
Abstract
Heavy metal Cadmium (Cd2+) pollution has become a severe environmental problem for aquatic organisms. In crustaceans, gills (Gi) and hepatopancreas (Hp) play a vital role in the toxicology. However, in Macrobrachium rosenbergill, there are few researches about gill and hepatopancreases responding to Cd2+ stress at a molecular level. In this study, transcriptomic analysis was applied to characterize gene expression profiles of gills and hepatopancreas of M. rosenbergill after Cd2+ exposure for 0 h, 3 h and 3 d. Six cDNA libraries (Gi 0 h, Gi 3 h, Gi 3 d, Hp 0 h, Hp 3 h, and Hp 3 d) were constructed and a total of 66,676 transcripts and 48,991 unigenes were annotated. Furthermore, differentially expressed genes (DEGs) were isolated by comparing the Cd2+ treated time-point libraries (3 h and 3 d group) with the control library (0 h group). The results showed that most of the DEGs were down-regulated after Cd2+ exposure and the number of DEGs among gill groups were significantly higher than those among hepatopancreas groups. GO functional and KEGG pathway analysis suggested many key DEGs in response to the Cd2+ stress, such as metallothionein and Hemocyanin. Additionally, a total of six DEGs were randomly selected to further identify their expressional profile by qPCR. The results indicated that these DEGs were involved in the response to Cd2+. This comparative transcriptome provides valuable molecular information on the mechanisms of responding to Cd2+ stress in M. rosenbergii, which lays the foundation for further understanding of heavy metal stress.
Similar content being viewed by others

Introduction
Over the past decades, aquatic heavy-metal pollution, such as copper (Cu2+), Zinc (Zn2+), and cadmium (Cd2+), has become one of the greatest concerns for aquatic environmental bio-monitoring worldwide1 due to its high toxicity, non-degradability, and subsequent bioaccumulation and biomagnification2,3,4,5. Cu2+ is an essential metal that participates in normal physiological process in crustaceans. But, the high concentrations of Cu2+ could induce the generation of reactive oxygen species (ROS), which lead to oxidative damage in many organisms6,7. Zn2+ is highly toxic for aquatic crustaceans and is a ubiquitous heavy metal in aquatic environment. High concentrations of Zn2+ inhibiting oxygen consumption had been reported in Litopenaeus vannamei8, Litopenaeus schmitti9, and Farfantepenaeus paulensis10. Among heavy metal pollutants, Cd2+ pollution ranks first in the world11, and it is toxic even at a very low concentration12. The toxic effects of cadmium have been well-documented in animals and humans. Cd2+ causes the accumulation of ROS which induce oxidization of biological macromolecules and results in various physiological damages to animal tissues and organs13,14. Furthermore, Cd2+ causes impairment of reproductive activity and disrupts endocrine function in fish15. Cd2+ induced cell apoptosis has been confirmed to be attributed to caspase-dependent and independent pathways of the mitochondria or endoplasmic reticulum (ER)16,17. Additionally, Cd2+ , a non-essential and potentially toxic metal, can be accumulated in humans via food chain18, which may result in morphological deformities, physiological dysfunctions and even death19. Previous researches showed that Cd2+ is known to accumulate in marine organisms and induced rapid genetic changes in many crustaceans, such as Sinopotamon henanense19 and Eriocheir sinensis20. Hence, it is essential to focus on the potential response mechanism caused by Cd2+ stress in crustaceans.
As an important respiratory organ, the gill (Gi) is involved in ion transport, acid–base balance and osmoregulation in crustaceans21,22. Due to the crustacean gills being exposed to the water in which they live, the gills play a vital role in the toxicological interactions, such as with heavy metals23. Furthermore, the hepatopancreas (Hp), a sensitive organ similar to the liver of higher organisms, is susceptible to be damaged by waterborne pollutants in crustaceans24,25,26. Therefore, gills and hepatopancreas are model organs for studying the response to heavy metal stress in crustaceans.
The giant freshwater prawn, Macrobrachium rosenbergii, is an important commercial prawn and widely cultured in China and other Pacific Rim countries27. As a freshwater cultured species, the prawn is susceptible to metal accumulation. Previous studies showed that structural changes of gills and hepatopancreas of M. rosenbergii could be caused by the Cu2+ accumulation, and the degree of damage observed was related to the elevated waterborne copper concentration24. Additionally, transcriptomic analysis of gills of M. rosenbergii showed that 19,417 and 8,989 differentially expressed genes (DEGs) were identified at 3 h and 48 h after Cu2+ exposure, respectively28, revealing that a large number of genes were involved in response to Cd2+ stress. Further research showed that the accumulation of Cd2+ also manifested histopathological changes in the gills and hepatopancreas of M. rosenbergii under Cd2+ exposure, and Cd2+ levels in tissues followed the order of: gills > hepatopancreas29. To date, however, limited researches were focused on the Cd2+-related stress response and regulatory gene in M. rosenbergii.
In this study, transcriptome sequencing of gills and hepatopancreas in M. rosenbergii was performed to analyze transcriptional responses under Cd2+ pollution. Many vital genes in response to the Cd2+ were identified. The study provided valuable and reliable data for aquaculture and environmental monitoring management, and elucidated the potential toxicological mechanism in M. rosenbergii.
Results
Transcriptome sequencing and functional gene annotation
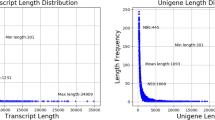
Six cDNA libraries were constructed for Illumina sequencing and the sequencing generated 47,932,697, 45,863,583, 46,324,011, 44,082,407, 43,621,438, and 46,621,968 clean reads for Gi 0 h, Gi 3 h, Gi 3 d, Hp 0 h, Hp 3 h, and Hp 3 d, respectively (Table 1). The clean reads were assembled, and generated 66,676 transcripts, which were further clustered into 48,991 unigenes. The unigenes comprised of 74,217,621 bases, and the average length, largest length and smallest length were 1514.92 bp, 36,523 bp and 201 bp, respectively (Table 2). A length distribution of the total number of transcripts and unigenes is shown in Fig. 1. It is clearly displayed that there were 30,034 contigs (61.31%) ranging from 201 to 1000 bp, 15,172 contigs (30.97%) ranging from 1001 to 4500 bp, and 3785 contigs (7.73%) over 4,500 bp in length. Likewise, there were 38,933 transcripts (33.16%) ranging from 201 to 1000 bp, 22,109 transcripts (21.2%) ranging from 1,001 to 4,500 bp, and 5,634 transcripts (8.45%) over 4,500 bp in length. The transcript average coverage is 1599.88 bp, indicating that a high-quality transcriptome was assembled. Therefore, the assembled contigs in our study provide a useful resource for future research on M. rosenbergii.

Length distribution of transcripts and unigenes. The X-axis indicates transcript and unigene size and the Y-axis indicates the number of transcript and unigene for each size.
We annotated all unigenes with reference to six functional databases (Table 3) and found that 14,812 (30.23%), 11,869 (24.23%), 16,093 (32.85%), 18,536 (37.84%), 14,384 (29.36%) and 16,463 (33.6%) unigenes were mapped to GO, KEGG, COG, NR, Swiss-Prot, and Pfam, respectively.
Identification of differentially expressed genes (DEGs)
Pearson correlation analysis showed good correlation among different replicates of the same sample, whereas significant differences were observed between the gill and hepatopancreas groups (Fig. 2). To identify genes displaying significant changes in expression level in the face of Cd2+ stress, we analyzed the expression level of each unigene by TPM method and found many DEGs by comparing the Cd2+ treated time-points libraries (3 h and 3 d group) with the control library (0 h group) (Supplementary Table S1). Compared to gill control group (Gi 0 h), a total of 6264 (2,010 upregulated and 4254 downregulated) and 5175 (2,186 upregulated and 2989 downregulated) DEGs were identified in the Gi 3 h group and Gi 3 d group (Fig. 3), respectively. Long duration of Cd2+ exposure (Gi 3 d group) caused 4222 genes to be differentially expressed compared with short duration (Gi 3 h group) (Fig. 3). Furthermore, Venn analysis showed that 3375 genes were differentially expressed at both time-points, while 2889 DEGs were regulated just at Gi 3 h group and 1,800 genes were altered just at Gi 3 d group (Fig. 4). These time-specific genes might help to illustrate the stress response at different time-points. Similarly, many DEGs also existed between Hp 0 h and Hp 3 h, Hp 0 h and Hp 3 d, as well as Hp 3 h and Hp 3 h groups (Figs. 3, 4). Additionally, the number of DEGs among gill groups were significantly higher than those among hepatopancreas groups.

Pearson correlation analysis among the samples from different biological replicates. The abscissa and ordinate represent the sample name, and the correlation coefficient is represented by color; deeper color represents a stronger correlation.

Differentially expressed genes (DEGs) in Cd2+ exposure and control groups. Y-axis represents the number of DEMs and X -axis represents different comparisons (Gi 0 h vs Gi 3 h, Gi 0 h vs Gi 3 d, Gi 3 h Vs Gi 3 d, Hp 0 h vs Hp 3 h, Hp 0 h vs Hp 3 d, and Hp 3 h vs Hp 3 d). Green dots represent up-regulated genes and red dots represent down-regulated genes. Gi, gill; HP hepatopancreas; 0 h, control; 3 h, Cd2+ treated 3 h; 3d, Cd2+ treated 3 days.

Venn diagram of differentially expressed genes (DEGs) in different comparisons. The overlaps represent the number of intersections of unigenes shared by the control group (0 h) and the two experimental groups (3 h and 3 d). Gi, gill; HP, hepatopancreas; 0 h, control; 3 h, Cd2+ treated 3 h; 3d, Cd2+ treated 3 days.
GO functional annotation and KEGG enrichment analysis of DEGs
To better understand the biological functions and gene interaction of DEGs, all DEGs (Gi 0 h vs Gi 3 h, Gi 0 h vs Gi 3 d, Hp 0 h vs Hp 3 h and Hp 0 h vs Hp 3 d) were annotated in GO terms (Fig. 5). Among the categories of biological process, cellular component, and molecular function, the top 2 enriched GO terms for each category were “cellular process and metabolic process”, “membrane part and cell part”, and “catalytic activity and binding”, respectively. A total of 59 genes were annotated to “response to oxidative stress” GO terms (Supplementary Table S2), such as catalase, peroxidase and NADH dehydrogenase. Subsequently, KEGG pathway analysis was performed to identify the functions of DEGs and biological pathways involved in metal stress response. The top 20 significantly enriched KEGG pathways in each comparison are shown in Fig. 6. For the gill groups, 13 of those pathways (Intestinal immune network for IgA production, ECM-receptor interaction, Platelet activation, Cardiac muscle contraction, Retrograde endocannabinoid signaling, Focal adhesion, PI3K-Akt signaling pathway, Non-alcoholic fatty liver disease (NAFLD), Oxidative phosphorylation, Parkinson disease, Thermogenesis, Huntington disease, and Alzheimer disease) were enriched in both Gi 3 h and Gi 3 d compared with Gi 0 h group, whereas for the hepatopancreas groups, only 4 pathways (Glycosaminoglycan degradation, Starch and sucrose metabolism, Amino sugar and nucleotide sugar metabolism, and Glycerolipid metabolism) were enriched in both Hp 3 h and Hp 3 d compared with Hp 0 h group. “Ribosome (272 genes)”, “Alzheimer disease (165 genes)”, “NOD-like receptor signaling pathway (21 genes)”, and “lysosome (52 genes)” were significantly the most gene enriched pathways in Gi 0 h vs Gi 3 h, Gi 0 h vs Gi 3 d, Hp 0 h vs Hp 3 h, and Hp 0 h vs Hp 3 d comparisons, respectively. These enriched pathways may play important roles in metal stress response. GO functional and KEGG analyses of DEGS (Gi 3 h vs Gi 3 d and Hp 3 h and Hp 3 d) are shown in Supplementary Figure S1.

Gene ontology functional analyses of differentially expressed genes. (a) Gi 0 h vs Gi 3 h and Gi 0 h vs Gi 3 d. (b) Hp 0 h vs Hp 3 h and Hp 0 h vs Hp 3 d. Each annotated sequence is assigned at least one GO term from biological process, cellular component or molecular function. The abscissa represents the GO terms and the ordinate represents the number of genes. Gi, gill; HP, hepatopancreas; 0 h, control; 3 h, Cd2+ treated 3 h; 3d, Cd2+ treated 3 days.

Kyoto Encyclopedia of Genes and Genomes functional analyses of differentially expressed genes between Gi 0 h and Gi 3 h (a), Gi 0 h and Gi 3 d (b), Hp 0 h and Hp 3 h (c), Hp 0 h and Hp 3 d (d). The ordinate shows the KEGG metabolic pathway and the broken line represents the number of genes. Gi, gill; HP, hepatopancreas; 0 h, control; 3 h, Cd2+ treated 3 h; 3d, Cd2 + treated 3 days. (The figures were created based on the KEGG pathway database www.kegg.jp/kegg/kegg1.html).
Validation of DEGs by qPCR
The general trend of six DEGs (MT, Hemo, IFRD1, Hsp 67B2, Zbed4, and GH) involved in metal transportation and stress response was in accordance with the results from RNA sequencing (Fig. 7). Obviously, the change range of expression level of many DEGs in gill treated groups is greater than those in hepatopancreas treated groups compared to control group. For example, the expression level of GH gene significantly increased by eight and seven times in gill at 3 h and 3 d group, respectively. In addition, the DEGs present various expression patterns in gill and hepatopancreas after Cd2+ exposure. For instance, the expression level of Hemo significantly increased in gill at 3 h and then decreased at 3d, but consistently increased in hepatopancreas under Cd2+ stress. In contrast, the expression level of Hsp67B2 gene significantly decreased in gill at 3 h and then increased at 3 d, but consistently increased in hepatopancreas under Cd2+ stress.

Validation of the relative expression level of six genes associated with Cd2+ stress using quantitative real time PCR. Blue bar represents the qPCR results, and red bar represents the RNA-Seq results. MT metallothionein, Hemo hemocyanin-like protein, IFRD1 interferon-related developmental regulator 1, Hsp67B2 heat shock protein 67B2-like, Zbed4 zinc finger BED domain-containing protein 4-like, GH gamma-glutamyl hydrolase-like.
Discussion
In crustaceans, the gill epithelium is generally regarded as a major organ of respiration and osmoregulation, and the first site to be exposed to environmental pollutants30. During waterborne exposure to heavy metals, gills act as a protective barrier between the internal and external environment23. Waterborne heavy metals are initially absorbed into epithelium cells of gill and transported into hemolymph, and finally infiltrated into internal organs23. Hepatopancreas is usually considered as a vital target organ for heavy metal toxicity and other environmental stresses in crustaceans and plays a major role in metal storage and in the detoxification process31. Additionally, crustaceans increase metabolic efficiency by promoting the digestive enzyme activities in hepatopancreas in response to heavy metal32. Therefore, the gill and hepatopancreas are considered as a good indicator of water quality, and a suitable model for studies of heavy metal pollution.
To better understand the molecular mechanisms of Cd2+ toxicity in M. rosenbergii., RNA-Seq was used to investigate gene expression differences of gill and hepatopancreas in response to Cd2+ exposure (0 h, 3 h, and 3 d). Six cDNA libraries were constructed and a total of 48,991 unigenes were functionally annotated. GO term enrichment and KEGG pathway enrichment were performed to find important genes and pathways during Cd2+ exposure in gill and hepatopancreas of M. rosenbergii. We analyzed DEGs by comparing the Cd2+ treated time-point libraries with the control library. The results showed that the number of down-regulated DEGs is larger than up-regulated DEGs (Fig. 3), indicating that gene expressions were mainly inhibited by Cd2+, which leads to impairments in M. rosenbergii. The results were similar with Sinopotamon henanense and Danio rerio under Cd2+ stress19,33. The number of DEGs among Gi groups were significantly higher than those among Hp groups (Fig. 3), suggesting that the gill has a stronger stress response than hepatopancreas in short time. Additionally, the number of DEGs in Gi groups decreased with the increment of exposure time, while in Hp groups, the number of DEGs increased with the increment of exposure time (Fig. 3). The above results might be attributed to the reason that the gill acts as the entry site and transient store organ of the heavy metal for a short period of exposure time, and Cd2+ is gradually transferred from the gills to hepatopancreas via the haemolymph with the prolongation of exposure time34.
Many genes related to oxidative stress were found in response to the Cd2+ stress (Supplementary Table S2), and present various expression patterns, as identified by qPCR (Fig. 7). For example, the expression level of metallothionein (MT) was significantly increased at 3 h, then decreased at 3d, which may be related to the accumulation of Cd2+. Many studies have shown that MT is critical to heavy metal detoxification35,36 in addition to storage of essential elements that are necessary for metalloenzymes37,38. Some studies have proven that the accumulation of heavy metal has significant time effects. For instance, in Oncorhynchus mykiss, Cu2+ uptake increased during the 1–2 h under radiolabelled copper exposure, and after 2 h, Cu2+ level significantly decreased in the gill39. A similar tendency was found in Acrossocheilus fasciatus40, in which the expression level of zinc-finger BED domain-containing protein (Zbed) was significantly decreased after exposure to Cd2+, which is also consistent with what has been observed in Mytilus galloprovincialis exposed to Cu2+41. In contrast, hemocyanin-like protein, a crucial immune protein in arthropods42,43,44, had significantly increased expression after exposure to Cd2+. Heavy metals are handled through separate metabolic pathways dependent on hemocyanin45. In addition, the expression level of heat shock proteins (Hsps), common stress-inducible proteins, has been known to increase under various stressors, such as oxidative stress, heavy metals, and viral infections46,47,48. For instance, Hsp70, Hsp40, and Hsp105, were significantly up-regulated in Eubalaena glacialis exposed to Cd2+. Interestingly, in M. rosenbergii, Hsp67B2 was consistently decreased in the hepatopancreas for three days under Cd2+ exposure, suggesting that Hsp67B2 may be suppressed by Cd2+ in this prawn. On the other hand, the expression level of IFRD1 was consistently increased in hepatopancreas under Cd2+ stress, which was consistent with the high upregulation of this gene in hepatopancreas of M. rosenbergii after virus infection49. IFRD1 protein has been proven to be involved in the regulation of inflammatory responses50, indicating that the increased expression of IFRD1 is intended to cure inflammation caused by Cd2+.
Nevertheless, further study is required to illustrate the regulatory mechanism of M. rosenbergii after exposure to Cd2+. The degree of histological damage of the gills and hepatopancreas under different concentrations and exposure days of Cd2+ is worth exploring in future research. Additionally, the effects of Cd2+ on the mitochondrion structure in the gill and on superoxide dismutase (SOD) activity still need to be investigated.
Conclusion
In conclusion, we successfully constructed comparative gill and hepatopancreas transcriptome datasets in Cd2+ treated groups and control group of M. rosenbergii. Thereafter, 48,991 unigenes were functionally annotated and a series of DEGs were isolated after Cd2+ exposure. Based on GO functional and KEGG pathway analyses, many DEGs that are potentially relevant to immune responses, antioxidant, and detoxification were identified.
Material and methods
Collection and maintenance of prawns
A total of nine female and nine male M. rosenbergii (23 ± 2.5 g) individuals used in this experiment were collected from Dinghe Aquatic Science and Technology Development Co. LTD (Jiangsu, China) and transported back to our laboratory. The prawns were maintained at 26 ± 2 °C in a 50-L aerated aquarium for three days before treatment. All animals were handled in accordance with guidelines established by the Animal Experiments Ethics Committee of Shanghai Ocean University for the care and use of laboratory animals.
Cadmium exposure experiment
Firstly, Cd2+ solution (50 mg/L) was prepared by dissolving 102 mg of CdCl2·2.5 H2O in 1 L deionized water. After temporary rearing, CdCl2 solution was added to the culture water and mixed immediately, so as to expose all the prawns to the Cd2+ (80 μg/L) based on the 96-h LC50 of Cd in M. rosenbergii51. Every day, the prawns were fed and the water was renewed by 50% to maintain water quality. Subsequently, the experimental prawns were anesthetized on ice and dissected. The gills (Gi) and hepatopancreas (Hp) were randomly sampled from six individuals (three males and three females) for each of the 3 time points: 0 h, 3 h and 3 d, after Cadmium exposure, and stored at − 80 °C immediately for the following RNA extraction.
Library construction and gene function annotation
Total RNA was extracted from gills and hepatopancreas using Trizol reagent (Invitrogen, USA). The purity and amount of the 36 RNA samples were assessed by NanoDrop2000C, and RNA integrity was verified by agarose electrophoresis. The RNA integrity was assessed by Agilent 2100 (RIN number > 6.5). For each group, equal amount of RNA from the six individuals were pooled to make a sample for library construction. Three replicates were conducted for each library for statistics and comparison. Subsequently, the mRNA was purified and submitted to synthesize cDNA. Finally, the resulting six libraries (Gi 0 h, Gi 3 h, Gi 3 d, Hp 0 h, Hp 3 h, and Hp 3 d) were sequenced on Illumina Hiseq platform at Maiorbio company (Shanghai, China). Raw reads were trimmed by deleting adapter, ploy-N and low-quality reads. Then, the remaining clean reads were assembled into longer contigs using Trinity. The longest transcripts of each gene were defined as unigenes. Assembled unigenes were annotated by comparison to six databases, including NCBI non-redundant protein database (NR), Swiss-Prot, Pfam, Cluster of Orthologous Groups of proteins (COG), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/pathway.html)52,53,54. The RNA-seq data have been deposited in the NCBI database under the accession number PRJNA707962.
Gene expression analysis
Gene expression values were calculated, using RSEM and measured as transcripts per kilobase per million mapped reads (TPM), for the six cDNA libraries. Correlation coefficients of samples were computed using Pearson correlation. DESeq2 (with parameters of: p-value < 0.05 and fold-change ≥ 2) was applied for analysis of differentially expressed genes (DEGs). The DEGs between the library pairs (Gi 0 h vs Gi 3 h, Gi 0 h vs Gi 3 d, Gi 3 h vs Gi 3 d Hp 0 h vs Hp 3 h, Hp 0 h vs Hp 3 d, and Hp 3 h vs Hp 3 d) were identified. Then GO and KEGG functional classification were performed to identify which DEGs were significantly enriched in GO terms and metabolic pathways.
Validation of DEGs expression profiles using quantitative real-time RT-PCR (qPCR)
To validate the Illumina sequencing results, the six pooled RNA samples originally used for transcriptome sequencing were analyzed by qPCR. Six randomly selected genes: metallothionein (MT), hemocyanin-like protein (Hemo), interferon-related developmental regulator 1 (IFRD1), heat shock protein 67B2 (Hsp 67B2), zinc finger BED domain-containing protein 4 (Zbed4), and gamma-glutamyl hydrolase (GH), were amplified by specific primers (Table 4). QPCR mixture (20 μL) contained 10 μL of PCR Master with SYRB green, 1 μL Cd2+ cDNA template (10 ng/ul), 0.25 μL of each primer (10 uM), and 8.5 μL H2O. The primers of β-actin were used as the internal control. The relative quantification of the six genes was calculated by the 2-△△CT method55. Analysis of qPCR results was performed in GraphPad Prism 8. All data were presented as means ± SD.
Approval statement
All experimental protocols were approved by the Key Laboratory of Freshwater Aquatic Genetic Resources, Ministry of Agriculture, Shanghai Ocean University in this paper.
ARRIVE guidelines statement
This study was carried out in compliance with the ARRIVE guidelines.
References
Idrus, F. A., Basri, M. M., Rahim, K. A. A., Abd Rahim, N. S. & Chong, M. D. Concentrations of cadmium, copper, and zinc in Macrobrachium rosenbergii (giant freshwater prawn) from natural environment. Bull. Environ. Contam. Toxicol. 100, 350–355. https://doi.org/10.1007/s00128-018-2270-3 (2018).
Borrego, J., Morales, J. A., de la Torre, M. L. & Grande, J. A. Geochemical characteristics of heavy metal pollution in surface sediments of the Tinto and Odiel river estuary (southwestern Spain). Environ. Geol. 41, 785–796. https://doi.org/10.1007/s00254-001-0445-3 (2002).
Rasmussen, A. D. & Andersen, O. Effects of cadmium exposure on volume regulation in the lugworm, Arenicola marina. Aquat. Toxicol. 48, 151–164. https://doi.org/10.1016/s0166-445x(99)00045-4 (2000).
Asuquo, F. E., Ewa-Oboho, I., Asuquo, E. F. & Udo, P. J. Fish species used as biomarker for heavy metal and hydrocarbon contamination for cross river, Nigeria. Environmentalist 24, 29–37. https://doi.org/10.1023/B:ENVR.0000046344.04734.39 (2004).
Papagiannis, I., Kagalou, I., Leonardos, J., Petridis, D. & Kalfakakou, V. Copper and zinc in four freshwater fish species from Lake Pamvotis (Greece). Environ. Int. 30, 357–362. https://doi.org/10.1016/j.envint.2003.08.002 (2004).
Zeeshan, M., Murugadas, A., Ghaskadbi, S., Rajendran, R. B. & Akbarsha, M. A. ROS dependent copper toxicity in Hydra-biochemical and molecular study. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 185, 1–12. https://doi.org/10.1016/j.cbpc.2016.02.008 (2016).
Schwarz, J. A., Mitchelmore, C. L., Jones, R., O’Dea, A. & Seymour, S. Exposure to copper induces oxidative and stress responses and DNA damage in the coral Montastraea franksi. Comp. Biochem. Physiol Part C: Toxicol. Pharmacol. 157, 272–279. https://doi.org/10.1016/j.cbpc.2012.12.003 (2013).
Wu, J. P. & Chen, H. C. Effects of cadmium and zinc on oxygen consumption, ammonium excretion, and osmoregulation of white shrimp (Litopenaeus vannamei). Chemosphere 57, 1591–1598. https://doi.org/10.1016/j.chemosphere.2004.07.033 (2005).
Barbieri, E. Use of oxygen consumption and ammonium excretion to evaluate the sublethal toxicity of cadmium and zinc on litopenaeus schmitti (burkenroad, 1936, crustacea). Water Environ. Res. 79, 641–646. https://doi.org/10.2175/106143006x136775 (2007).
Barbieri, E. Effects of zinc and cadmium on oxygen consumption and ammonium excretion in pink shrimp (Farfantepenaeus paulensis, Pérez-Farfante, 1967, Crustacea). Ecotoxicology 18, 312–318. https://doi.org/10.1007/s10646-008-0285-y (2009).
Su, H. et al. Transcriptome analysis of the ovary of beet armyworm Spodoptera exigua under different exposures of cadmium stress. Chemosphere 251, 126372. https://doi.org/10.1016/j.chemosphere.2020.126372 (2020).
Jones, M. M. & C, C. Cadmium, a unique metal. Toxicol. J. 62, 1–25 (1990).
Thévenod, F. Cadmium and cellular signaling cascades: To be or not to be?. Toxicol. Appl. Pharmacol. 238, 221–239. https://doi.org/10.1016/j.taap.2009.01.013 (2009).
Revathi, P., Vasanthi, L. A. & Munuswamy, N. Effect of cadmium on the ovarian development in the freshwater prawn Macrobrachium rosenbergii (De Man). Ecotoxicol. Environ. Saf. 74, 623–629. https://doi.org/10.1016/j.ecoenv.2010.08.027 (2011).
Novais, S. C., Coen, W. D. & Amorim, M. J. B. Transcriptional responses in Enchytraeus albidus (Oligochaeta): Comparison between cadmium and zinc exposure and linkage to reproduction effects. Environ. Toxicol. Chem. 31, 2289–2299. https://doi.org/10.1002/etc.1946 (2012).
Biagioli, M., Wtjen, W., Beyersmann, D., Zoncu, R. & Bucci, S. Cadmium-induced apoptosis in murine fibroblast is suppressed by Bcl-2. Arch. für Toxikologie 75, 313–320. https://doi.org/10.1007/s002040100236 (2001).
Mao, W. P. et al. Cadmium induces apoptosis in human embryonic kidney (HEK) 293 cells by caspase-dependent and -independent pathways acting on mitochondria. Toxicol. In Vitro 21, 343–354. https://doi.org/10.1016/j.tiv.2006.09.004 (2007).
Gaspic, Z. K., Zvonaric, T., Vrgoc, N., Odzak, N. & Baric, A. Cadmium and lead in selected tissues of two commercially important fish species from the Adriatic Sea. Water Res. 36, 5023–5028. https://doi.org/10.1016/s0043-1354(02)00111-2 (2002).
Sun, M., Li, Y. T., Liu, Y., Lee, S. C. & Wang, L. Transcriptome assembly and expression profiling of molecular responses to cadmium toxicity in hepatopancreas of the freshwater crab Sinopotamon henanense. Sci. Rep. 6, 19405. https://doi.org/10.1038/srep19405 (2016).
Tang, D., Guo, H., Shi, X. & Wang, Z. Comparative transcriptome analysis of the gills from the chinese mitten crab (Eriocheir japonica sinensis) exposed to the heavy metal cadmium. Turk. J. Fish. Aquat. Sci. 20, 467–479. https://doi.org/10.4194/1303-2712-v20_6_05 (2020).
P., W. , et al. The organization and control of metabolism in the crustacean gill. Comp. Biochem. Physiol. 33, 529–548. https://doi.org/10.1016/0010-406X(70)90369-5 (1970).
Henry, R. P., Lucu, C., Onken, H. & Weihrauch, D. Multiple functions of the crustacean gill: osmotic/ionic regulation, acid-base balance, ammonia excretion, and bioaccumulation of toxic metals. Front. Physiol. 3, 431. https://doi.org/10.3389/fphys.2012.00431 (2012).
Agoes, et al. Impact of copper on the structure of gills and epipodites of the shrimp Penaeus japonicus (Decapoda). J. Crustacean Biol. 2, 209–223. https://doi.org/10.2307/1549227 (1999).
Li, N., Zhao, Y. & Yang, J. Impact of waterborne copper on the structure of gills and hepatopancreas and its impact on the content of metallothionein in juvenile giant freshwater prawn Macrobrachium rosenbergii (Crustacea: Decapoda) (vol 52, pg 73, 2009). Arch. Environ. Contam. Toxicol 56, 811–812. https://doi.org/10.1007/s00244-009-9306-y (2009).
Vogt, G. Monitoring of environmental pollutants such as pesticides in prawn aquaculture by histological diagnosis. Aquaculture 67, 157–164. https://doi.org/10.1016/0044-8486(87)90021-4 (1987).
Baticados, M. C. L. & Tendencia, E. A. Effects of gusathion a on the survival and shell quality of juvenile penaeus monodon. Aquaculture 93, 9–19. https://doi.org/10.1016/0044-8486(91)90201-H (1991).
Sung, H. H., Kao, W. Y. & Su, Y. J. Effects and toxicity of phthalate esters to hemocytes of giant freshwater prawn, Macrobrachium rosenbergii. Aquat. Toxicol. 64, 25–37. https://doi.org/10.1016/s0166-445x(03)00011-0 (2003).
Guo, H., Deng, M., Liang, J., Lu, W. & Shen, Y. Gill transcriptome alterations in Macrobrachium rosenbergii under copper exposure. Chemosphere 233, 796–808. https://doi.org/10.1016/j.chemosphere.2019.06.025 (2019).
Kaoud, H. A. & Rezk, A. Effect of exposure to cadmium on the tropical freshwater prawn Macrobrachium rosenbergii. Afr. J. Aquat. Sci 36, 253–260. https://doi.org/10.2989/16085914.2011.636899 (2011).
Burnett, L. E. & McMahon, B. R. Facilitation of CO2 excretion by carbonic anhydrase located on the surface of the basal membrane of crab gill epithelium. Respir. Physiol. 62, 341–348. https://doi.org/10.1016/0034-5687(85)90089-1 (1985).
White, S. L. & Rainbow, P. S. Accumulation of cadmium by Palaemon elegans (Crustacea: Decapoda). Mar. Ecol. Prog. Ser. 32, 17–25. https://doi.org/10.3354/meps032017 (1986).
Xuan, R. et al. Oxygen consumption and metabolic responses of freshwater crab Sinopotamon henanense to acute and sub-chronic cadmium exposure. Ecotoxicol. Environ. Saf. 89, 29–35. https://doi.org/10.1016/j.ecoenv.2012.10.022 (2013).
Gao, M., Lv, M., Liu, Y. & Song, Z. Transcriptome analysis of the effects of Cd and nanomaterial-loaded Cd on the liver in zebrafish. Ecotoxicol. Environ. Saf. 164, 530–539. https://doi.org/10.1016/j.ecoenv.2018.08.068 (2018).
Bjerregaard, P. Influence of physiological condition on cadmium transport from haemolymph to hepatopancreas in Carcinus maenas. Mar. Biol. 106, 199–209. https://doi.org/10.1007/BF01314801 (1990).
Mouneyrac, C. et al. Trace-metal detoxification and tolerance of the estuarine worm Hediste diversicolor chronically exposed in their environment. Mar. Biol. 143, 731–744. https://doi.org/10.1007/s00227-003-1124-6 (2003).
Sterenborg, I. & Roelofs, D. Field-selected cadmium tolerance in the springtail Orchesella cincta is correlated with increased metallothionein mRNA expression. Insect Biochem. Mol. Biol. 33, 741–747. https://doi.org/10.1016/s0965-1748(03)00070-5 (2003).
Brouwer, M., Winge, D. R. & Gray, W. R. Structural and functional diversity of copper-metallothioneins from the American lobster Homarus americanus. J. Inorg. Biochem. 35, 289–303. https://doi.org/10.1016/0162-0134(89)84018-8 (1989).
Viarengo, A. & Nott, J. A. Mechanisms of heavy metal cation homeostasis in marine invertebrates. Comp. Biochem. Physiol Part C Toxicol. Pharmacol. 104, 355–372. https://doi.org/10.1016/0742-8413(93)90001-2 (1993).
Sappal, R. et al. Copper and hypoxia modulate transcriptional and mitochondrial functional-biochemical responses in warm acclimated rainbow trout (Oncorhynchus mykiss). Environ. Pollut. 211, 291–306. https://doi.org/10.1016/j.envpol.2015.11.050 (2016).
Liu, G.-D. et al. Molecular cloning, characterization and expression analysis of metallothionein in the liver of the teleost Acrossocheilus fasciatus exposed to cadmium chloride. Environ. Toxicol. Pharmacol. 53, 1–9. https://doi.org/10.1016/j.etap.2017.03.020 (2017).
Gomes, T., Chora, S., Pereira, C. G., Cardoso, C. & Bebianno, M. J. Proteomic response of mussels Mytilus galloprovincialis exposed to CuO NPs and Cu2+: An exploratory biomarker discovery. Aquat. Toxicol. 155, 327–336. https://doi.org/10.1016/j.aquatox.2014.07.015 (2014).
Yan, F. et al. Identification and agglutination properties of hemocyanin from the mud crab (Scylla serrata). Fish Shellfish Immunol. 30, 354–360. https://doi.org/10.1016/j.fsi.2010.11.004 (2011).
Decker, H. et al. Minireview: Recent progress in hemocyanin research. Integr. Comp. Biol. 47, 631–644. https://doi.org/10.1093/icb/icm063 (2007).
Decker, H. & Jaenicke, E. Recent findings on phenoloxidase activity and antimicrobial activity of hemocyanins. Dev. Comp. Immunol. 28, 673–687. https://doi.org/10.1016/j.dci.2003.11.007 (2004).
Pedrini-Martha, V. et al. Responsiveness of metallothionein and hemocyanin genes to cadmium and copper exposure in the garden snail Cornu aspersum. J. Exp. Zool., Part A https://doi.org/10.1002/jez.2425 (2020).
Singh, M. P., Reddy, M. M. K., Mathur, N., Saxena, D. K. & Chowdhuri, D. K. Induction of hsp70, hsp60, hsp83 and hsp26 and oxidative stress markers in benzene, toluene and xylene exposed Drosophila melanogaster: Role of ROS generation. Toxicol. Appl. Pharmacol. 235, 226–243. https://doi.org/10.1016/j.taap.2008.12.002 (2009).
Gupta, S. C., Sharma, A., Mishra, M., Mishra, R. K. & Chowdhuri, D. K. Heat shock proteins in toxicology: How close and how far?. Life Sci. 86, 377–384. https://doi.org/10.1016/j.lfs.2009.12.015 (2010).
Singh, M. P. et al. Effects of co-exposure of benzene, toluene and xylene to Drosophila melanogaster: Alteration in hsp70, hsp60, hsp83, hsp26, ROS generation and oxidative stress markers. Chemosphere 79, 577–587. https://doi.org/10.1016/j.chemosphere.2010.01.054 (2010).
Arockiaraj, J. et al. First report on interferon related developmental regulator-1 from Macrobrachium rosenbergii: Bioinformatic analysis and gene expression. Fish Shellfish Immunol. 32, 929–933. https://doi.org/10.1016/j.fsi.2012.02.011 (2012).
Micheli, L. et al. Pc4/tis7/ifrd1 stimulates skeletal muscle regeneration and is involved in myoblast differentiation as a regulator of myod and nf-κb. J. Biol. Chem. 286, 5691–5707. https://doi.org/10.1074/jbc.M110.162842 (2010).
Kaoud, H. A. & Rezk, A. Effect of exposure to cadmium on the tropical freshwater prawn Macrobrachium rosenbergii. Afr. J. Aquat. Sci. 36, 253–260. https://doi.org/10.2989/16085914.2011.636899 (2011).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551. https://doi.org/10.1093/nar/gkaa970 (2021).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR. Methods 25, 402–408. https://doi.org/10.1006/meth.2001.1262 (2002).
Acknowledgements
This research was supported by the Natural Science Foundation of China (project number 31902348).
Author information
Authors and Affiliations
Contributions
K.M. and X.L. conceived and designed the experiments; X.L. and H.Q. performed the experiments; J.F., Z.G., and H.B. analyzed the data; J.F., H.J., B.Y. and J.G. contributed reagents/materials/analysis tools; X.L. and K.M. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
Author Jinhua Gong was the general manager of Dinghe Aquatic Science and Technology Development Co., LTD. All authors declare that the research was conducted in the absence of any commercial or financial relationships.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.

Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Liu, X., Jiang, H., Ye, B. et al. Comparative transcriptome analysis of the gills and hepatopancreas from Macrobrachium rosenbergii exposed to the heavy metal Cadmium (Cd2+). Sci Rep 11, 16140 (2021). https://doi.org/10.1038/s41598-021-95709-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-95709-w
- Springer Nature Limited