondrion-rich chloride cells of eurythermic teleost fish: Cold shock vs. cold acclimation. Comp Biochem Physiol A Mol Integr Physiol. 2012, 162 (3): 234-244. 10.1016/j.cbpa.2012.03.010.
Yadav S: Heavy metals toxicity in plants: an overview on the role of glutathione and phytochelatins in heavy metal stress tolerance of plants. S Afr J Bot. 2010, 76 (2): 167-179. 10.1016/j.sajb.2009.10.007.
Espinoza HM, Williams CR, Gallagher EP: Effect of cadmium on glutathione S-transferase and metallothionein gene expression in coho salmon liver, gill and olfactory tissues. Aquat Toxicol. 2012, 110: 37-44.
Srikanth K, Pereira E, Duarte A, Ahmad I: Glutathione and its dependent enzymes’ modulatory responses to toxic metals and metalloids in fish—a review. Environ Sci Pollut Res Int. 2013, 20: 2133-2149. 10.1007/s11356-012-1459-y.
Choi CY, An KW, An MI: Molecular characterization and mRNA expression of glutathione peroxidase and glutathione S-transferase during osmotic stress in olive flounder (Paralichthys olivaceus). Comp Biochem Physiol A Mol Integr Physiol. 2008, 149 (3): 330-337. 10.1016/j.cbpa.2008.01.013.
Zanette J, de Almeida EA, da Silva AZ, Guzenski J, Ferreira JF, Di Mascio P, Marques MRF, Bainy ACD: Salinity influences glutathione S-transferase activity and lipid peroxidation responses in the Crassostrea gigas oyster exposed to diesel oil. Sci Total Environ. 2011, 409 (10): 1976-1983. 10.1016/j.scitotenv.2011.01.048.
Zhou J, Wang W-N, Wang A-L, He W-Y, Zhou Q-T, Liu Y, Xu J: Glutathione S-transferase in the white shrimp Litopenaeus vannamei: Characterization and regulation under pH stress. Comp Biochem Physiol C Toxi Pharmacol. 2009, 150 (2): 224-230. 10.1016/j.cbpc.2009.04.012.
Vidal-Liñán L, Bellas J: Practical procedures for selected biomarkers in mussels, Mytilus galloprovincialis—Implications for marine pollution monitoring. Sci Total Environ. 2013, 461: 56-64.
Fabrini R, Bocedi A, Del Grosso E, Morici L, Federici G, Palleschi A, Ricci G: Erythrocyte glutathione transferase: A novel biomarker to check environmental pollution hazardous for humans. Biochem Biophys Res Commun. 2012, 426 (1): 71-75. 10.1016/j.bbrc.2012.08.037.
Lavado R, Aparicio-Fabre R, Schlenk D: Effects of salinity acclimation on the pesticide-metabolizing enzyme flavin-containing monooxygenase (FMO) in rainbow trout (Oncorhynchus mykiss). Comp Biochem Physiol Toxi Pharmacol : CBP. 2013, 157 (1): 9-15. 10.1016/j.cbpc.2012.08.004.
Xu J, Ji P, Zhao Z, Zhang Y, Feng J, Wang J, Li J, Zhang X, Zhao L, Liu G: Genome-wide SNP discovery from transcriptome of four common carp strains. PLoS ONE. 2012, 7 (10): e48140-10.1371/journal.pone.0048140.
Robinson MD, McCarthy DJ, Smyth GK: edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010, 26 (1): 139-140. 10.1093/bioinformatics/btp616.
Acknowledgements
This study was supported by the grants from National Natural Science Foundation of China (No. 31101893), National High-tech R&D Program of China (2011AA100401), China Ministry of Agriculture “948” Program (No. 2013-Z12) and Research Foundation of Chinese Academy of Fishery Sciences (2013C011).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JX and QL contributed equally and their contribution accounts for the major part of this study. PX conceived the study, and drafted the manuscript. JX and QL worked on sample collection and RNA sequencing, and participated in major bioinformatic analysis and manuscript preparation. LX and JL worked on DEG pipeline development. SW and YJ participated in gene pathway analysis. ZZ, YZ and CD participated in sample collection. XS participated in discussions and provided advices. All authors read and approved the final manuscript.
Jian Xu, Qiang Li contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Xu, J., Li, Q., Xu, L. et al. Gene expression changes leading extreme alkaline tolerance in Amur ide (Leuciscus waleckii) inhabiting soda lake. BMC Genomics 14, 682 (2013). https://doi.org/10.1186/1471-2164-14-682
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-682