
Abstract
Background
Genomic analysis of bacteriophages infecting the Burkholderia cepacia complex (BCC) is an important preliminary step in the development of a phage therapy protocol for these opportunistic pathogens. The objective of this study was to characterize KL1 (vB_BceS_KL1) and AH2 (vB_BceS_AH2), two novel Burkholderia cenocepacia-specific siphoviruses isolated from environmental samples.
Results
KL1 and AH2 exhibit several unique phenotypic similarities: they infect the same B. cenocepacia strains, they require prolonged incubation at 30°C for the formation of plaques at low titres, and they do not form plaques at similar titres following incubation at 37°C. However, despite these similarities, we have determined using whole-genome pyrosequencing that these phages show minimal relatedness to one another. The KL1 genome is 42,832 base pairs (bp) in length and is most closely related to Pseudomonas phage 73 (PA73). In contrast, the AH2 genome is 58,065 bp in length and is most closely related to Burkholderia phage BcepNazgul. Using both BLASTP and HHpred analysis, we have identified and analyzed the putative virion morphogenesis, lysis, DNA binding, and MazG proteins of these two phages. Notably, MazG homologs identified in cyanophages have been predicted to facilitate infection of stationary phase cells and may contribute to the unique plaque phenotype of KL1 and AH2.
Conclusions
The nearly indistinguishable phenotypes but distinct genomes of KL1 and AH2 provide further evidence of both vast diversity and convergent evolution in the BCC-specific phage population.
Similar content being viewed by others


Background
The clinical administration of bacteriophages, referred to as phage therapy, has now been used to treat bacterial infections for nearly a century. Although this type of therapy had been largely abandoned outside of Eastern Europe since antibiotics became available in the 1940s, the emergence of antibiotic-resistant pathogens has re-established phage therapy as a viable antibacterial treatment[1]. Recent studies have shown that phages and phage components are effective both in animal models (against species such as Staphylococcus, Pseudomonas, Klebsiella, Escherichia, Salmonella, and Campylobacter) and in human clinical trials[2–8]. Advances in phage delivery and storage (such as nebulization, lyophilization, and spray drying for respiratory phage therapy) and genomic characterization (including high-throughput sequencing and annotation) have made phage therapy more feasible with respect to both logistics and safety[9–12].
One group of bacteria that is thought to be an excellent target for phage therapy is the Burkholderia cepacia complex (BCC). These bacterial species, which primarily infect patients with cystic fibrosis (CF), are problematic because they can cause serious illness (including, in up to 20% of cases, a fatal necrotizing pneumonia referred to as ‘cepacia syndrome’), they are capable of patient-to-patient spread (particularly in settings such as CF centers), and, perhaps most importantly, they are highly antibiotic resistant[13–16]. Very few antibiotics are active against the BCC, even in combination: Zhou et al.[16] tested a panel of antibiotics against BCC clinical isolates and determined that less than half of the strains were susceptible to even the most effective drugs. Clinically, the most commonly isolated BCC species are Burkholderia multivorans and Burkholderia cenocepacia, with the latter thought to be the most pathogenic[17]. BCC phage therapy trials have focused on this species and, to date, phages have been shown to be effective against B. cenocepacia in both invertebrate and mammalian infection models[18–20].
As B. cenocepacia infections are some of the most problematic for the CF community, the isolation and characterization of novel phages that infect this species remains a priority. Many of these phages have been isolated in recent years, but only some have been fully sequenced (reviewed in[21, 22]). Here, we describe the isolation and characterization of KL1 and AH2, two novel B. cenocepacia-specific phages with identical host ranges and unique growth characteristics, but strikingly dissimilar genomes.
Results and discussion
Isolation, host range and morphology
KL1 was isolated from sewage using B. cenocepacia K56-2 as a host. In contrast to enterobacteria phages, which are commonly found in sewage[23], this is the first report of BCC phage isolation from this source. AH2 was isolated from Nandina sp. (also known as heavenly bamboo) soil using B. cenocepacia C6433. BCC phages have commonly been isolated from both rhizospheres and soil samples, including that of onion and Dracaena sp.[18, 20, 24–27].
KL1 and AH2 are very similar with respect to both host range and growth characteristics. These phages have a relatively narrow tropism, infecting B. cenocepacia K56-2, C6433, 715J, and K63-3. Both KL1 and AH2 exhibit a pattern of lysis that is unique in our collection of BCC-specific phages: although high titre stocks of these phages are very concentrated (up to 1011 plaque forming units [PFU]/ml), these phages do not produce clear lysis in agar overlays after 16 h incubation like other phages that we have previously characterized[18, 19, 26, 28–30]. Instead, turbid or no clearing is observed at high titres, with mottling or individual plaques observed at lower titres (approximately 107 PFU/ml or less). At low titres, incubation at 30°C for greater than 16 h is required for plaque formation, but plaques are not observed if incubation is at 37°C (Figure1). Individual plaques are turbid with a diameter of 0.5-2 mm (larger plaques may have a punctate appearance). When tested with a panel of K56-2 mutants with progressive deficiencies in lipopolysaccharide (LPS) structure (from the O-antigen to the core)[31, 32], both phages were able to infect each mutant, suggesting that neither KL1 nor AH2 uses LPS as a major receptor.

Development and morphology of KL1 and AH2 plaques. Phages were plated in half-strength Luria-Bertani (½ LB) agar overlays with a 16 h liquid culture of Burkholderia cenocepacia C6433. Plates were incubated at 30°C or 37°C and photographed after 16, 24, and 48 h. C6433 30°C plates (center) are representative of growth at both 30°C and 37°C.
Both KL1 and AH2 belong to the order Caudovirales and family Siphoviridae as determined by electron microscopy. The KL1 virion has a non-contractile tail approximately 160 nm in length and a capsid approximately 55 nm in diameter (Figure2A). The AH2 virion is slightly larger, with a non-contractile tail approximately 220 nm in length and a capsid approximately 60 nm in diameter (Figure2B). The stacked rings comprising the tail structure are visible in the AH2 micrograph (Figure2B).

KL1 (A) and AH2 (B) virion morphology. Phages were stained with 2% phosphotungstic acid and visualized at 180,000-fold magnification by transmission electron microscopy. Scale bars represent 50 nm.
Genome characterization
Despite the similarities in phenotype between KL1 and AH2 with respect to host range and growth characteristics, the genomes of these two phages are dissimilar. Restriction fragment length polymorphism (RFLP) analysis shows distinct banding patterns of EcoRI-digested KL1 and AH2 genomic DNA, suggesting that their sequences are substantially different (Figure3). This prediction is confirmed by the results of whole genome pyrosequencing (discussed below) and is illustrated in Figure4A: in a Circos plot of a PROmer comparison of these two phages, no regions of similarity at the protein level are observed under the parameters used.

RFLP analysis of KL1 and AH2 genomic DNA. 5 μg of genomic DNA were digested overnight with EcoRI and separated on a 0.8% agarose gel. The DNA in the ambient gel (left) was not heated, while the DNA in the 80°C gel (right) was incubated 20 min at 80°C and chilled on ice prior to loading. Arrows indicate bands containing cos site DNA. L: 1 Kb Plus DNA Ladder (Invitrogen).

Circos plots of KL1 and AH2 PROmer comparisons. Green ribbons indicate regions of similarity between two genomes at the protein level. Each region is on the same strand in both genomes. The scale (in kbp) is shown on the periphery of the plots. PROmer parameters: breaklen = 60, maxgap = 30, mincluster = 20, minmatch = 6. A) KL1/AH2 comparison; B) KL1/Pseudomonas phage 73 (PA73) comparison; C) AH2/Burkholderia phage BcepNazgul comparison.
The KL1 genome is 42,832 base pairs (bp) in length and has a 54.6% GC content. This percentage is lower than that for most Burkholderia-specific phages, which tend to have GC contents between 60–65% (excluding phages such as BcepB1A [54.5%], BcepF1 [55.9%], and BcepGomr [56.3%]). We were unable to identify a KL1 cos site following incubation of the DNA at 80°C, as the RFLP profiles appeared identical both before and after heating (Figure3). KL1 is predicted to encode 55 proteins, all of which have an ATG start codon, except for gp2 which has a GTG codon (Figure5, Table1).

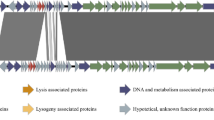
Genome maps of KL1 and AH2. Genes transcribed in the forward direction are shown above and those transcribed in the reverse direction are shown below. The scale (in kbp) is shown below the maps. Legend: light blue, lysis; purple, capsid morphogenesis and DNA packaging; pink, tail morphogenesis; red, DNA binding; green, MazG; gray, unknown function.
KL1 is most similar to Pseudomonas phage 73 (PA73; NC_007806), a siphovirus that infects Pseudomonas aeruginosa[33]. These phages are similar with respect to genome length (42,999 bp for PA73 and 42,832 bp for KL1), GC content (53.6% for PA73 and 54.6% for KL1), and predicted number of proteins (52 for PA73 and 55 for KL1). BLASTN comparison of KL1 and PA73 indicates that these sequences are similar over 69% of the KL1 genome. KL1 encodes a protein most similar to each PA73 protein from ORF001–ORF052 (excluding 12 proteins) (Table1). Most PA73 proteins show limited similarity to others in the NCBI database and have not been assigned a putative function[33]. Of the 9 PA73 proteins with predicted functions, all but one (peptidyl-tRNA hydrolase [peptide chain release factor]) is similar to a KL1 protein: holin, terminase large subunit, head morphogenesis protein, tail tape measure protein, DNA polymerase, superfamily II helicase/restriction enzyme, helicase (annotated here as recombinase), and dCMP deaminase (KL1 gp2, gp7, gp9, gp21, gp27, gp30, gp33, and gp52, respectively) (Table1). Of the KL1 proteins most similar to a PA73 protein, the most similar is gp33 (91% identity with ORF032) and the least similar is gp24 (36% identity with ORF023) (Table1). In a Circos plot of a PROmer comparison of these phages, the majority of the two genomes are similar at the protein level (Figure4B).
The AH2 genome is 58,065 bp in length and has a 61.3% GC content. Incubation of the DNA at 80°C caused a shift in the RFLP profile (Figure3), suggesting the presence of a cos site. Sequencing of the shifted fragments indicates that AH2 has a 12 bp 5’ overhang cos site with a sequence almost identical (1 bp difference) to that of Burkholderia phage BcepNazgul (NC_005091). AH2 is predicted to encode 78 proteins (Figure5, Table2). The majority of the start codons (70) are ATG, 6 are GTG and 2 are TTG (Table2).
AH2 is most similar to BcepNazgul, a siphovirus isolated from soil that infects Burkholderia ambifaria. Like PA73 and KL1, these phages are similar with respect to genome length (57,455 bp for BcepNazgul and 58,065 bp for AH2), GC content (60.6% for BcepNazgul and 61.3% for AH2), and predicted number of proteins (73 for BcepNazgul and 78 for AH2). In contrast to KL1 (which is closely related to a single phage), AH2 encodes proteins similar to those from a variety of bacteria and phages (Table2) and so is less closely related to BcepNazgul than KL1 is to PA73. BLASTN comparison of AH2 and BcepNazgul indicates that these sequences are similar over 16% of the AH2 genome. Twenty-one AH2 proteins are most similar to a BcepNazgul protein (Table2) and 39 show some similarity based on BLASTP analysis. Of the AH2 proteins most similar to a BcepNazgul protein, the most similar is gp12 (74% identity with Nazgul10) and the least similar is gp20 (24% identity with Nazgul21) (Table2). In a Circos plot of a PROmer comparison of these phages, the most similar regions at the protein level correspond to AH2 gp12, gp71, gp78 (similar to BcepNazgul Nazgul10, helicase, and DR0530-like primase, respectively) and a portion of the putative capsid morphogenesis and DNA packaging module (Figure4C).
Module analysis
Overview
We have identified the proteins encoded by KL1 and AH2 as belonging to four different functional categories: virion morphogenesis (including capsid morphogenesis/DNA packaging and tail morphogenesis), lysis, DNA binding (the largest and broadest category), and MazG (a pyrophosphohydrolase[34]). Although the proteins encoded by each phage perform many of the same functions (e.g. both KL1 gp11 and AH2 gp62 are predicted to be major capsid proteins) (Tables1 and2), the proteins themselves are dissimilar. As we discuss below, the finding that KL1 and AH2 can create nearly identical phenotypes with two dissimilar sets of proteins may be compelling evidence for convergent evolution occurring in these BCC-specific phages.
Virion morphogenesis
Although we have determined that KL1 is a siphovirus (Figure2A), the identity of many of the structural genes remains unknown. As discussed above, KL1 is most closely related to PA73, a phage whose proteins have largely uncharacterized functions. Based on BLASTP analysis, we have been able to predict the identity of only eight KL1 structural proteins: three involved in capsid morphogenesis and DNA packaging and five involved in tail morphogenesis. Gp7 (terminase large subunit) and gp9 (head morphogenesis protein) are similar to PA73 ORF006 and ORF008, respectively, both of which have been assigned putative functions in the PA73 annotation (Table1). Gp11 (major capsid protein) is similar to the major capsid proteins of Escherichia phage K1H and Listonella phage ϕHSIC. Gp20 is similar to tail proteins from multiple Escherichia phages including K1G, K1H, and K1ind1-K1ind3. Gp21 is predicted to be the tail tape measure as it is the largest protein encoded by KL1 (1272 amino acids [aa]) and it is similar to the predicted PA73 tape measure protein ORF020 (Table1). Finally, gp24-gp26 are similar to BcepNazgul tail assembly proteins. Using HHpred analysis, we were able to identify an additional three proteins at a probability threshold of 75%. Gp8 is similar to bacteriophage SPP1 portal protein (99.44% probability), gp15 is similar to λ gpFII head-tail joining protein (82.86% probability), and gp16 is similar to λ gpU minor tail protein (77.70% probability) (Additional file 1: Table S1).
In comparison with KL1, the structural proteins of AH2 are well defined. Genes 62–68 make up the capsid morphogenesis and DNA packaging module, containing genes encoding the major capsid protein, decorator protein, prohead protease, portal protein, head-tail joining protein, and terminase subunits (large and small) (Table2). Each of these proteins is similar to a BcepNazgul protein, with percent identities between 49-58%. Several genes between 47 and 56 are similar to genes encoding BcepNazgul conserved tail assembly proteins, tape measure protein, and pre-tape measure frameshift protein G-T (with percent identities between 26-38%). Two additional AH2 tail proteins were identified using BLASTP (gp46, similar to Pseudomonas psychrotolerans L19 phage tail fiber protein) or HHpred (gp58, similar to λ gpU minor tail protein) analysis (Additional file 2: Table S2). Hypothetical proteins encoded in this region are likely to be involved in tail morphogenesis based on the proximity of their genes to this module.
Most tailed phages encode two tail proteins proximal to the tail tape measure gene by way of a −1 translational frameshift[35]. We have previously identified these frameshifted genes in the BCC-specific phages KS9, KS5, KS14, and KL3[19, 29]. Using FSFinder and manual scanning for XXXYYYZ motifs, we predict that KL1 gp18/gp19 and AH2 gp55/gp56 are expressed using this mechanism. The predicted frameshift site in KL1 is GGGAAAC, immediately upstream of the gp18 TGA stop codon (Figure6 andAdditional file 3: Figure S3). A −1 ribosomal shift following the terminal C will allow for expression of the 264 aa gp19 and the 142 aa gp18 from the same start codon (Figure6). Although most phages encode their frameshifted proteins immediately upstream of the tail tape measure gene, KL1 encodes an intervening tail protein, gp20 (Table1, Figure5). This organization is similar to that of Escherichia coli phage HK97, Bacillus subtilis phage SPP1, Methanobacterium thermoautotrophicum phage ψM2, Methanothermobacter wolfei phage ψM100, Lactococcus phages c2 and BIL67, and Natrialba magadii phage ϕch1[35]. The predicted frameshift site in AH2 is AAAAAAG (Figure6 andAdditional file 3: Figure S3), the same sequence used by E. coli phage VT1-Sakai, M. thermoautotrophicum phage ψM2, Staphylococcus aureus phages PVL and PV83, Lactococcus lactis phage ul36, and Borrelia burgdorferi prophage Borreliapro[35]. In the case of AH2, a −1 shift of the ribosome following the G in this sequence will allow for the 228 aa gp55 to be expressed instead of the 146 aa gp56 (Figure6). Using BLASTP or HHpred searches, we were unable to identify the KL1 or AH2 major tail proteins. However, we predict that these proteins may be gp17 in KL1 and gp57 in AH2 as the major tail genes are generally positioned upstream of the frameshifted protein genes[35]. Although not present in all sequences, RNA secondary structures are often found downstream of frameshift sites[19, 29, 35, 36]. Mfold analysis of the 35 bases downstream of the putative KL1 and AH2 sites suggests that stem-loop structures could form in both of these regions (Additional file 3: Figure S3).

Sequences of the KL1 and AH2 predicted translational frameshift sites. For each phage, the first row shows the DNA sequence (with the predicted frameshift site underlined); the second row shows the amino acid sequence in the original frame (the KL1 gp18 stop codon is represented by an asterisk); the third row shows the amino acid sequence in the −1 frame; the fourth row shows the amino acid sequence of the frameshifted protein.
Lysis
In KL1, we have identified the genes putatively encoding the holin, lysin, Rz and Rz1 lysis proteins. In a BLASTP search, gp2 shows similarity to putative holin proteins of PA73 and BcepNazgul. TMHMM analysis of this protein indicates that it has two transmembrane domains, so gp2 is predicted to be a class II holin[37]. Gp3 is similar to the endolysin of Erwinia phage vB_EamP-S6 (HQ728266) and contains lysozyme and peptidoglycan-binding conserved domains. Although gp4 does not show similarity to any Rz proteins in the NCBI database, it is predicted to contain a single N-terminal transmembrane domain, characteristic of Rz proteins[38]. Gp5 is predicted to be the KL1 Rz1 protein as it is similar to BcepNazgul Rz1 and LipoP analysis identifies a signal peptidase II cleavage site between positions 17 and 18 (resulting in a 70 aa protein with 4 proline residues [5.7% proline]). The proportion of prolines in the predicted Rz1 lipoprotein is low compared to previously identified Rz1 proteins in BCC phages[19, 29, 39].
The same lysis proteins were identified in AH2. Like KL1 gp2, the putative AH2 holin gp43 is similar to the BcepNazgul holin, has two transmembrane domains, and is predicted to be a class II holin. Although gp42 shows no similarity to endolysins in a BLASTP search, HHpred analysis reveals similarity to both eukaryotic and prokaryotic lysozyme proteins. Gp41 is predicted to be the AH2 Rz protein as it has a single N-terminal transmembrane domain. Although manual annotation has been required for identification of the Rz1 gene in KL1 and in our previous studies[19, 29], we predict that the GeneMark-assigned gp40 is the AH2 Rz1 protein. Gp40 is similar to BcepNazgul Rz1 and has a signal peptidase II cleavage site between amino acids 15 and 16. Similar to the predicted KL1 Rz1, the proportion of prolines present in this protein is relatively low (3/61 or 4.9%). It is unclear from this analysis what protein(s) may contribute to the unique plaque phenotype observed in both of these phages. Aside from the low proportion of proline found in the putative Rz1 proteins, KL1 and AH2 appear to have relatively standard lysis modules, suggesting that unique (and as yet unidentified) proteins may be responsible for controlling lysis timing in each phage.
DNA binding
Of the 8 KL1 proteins similar to a PA73 protein with an assigned function, half of these are DNA- or nucleotide-binding proteins: DNA polymerase (gp27), superfamily II helicase/restriction enzyme (gp30), helicase (annotated here as recombinase [gp33]), and dCMP deaminase (gp52) (Table1). In addition, KL1 encodes a putative DNA polymerase III β subunit (gp28), exonuclease (gp31), transcriptional regulator (gp36), primase (gp37), and Vsr endonuclease (gp44) (Table1 andAdditional file 1: Table S1). In a multi-genome analysis performed by Lopes et al.[40], it was determined that PA73 ORF032 is distantly related to Lactococcus phage ϕ31 Sak4 recombinase. When this protein was expressed in E. coli, it exhibited recombinase activity, but was found to be less efficient than λ Redβ[40]. Furthermore, PA73 encodes an exonuclease, as is found in characterized phage recombinase pairs such as Redαβ in λ and RecET in rac[40]. KL1 gp33 is most closely related to PA73 ORF032 and, with 91% identity, is the KL1 protein most similar to a PA73 protein. In addition, KL1 gp31 has 65% identity with PA73 ORF030 and both of these proteins are similar to λ Redα (99.21% probability for gp31 and 99.17% probability for ORF030) (Table1 andAdditional file 1: Table S1). It is interesting to note that, despite the relatively limited similarity between KL1 and other previously sequenced BCC-specific phages, both gp31 and gp33 are similar to proteins from Burkholderia phage BcepGomr (BcepGomrgp43 and BcepGomrgp45, respectively)[40]. Although further characterization of these proteins is required in both KL1 and BcepGomr, it is possible that these exonucleases and Sak4-like recombinases represent a conserved recombination system in certain BCC-specific phages.
AH2 encodes DNA replication, modification, and repair proteins including a putative DNA polymerase III β subunit (gp20), Vsr endonuclease (gp32), excinuclease (gp33), restriction endonuclease/methylase pair (gp34/gp35), integrase (gp37), helicase (gp71), resolvase (gp72), DNA polymerase (gp73), single-stranded DNA binding protein (gp74), Cas4 superfamily exonuclease (gp75), and primase (gp78) (Table2). Other putative DNA binding proteins are predicted to be involved in transcriptional regulation. Gp28 is similar to partitioning and regulation proteins from Thermus thermophilus (100% probability) and E. coli (99.86% probability) (Additional file 2: Table S2). The gp30 and gp31 predicted proteins belong to the helix-turn-helix MerR superfamily and the pyocin activator superfamily, respectively. Both of these proteins, in addition to gp69, also show similarity to excisionase proteins (Additional file 2: Table S2). Gp70 and gp77 are similar to the lysogeny control proteins CI from enterobacteria phage 186 (99.87% probability) and Cro from Xylella fastidiosa Ann-1 (96.60% probability), respectively (Additional file 2: Table S2).
AH2 gp32-gp35 are predicted to be part of a DNA protection and repair module. Vsr (very short patch repair) endonucleases are involved in the repair of 5-methylcytosine to thymine deamination[41]. Previously, we identified a Vsr endonuclease in the BCC-specific phage KL3 that, along with an EcoRII-C endonuclease/methylase pair, was predicted to be part of a novel non-self DNA degradation and self DNA protection/repair module[29]. Our model proposed that non-KL3 DNA (i.e. that of the host or a superinfecting phage) would be degraded by the endonuclease (gp45), while KL3 DNA would be protected by the methylase (gp47) (converting cytosine to 5-methylcytosine). Vsr endonuclease (gp46) and very short patch repair would then prevent the accumulation of mutations caused by 5-methylcytosine deamination[29].
The DNA protection and repair system of AH2 is analogous to that of KL3. AH2 gp32 has 51% identity with the KL3 Vsr endonuclease and is similar to E. coli Vsr endonuclease (100% probability) (Additional file 2: Table S2). AH2 also encodes an endonuclease/methylase pair: gp34 is similar to Kluyvera ascorbata KasI (64% identity) while gp35 is similar to K. ascorbata M.KasI, Brevundimonas diminuta ATCC 11568 cytosine-specific methyltransferase NlaX, and Acetobacter pomorum DM001 modification methylase HpaII (63-66% identity). Gp35 also has several methylase conserved domains, including Dcm (an enzyme that produces 5-methylcytosine bases at sites recognized by Vsr endonuclease)[41]. Gp33 is similar to Thermotoga maritima UvrABC system protein C (98.35% probability) and could function together with UvrAB in nucleotide excision repair (Additional file 2: Table S2)[42]. Although further experiments are required to identify the recognition sites of gp34 and gp35, we predict that this module may function as follows: gp34 cleaves non-self DNA, while self DNA is protected by gp35 methylation and subsequent gp32 repair (with gp33 participating in nucleotide excision repair). Although the identity and arrangement of genes in this module is different in AH2 than in KL3, the identification of a similar module in an unrelated BCC-specific phage suggests that these genes may be widely used for DNA protection and repair in this group of viruses.
MazG
A notable protein encoded by both KL1 and AH2 is MazG. MazG is a pyrophosphohydrolase that acts on ppGpp, one of the signaling molecules in bacteria produced during the stringent response[43]. When bacterial cells are in an amino acid-limited environment, RelA synthesizes pppGpp, the precursor of ppGpp, and the latter activates the expression of genes required for cell survival (such as rpoS) and represses genes required for protein and DNA synthesis (reviewed in[44]). Recently, there has been a great deal of interest in marine phages (especially cyanophages) that encode MazG homologs, such as Prochlorococcus phages P-SSM2 and P-SSM4, Synechococcus phage S-PM2, Prochlorococcus and Synechococcus phage Syn9, Roseobacter phage SIO1, Pseudoalteromonas phage H105/1, almost one-fifth of the cyanophages tested by Bryan et al.[45], and all of the cyanophages analyzed by Sullivan et al.[46][47–51]. It has been suggested that these MazG-encoding phages are better able to infect and propagate within their hosts, which are found in nutrient-limited water. By inactivating ppGpp, these phages can promote the expression of genes that would usually be expressed by an exponential phase cell under nutrient-rich conditions, such as those required for protein and DNA synthesis[52]. There are few published reports of the mazG gene in non-marine phages, but it has been previously identified in Myxococcus phage Mx8 and mycobacteriophage L5[45].
The putative MazG proteins encoded by KL1 and AH2 are gp35 and gp25, respectively. KL1 gp35 is similar to putative MazG proteins from phages infecting Synechococcus (including S-CRM01, S-SM2, and S-ShM2), Prochlorococcus (including P-HM1, P-HM2, and P-SSM2), and Bacillus (0305ϕ8-36), as well as to PA73 hypothetical protein ORF034 (Table1). AH2 gp25 is similar to putative Clostridium MazG proteins and to the Burkholderia phage proteins ϕE255 gp37, BcepMu gp06, and BcepB1A gp71. Both gp35 and gp25 are similar to E. coli MazG (100% and 99.76% probability, respectively) (Additional file 1: Table S1 andAdditional file 2: Table S2). Because BCC bacteria found in soil and water are likely to be nutrient-limited (similar to cyanobacteria), MazG proteins in BCC-specific phages may help to facilitate infection in the environment. This protein may also be involved in the unique plaque phenotype of these phages, as the appearance of plaques at low titre after >16 h incubation (at which time the bacterial lawn appears intact) (Figure1) suggests that lysis of stationary phase cells may be occurring. Such a trait would be especially important for clinical use, as phage activity may be increased against stationary and/or biofilm cells found in the CF lung.
MazG may also have an effect with respect to BCC pathogenicity. Synthesis of ppGpp has been associated with virulence in species such as Legionella, Listeria, Pseudomonas, Salmonella, Mycobacterium, and Vibrio (although the association in this species has been controversial)[53–59]. In P. aeruginosa, relA mutants are less virulent than the wildtype when tested in the Drosophila melanogaster model[55] and relA spoT mutants have reduced antibiotic tolerance[60]. Because MazG activity may mimic the effects of these mutations, it is possible that phage-encoded MazG could modulate the virulence and/or antibiotic tolerance of a lysogen. Further experiments are required to determine if the putative KL1 and AH2 MazG proteins have pyrophosphohydrolase activity, if these genes are expressed in lysogens, and if MazG expression has an effect on pathogenicity.
Convergent evolution
Although there have been relatively few papers published on the subject, the occurrence of convergent evolution in bacteriophages has been documented previously. Most studies examine the phenomenon at the molecular level by identifying identical base pair and amino acid changes that occur in different phage lineages under the same environmental conditions[61–64]. Structural examples of convergent evolution, such as the Caudovirales tail and the tectivirus pseudo-tail, have been reviewed previously[65]. Given the ever-increasing number of completed phage genome sequences, it is expected that many more examples remain to be identified (particularly at the whole genome level). Furthermore, there are likely many examples in the literature of phages with similar phenotypes but dissimilar genomes that have not explicitly been identified as examples of convergent evolution, perhaps because they exhibit what is considered to be a “standard” plaque phenotype.
We predict that KL1 and AH2 represent examples of phage convergent evolution at the whole genome level. As discussed above, these two phages exhibit a plaque phenotype that is both similar and unique in comparison to all other BCC-specific phages that we have characterized previously. Because of these characteristics, KL1 and AH2 were thought to be the same phage prior to RFLP and genomic analysis. However, these phages appear to have convergently evolved because, as discussed throughout, their genomes are almost entirely dissimilar (Figure4A). The relative rarity of this phenotype among characterized phages of the BCC and other species may be at least partially explained by sampling bias. Standard phage isolation protocols most readily identify those phages that have easily visible plaques on multiple hosts after overnight incubation at a broad range of temperatures. Phages such as KL1 and AH2 may be missed because of poorly visible plaques, incompatible hosts, insufficient incubation times, incorrect temperatures, titres that are too high or too low, overgrowth of bacteria, and/or competition by more rapidly lysing phages. As novel phages continue to be isolated from environmental samples using diverse bacterial hosts, the prevalence, distribution, and genetic basis of this phenotype should become more apparent.
Several mechanisms could explain the delayed plaque formation observed here, including long latent periods or lysis inhibition (both with concomitantly large burst sizes)[66], preferential infection of stationary phase cells, or the gradual release of diffusible lytic enzymes from small plaques. In order to differentiate these possibilities, we performed one-step growth curves for both phages using either exponential or stationary phase C6433 as a host. Using a variation of a standard protocol (described in Methods), the phage titres unexpectedly remained stable (within one order of magnitude) over a 4 h period. Given the uninformative nature of these results, we have thus far been unable to identify the mechanism(s) responsible for the plaque phenotype. Taking into consideration the very specific conditions required for the observation of KL1 or AH2 plaques on solid medium, we predict that the infection kinetics in liquid culture may be highly dependent upon host (both strain and growth phase), incubation time, temperature, titre, and potentially other factors (such as medium) that are not accounted for using standard one-step growth curve protocols.
Conclusions
A recent publication by Ceyssens et al.[67] provides an interesting counterpoint to our study. While we identified KL1 and AH2 as phages that were phenotypically-similar but genomically-distinct, this group analyzed a set of Pseudomonas phages that were phenotypically-distinct but genomically-similar. They found that, among ϕKMV-like viruses with between 83-97% nucleotide identity, there were significant differences observed with respect to latent period, host range, and antibody reactivity[67]. We have made similar observations with our collection of BCC-specific phages: two phages can have distinct phenotypes with respect to liquid clearing and host range while at the same time having almost identical genomes[22]. Taken together, the observations made by Ceyssens et al.[67] and those discussed in this study provide a) novel examples of both divergent and convergent phage evolution and b) further evidence of the broad diversity of phages that infect Gram-negative opportunistic pathogens.
Methods
Bacterial strains and growth conditions
Burkholderia cenocepacia strains K56-2 and C6433, part of the Burkholderia cepacia complex experimental strain panel[68, 69], were used for phage isolation and propagation. Strains used for host range analysis (also part of the panel) were acquired from the Belgium Coordinated Collection of Microorganisms LMG Bacteria Collection (Ghent, Belgium) and the Canadian Burkholderia cepacia complex Research and Referral Repository (Vancouver, BC). Strains were grown aerobically overnight at 30°C on half-strength Luria-Bertani (½ LB) solid medium or in ½ LB broth with shaking. Lysates for DNA isolation were prepared from soft agar overlays made with ½ LB medium containing agarose instead of agar.
Phage isolation and propagation
KL1 and AH2 were isolated from sewage and Nandina sp. soil, respectively, using standard extraction protocols[26]. Environmental samples were incubated with shaking at 30°C in a slurry of ½ LB broth, suspension medium (SM) (50 mM Tris–HCl [pH 7.5], 100 mM NaCl, 10 mM MgSO4, 0.01% gelatin solution), and BCC liquid culture (K56-2 for KL1 isolation and C6433 for AH2 isolation). Solids were pelleted by centrifugation and the supernatant was filter-sterilized, plated in soft agar overlays with the BCC strain used in the extraction, and incubated overnight at 30°C and >24 h at room temperature. Plaques were picked using a sterile Pasteur pipette and transferred into 1 ml SM. Phage propagation was performed using soft agar overlays: 100 μl liquid culture and 100 μl phage stock (diluted in SM if necessary) were incubated 20 min at room temperature, mixed with 3 ml 0.7% ½ LB top agar, overlaid on a plate of ½ LB solid medium, and incubated at 30°C and room temperature until plaque formation was complete. High titre stocks were made by transferring multiple plaques into SM or by overlaying plates with SM and incubating 4–8 h at 4°C on a platform rocker.
Lysis characterization
Host ranges were performed using soft agar overlays (as described above) or by spotting 10 μl aliquots of phage stock (at multiple dilutions) onto a freshly-plated soft agar overlay containing 100 μl liquid culture. K56-2 LPS mutant[31, 32] host ranges were performed similarly using wildtype K56-2, RSF19 (wbxE::pRF201), XOA7 (waaL::pGPΩ Tp), XOA15 (wabR::pGPΩ Tp), XOA17 (wabS::pGPApTp), XOA8 (wabO::pGPΩ Tp), and CCB1 (waaC::pGPΩ Tp) (kindly provided by Miguel Valvano).
One-step growth curves were performed using a variation of a standard protocol[39]. One hundred microliters of diluted phage lysate containing 106 PFU of KL1 or AH2 was mixed with 108 colony forming units of C6433 (900 μl 5 h liquid culture [for exponential phase curves] or 100 μl 16 h liquid culture diluted in 800 μl spent ½ LB broth [for stationary phase curves]). The suspension was incubated 15 minutes at 30°C, diluted 1:1000 into a flask containing ½ LB broth (exponential) or spent ½ LB broth (stationary), and incubated without shaking at 30°C. One milliliter samples were withdrawn at one hour intervals for 4 h. Two 100 μl samples were plated immediately in soft agar overlays with C6433. One hundred microliters of chloroform was then added to the sample, mixed 5 s on a vortexer, and separated by centrifugation for 1 min at 13,000 rpm. Two 100 μl chloroform-treated samples were then plated immediately in soft agar overlays with C6433. Plates were incubated 48 h at 30°C prior to plaque enumeration. Experiments were performed in triplicate for each condition (KL1 exponential or stationary phase, AH2 exponential or stationary phase).
Electron microscopy
Filter-sterilized high titre stocks of KL1 and AH2 were used for electron microscopy. 5–10 μl of phage lysate was deposited onto a carbon-coated copper grid and incubated 5 min at room temperature. Following adsorption of excess lysate onto a filter paper, the grids were stained with 2% phosphotungstic acid for 2 min. Grids were viewed using a Philips/FEI (Morgagni) transmission electron microscope with charge-coupled device camera (University of Alberta Department of Biological Sciences Advanced Microscopy Facility).
DNA isolation, RFLP analysis, and sequencing
Phage DNA was isolated using polyethylene glycol precipitation and guanidine thiocyanate lysis. One hundred milliliters of phage lysate (propagated on C6433) was collected by overlaying turbid-clear or mottled ½ LB agarose plates with SM and incubating at 4°C 4–8 h on a platform rocker. Following the addition of chloroform, debris in the lysate was pelleted by centrifugation for 10 min at 10,000 rcf and 4°C and the supernatant was filter-sterilized with a Millex-HA 0.45 μm syringe driven filter unit (Millipore, Billerica, MA). Fifty milliliter aliquots of the supernatant were incubated at 37°C ≥40 min with 10 μl DNase I, 10 μl DNase I buffer and 6 μl RNase (Fermentas, Burlington, ON) to degrade contaminating bacterial nucleic acids. Following centrifugation for 10 min at 4000 rcf and 4°C, phages in the supernatant were precipitated in 1 M NaCl and 10% w/v PEG 8000 at 4°C. The precipitated phages were pelleted by centrifugation for 20 min at 10,000 rcf and 4°C and resuspended in 1.6 ml SM. To eliminate residual DNase I activity, the phage suspension was incubated at 37°C 10 min with 40 μl 20 mg/ml proteinase K. Following extraction of the phages with an equal volume of chloroform and the addition of EDTA to 100 mM, ½ volume of 6 M guanidine thiocyanate was added to disrupt the capsids and release the phage DNA. DNA was then purified using the GENECLEAN Turbo Kit (Qbiogene, Irvine, CA). Phage DNA was quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA).
RFLP analysis was performed using 5 μg of phage DNA digested overnight at 37°C with EcoRI (Invitrogen, Carlsbad, CA). For cos site screening, 5 μg EcoRI digests were incubated 20 min at 80°C, cooled on ice, and separated on 0.8% agarose gels in 1x TAE (pH 8.0). Bands present only in the heated sample were excised from the gel, purified using a GENECLEAN III kit (Qbiogene), cloned into pJET1.2 (Fermentas), and sequenced to identify the cos site. Preliminary sequencing of EcoRI phage DNA fragments cloned into pUC19 was performed as described previously[19, 29]. For complete genome sequencing, phage DNA was submitted to 454 Life Sciences (Branford, CT) for pyrosequencing. The genome sequences of KL1 and AH2 have been deposited in GenBank with the accession numbers JF939047 and JN564907. Sequence start sites for these files were chosen based on alignment with PA73 for KL1 and at the cos site for AH2.
Bioinformatics analysis
Annotation of the genome sequences and determination of GC contents were performed using GeneMark (http://exon.biology.gatech.edu/gmhmm2_prok.cgi)[70]. Manual annotations were performed for KL1 5 (encoding Rz1) and KL1 19/AH2 55 (encoding translationally-frameshifted tail proteins). Homology searches and conserved domain searches were performed using HHpred (http://toolkit.tuebingen.mpg.de/hhpred)[71] and NCBI’s BLASTN/BLASTP (for full genomes and individual proteins, respectively) (http://blast.ncbi.nlm.nih.gov)[72] and Conserved Domain Search (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi)[73]. FSFinder was used for translational frameshift identification (http://wilab.inha.ac.kr/fsfinder)[74]. Mfold was used for stem-loop structure identification (http://mfold.rna.albany.edu/?q=mfold)[75]. Sequence comparisons were visualized using Circos (http://circos.ca)[76] and PROmer (http://mummer.sourceforge.net)[77] with the following parameters: breaklen = 60, maxgap = 30, mincluster = 20, minmatch = 6. Lysis protein analysis was performed using TMHMM for transmembrane region identification (http://www.cbs.dtu.dk/services/TMHMM)[78] and LipoP for signal peptidase II cleavage site identification (http://www.cbs.dtu.dk/services/LipoP)[79].
References
Merril CR, Scholl D, Adhya SL: The prospect for bacteriophage therapy in Western medicine. Nat Rev Drug Discov. 2003, 2: 489-497. 10.1038/nrd1111.
Daniel A, Euler C, Collin M, Chahales P, Gorelick KJ, Fischetti VA: Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus . Antimicrob Agents Chemother. 2010, 54: 1603-1612. 10.1128/AAC.01625-09.
Morello E, Saussereau E, Maura D, Huerre M, Touqui L, Debarbieux L: Pulmonary bacteriophage therapy on Pseudomonas aeruginosa cystic fibrosis strains: First steps towards treatment and prevention. PLoS One. 2011, 6: e16963-10.1371/journal.pone.0016963.
Kumari S, Harjai K, Chhibber S: Bacteriophage versus antimicrobial agents for the treatment of murine burn wound infection caused by Klebsiella pneumoniae B5055. J Med Microbiol. 2011, 60: 205-210. 10.1099/jmm.0.018580-0.
Oliveira A, Sereno R, Azeredo J: In vivo efficiency evaluation of a phage cocktail in controlling severe colibacillosis in confined conditions and experimental poultry houses. Vet Microbiol. 2010, 146: 303-308. 10.1016/j.vetmic.2010.05.015.
Waseh S, Hanifi-Moghaddam P, Coleman R, Masotti M, Ryan S, Foss M, MacKenzie R, Henry M, Szymanski CM, Tanha J: Orally administered P22 phage tailspike protein reduces Salmonella colonization in chickens: Prospects of a novel therapy against bacterial infections. PLoS One. 2010, 5: e13904-10.1371/journal.pone.0013904.
Carvalho CM, Gannon BW, Halfhide DE, Santos SB, Hayes CM, Roe JM, Azeredo J: The in vivo efficacy of two administration routes of a phage cocktail to reduce numbers of Campylobacter coli and Campylobacter jejuni in chickens. BMC Microbiol. 2010, 10: 232-10.1186/1471-2180-10-232.
Wright A, Hawkins CH, Änggård EE, Harper DR: A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa ; A preliminary report of efficacy. Clin Otolaryngol. 2009, 34: 349-357. 10.1111/j.1749-4486.2009.01973.x.
Golshahi L, Seed KD, Dennis JJ, Finlay WH: Toward modern inhalational bacteriophage therapy: Nebulization of bacteriophages of Burkholderia cepacia complex. J Aerosol Med Pulm Drug Deliv. 2008, 21: 351-359. 10.1089/jamp.2008.0701.
Golshahi L, Lynch KH, Dennis JJ, Finlay WH: In vitro lung delivery of bacteriophages KS4-M and ΦKZ using dry powder inhalers for treatment of Burkholderia cepacia complex and Pseudomonas aeruginosa infections in cystic fibrosis. J Appl Microbiol. 2011, 110: 106-117. 10.1111/j.1365-2672.2010.04863.x.
Matinkhoo S, Lynch KH, Dennis JJ, Finlay WH, Vehring R: Spray-dried respirable powders containing bacteriophages for the treatment of pulmonary infections. J Pharm Sci. 2011, 100: 5197-5205. 10.1002/jps.22715.
Henn MR, Sullivan MB, Stange-Thomann N, Osburne MS, Berlin AM, Kelly L, Yandava C, Kodira C, Zeng Q, Weiand M, Sparrow T, Saif S, Giannoukos G, Young SK, Nusbaum C, Birren BW, Chisholm SW: Analysis of high-throughput sequencing and annotation strategies for phage genomes. PLoS One. 2010, 5: e9083-10.1371/journal.pone.0009083.
Isles A, Maclusky I, Corey M: Pseudomonas cepacia infection in cystic fibrosis: An emerging problem. J Pediatr. 1984, 104: 206-210. 10.1016/S0022-3476(84)80993-2.
Ryley HC, Doull IJM: Burkholderia cepacia complex infection in patients with cystic fibrosis: Laboratory investigations, epidemiology and clinical management. Rev Med Microbiol. 2003, 14: 15-24. 10.1097/00013542-200301000-00002.
LiPuma JJ, Dasen SE, Nielson DW, Stern RC, Stull TL: Person-to-person transmission of Pseudomonas cepacia between patients with cystic fibrosis. Lancet. 1990, 336: 1094-1096. 10.1016/0140-6736(90)92571-X.
Zhou J, Chen Y, Tabibi S, Alba L, Garber E, Saiman L: Antimicrobial susceptibility and synergy studies of Burkholderia cepacia complex isolated from patients with cystic fibrosis. Antimicrob Agents Chemother. 2007, 51: 1085-1088. 10.1128/AAC.00954-06.
Baldwin A, Mahenthiralingam E, Drevinek P, Pope C, Waine DJ, Henry DA, Speert DP, Carter P, Vandamme P, LiPuma JJ, Dowson CG: Elucidating global epidemiology of Burkholderia multivorans in cases of cystic fibrosis by multilocus sequence typing. J Clin Microbiol. 2008, 46: 290-295. 10.1128/JCM.01818-07.
Seed KD, Dennis JJ: Experimental bacteriophage therapy increases survival of Galleria mellonella larvae infected with clinically relevant strains of the Burkholderia cepacia complex. Antimicrob Agents Chemother. 2009, 53: 2205-2208. 10.1128/AAC.01166-08.
Lynch KH, Seed KD, Stothard P, Dennis JJ: Inactivation of Burkholderia cepacia complex phage KS9 gp41 identifies the phage repressor and generates lytic virions. J Virol. 2010, 84: 1276-1288. 10.1128/JVI.01843-09.
Carmody LA, Gill JJ, Summer EJ, Sajjan US, Gonzalez CF, Young RF, LiPuma JJ: Efficacy of bacteriophage therapy in a model of Burkholderia cenocepacia pulmonary infection. J Infect Dis. 2010, 201: 264-271. 10.1086/649227.
Semler DD, Lynch KH, Dennis JJ: The promise of bacteriophage therapy for Burkholderia cepacia complex respiratory infections. Front Cell Inf Microbio. 2012, 1: 27-
Lynch KH, Dennis JJ: Cangene gold medal award lecture - genomic analysis and modification of Burkholderia cepacia complex bacteriophages. Can J Microbiol. 2012, 58: 221-235. 10.1139/w11-135.
Imamovic L, Ballesté E, Jofre J, Muniesa M: Quantification of Shiga toxin-converting bacteriophages in wastewater and in fecal samples by real-time quantitative PCR. Appl Environ Microbiol. 2010, 76: 5693-5701. 10.1128/AEM.00107-10.
Langley R, Kenna DT, Vandamme P, Ure R, Govan JRW: Lysogeny and bacteriophage host range within the Burkholderia cepacia complex. J Med Microbiol. 2003, 52: 483-490. 10.1099/jmm.0.05099-0.
Summer EJ, Gonzalez CF, Bomer M, Carlile T, Embry A, Kucherka AM, Lee J, Mebane L, Morrison WC, Mark L, King MD, LiPuma JJ, Vidaver AK, Young R: Divergence and mosaicism among virulent soil phages of the Burkholderia cepacia complex. J Bacteriol. 2006, 188: 255-268. 10.1128/JB.188.1.255-268.2006.
Seed KD, Dennis JJ: Isolation and characterization of bacteriophages of the Burkholderia cepacia complex. FEMS Microbiol Lett. 2005, 251: 273-280. 10.1016/j.femsle.2005.08.011.
Gill JJ, Summer EJ, Russell WK, Cologna SM, Carlile TM, Fuller AC, Kitsopoulos K, Mebane LM, Parkinson BN, Sullivan D, Carmody LA, Gonzalez CF, LiPuma JJ, Young R: Genomes and characterization of phages Bcep22 and BcepIL02, founders of a novel phage type in Burkholderia cenocepacia . J Bacteriol. 2011, 193: 5300-5313. 10.1128/JB.05287-11.
Goudie AD, Lynch KH, Seed KD, Stothard P, Shrivastava S, Wishart DS, Dennis JJ: Genomic sequence and activity of KS10, a transposable phage of the Burkholderia cepacia complex. BMC Genomics. 2008, 9: 615-10.1186/1471-2164-9-615.
Lynch KH, Stothard P, Dennis JJ: Genomic analysis and relatedness of P2-like phages of the Burkholderia cepacia complex. BMC Genomics. 2010, 11: 599-10.1186/1471-2164-11-599.
Lynch KH, Stothard P, Dennis JJ: Characterization of DC1, a broad-host-range Bcep22-like podovirus. Appl Environ Microbiol. 2012, 78: 889-891. 10.1128/AEM.07097-11.
Loutet SA, Flannagan RS, Kooi C, Sokol PA, Valvano MA: A complete lipopolysaccharide inner core oligosaccharide is required for resistance of Burkholderia cenocepacia to antimicrobial peptides and bacterial survival in vivo. J Bacteriol. 2006, 188: 2073-2080. 10.1128/JB.188.6.2073-2080.2006.
Ortega X, Silipo A, Saldfas MS, Bates CC, Molinaro A, Valvano MA: Biosynthesis and structure of the Burkholderia cenocepacia K56-2 lipopolysaccharide core oligosaccharide: Truncation of the core oligosaccharide leads to increased binding and sensitivity to polymyxin B. J Biol Chem. 2009, 284: 21738-21751. 10.1074/jbc.M109.008532.
Kwan T, Liu J, DuBow M, Gros P, Pelletier J: Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J Bacteriol. 2006, 188: 1184-1187. 10.1128/JB.188.3.1184-1187.2006.
Zhang J, Inouye M: MazG, a nucleoside triphosphate pyrophosphohydrolase, interacts with Era, an essential GTPase in Escherichia coli . J Bacteriol. 2002, 184: 5323-5329. 10.1128/JB.184.19.5323-5329.2002.
Xu J, Hendrix RW, Duda RL: Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol Cell. 2004, 16: 11-21. 10.1016/j.molcel.2004.09.006.
Alam SL, Atkins JF, Gesteland RF: Programmed ribosomal frameshifting: Much ado about knotting!. Proc Natl Acad Sci USA. 1999, 96: 14177-14179. 10.1073/pnas.96.25.14177.
Young R, Wang I-N, Roof WD: Phages will out: Strategies of host cell lysis. Trends Microbiol. 2000, 8: 120-128. 10.1016/S0966-842X(00)01705-4.
Summer EJ, Berry J, Tran TAT, Niu L, Struck DK, Young R: Rz/Rz1 lysis gene equivalents in phages of Gram-negative hosts. J Mol Biol. 2007, 373: 1098-1112. 10.1016/j.jmb.2007.08.045.
Summer EJ, Gonzalez CF, Carlisle T, Mebane LM, Cass AM, Savva CG, LiPuma J, Young R: Burkholderia cenocepacia phage BcepMu and a family of Mu-like phages encoding potential pathogenesis factors. J Mol Biol. 2004, 340: 49-65. 10.1016/j.jmb.2004.04.053.
Lopes A, Amarir-Bouhram J, Faure G, Petit M-A, Guerois R: Detection of novel recombinases in bacteriophage genomes unveils Rad52, Rad51 and Gp2.5 remote homologs. Nucleic Acids Res. 2010, 38: 3952-3962. 10.1093/nar/gkq096.
Hennecke F, Kolmar H, Brundl K, Fritz H-J: The vsr gene product of E. coli K-12 is a strand- and sequence-specific DNA mismatch endonuclease. Nature. 1991, 353: 776-778. 10.1038/353776a0.
Lin J-J, Sancar A: The C-terminal half of UvrC protein is sufficient to reconstitute (A)BC excinuclease. Proc Natl Acad Sci USA. 1991, 88: 6824-6828. 10.1073/pnas.88.15.6824.
Gross M, Marianovsky I, Glaser G: MazG - A regulator of programmed cell death in Escherichia coli . Mol Microbiol. 2006, 59: 590-601. 10.1111/j.1365-2958.2005.04956.x.
Magnusson LU, Farewell A, Nyström T: ppGpp: A global regulator in Escherichia coli . Trends Microbiol. 2005, 13: 236-242. 10.1016/j.tim.2005.03.008.
Bryan MJ, Burroughs NJ, Spence EM, Clokie MRJ, Mann NH, Bryan SJ: Evidence for the intense exchange of MazG in marine cyanophages by horizontal gene transfer. PLoS One. 2008, 3: e2048-10.1371/journal.pone.0002048.
Sullivan MB, Huang KH, Ignacio-Espinoza JC, Berlin AM, Kelly L, Weigele PR, DeFrancesco AS, Kern SE, Thompson LR, Young S, Yandava C, Fu R, Krastins B, Chase M, Sarracino D, Osburne MS, Henn MR, Chisholm SW: Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol. 2010, 12: 3035-3056. 10.1111/j.1462-2920.2010.02280.x.
Sullivan MB, Coleman ML, Weigele P, Rohwer F, Chisholm SW: Three Prochlorococcus cyanophage genomes: Signature features and ecological interpretations. PLoS Biol. 2005, 3: e144-10.1371/journal.pbio.0030144.
Mann NH, Clokie MRJ, Millard A, Cook A, Wilson WH, Wheatley PJ, Letarov A, Krisch HM: The genome of S-PM2, a "photosynthetic" T4-type bacteriophage that infects marine Synechococcus strains. J Bacteriol. 2005, 187: 3188-3200. 10.1128/JB.187.9.3188-3200.2005.
Weigele PR, Pope WH, Pedulla ML, Houtz JM, Smith AL, Conway JF, King J, Hatfull GF, Lawrence JG, Hendrix RW: Genomic and structural analysis of Syn9, a cyanophage infecting marine Prochlorococcus and Synechococcus . Environ Microbiol. 2007, 9: 1675-1695. 10.1111/j.1462-2920.2007.01285.x.
Angly F, Youle M, Nosrat B, Srinagesh S, Rodriguez-Brito B, McNairnie P, Deyanat-Yazdi G, Breitbart M, Rohwer F: Genomic analysis of multiple Roseophage SIO1 strains. Environ Microbiol. 2009, 11: 2863-2873. 10.1111/j.1462-2920.2009.02021.x.
Duhaime MB, Wichels A, Waldmann J, Teeling H, Glöckner FO: Ecogenomics and genome landscapes of marine Pseudoalteromonas phage H105/1. ISME J. 2011, 5: 107-121. 10.1038/ismej.2010.94.
Clokie MRJ, Mann NH: Marine cyanophages and light. Environ Microbiol. 2006, 8: 2074-2082. 10.1111/j.1462-2920.2006.01171.x.
Hammer BK, Swanson MS: Co-ordination of Legionella pneumophila virulence with entry into stationary phase by ppGpp. Mol Microbiol. 1999, 33: 721-731. 10.1046/j.1365-2958.1999.01519.x.
Taylor CM, Beresford M, Epton HAS, Sigee DC, Shama G, Andrew PW, Roberts IS: Listeria monocytogenes relA and hpt mutants are impaired in surface-attached growth and virulence. J Bacteriol. 2002, 184: 621-628. 10.1128/JB.184.3.621-628.2002.
Erickson DL, Lines JL, Pesci EC, Venturi V, Storey DG: Pseudomonas aeruginosa relA contributes to virulence in Drosophila melanogaster . Infect Immun. 2004, 72: 5638-5645. 10.1128/IAI.72.10.5638-5645.2004.
Jeong J-H, Song M, Park S-I, Cho K-O, Joon HR, Choy HE: Salmonella enterica serovar Gallinarum requires ppGpp for internalization and survival in animal cells. J Bacteriol. 2008, 190: 6340-6350. 10.1128/JB.00385-08.
Klinkenberg LG, Lee J-H, Bishai WR, Karakousis PC: The stringent response is required for full virulence of Mycobacterium tuberculosis in guinea pigs. J Infect Dis. 2010, 202: 1397-1404. 10.1086/656524.
Haralalka S, Nandi S, Bhadra RK: Mutation in the relA gene of Vibrio cholerae affects in vitro and in vivo expression of virulence factors. J Bacteriol. 2003, 185: 4672-4682. 10.1128/JB.185.16.4672-4682.2003.
Silva AJ, Benitez JA: A Vibrio cholerae relaxed (relA) mutant expresses major virulence factors, exhibits biofilm formation and motility, and colonizes the suckling mouse intestine. J Bacteriol. 2006, 188: 794-800. 10.1128/JB.188.2.794-800.2006.
Nguyen D, Joshi-Datar A, Lepine F, Bauerle E, Olakanmi O, Beer K, McKay G, Siehnel R, Schafhauser J, Wang Y, Britigan BE, Singh PK: Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science. 2011, 334: 982-986. 10.1126/science.1211037.
Bull JJ, Badgett MR, Wichman HA, Huelsenbeck JP, Hillis DM, Gulati A, Ho C, Molineux IJ: Exceptional convergent evolution in a virus. Genetics. 1997, 147: 1497-1507.
Wichman HA, Badgett MR, Scott LA, Boulianne CM, Bull JJ: Different trajectories of parallel evolution during viral adaptation. Science. 1999, 285: 422-424. 10.1126/science.285.5426.422.
Wichman HA, Scott LA, Yarber CD, Bull JJ: Experimental evolution recapitulates natural evolution. Phil Trans R Soc Lond. 2000, 355: 1677-1684. 10.1098/rstb.2000.0731.
Bollback JP, Huelsenbeck JP: Parallel genetic evolution within and between bacteriophage species of varying degrees of divergence. Genetics. 2009, 181: 225-234.
Ackermann H-W: 5500 Phages examined in the electron microscope. Arch Virol. 2007, 152: 227-243. 10.1007/s00705-006-0849-1.
Abedon ST: Phage evolution and ecology. Adv Appl Microbiol. 2009, 67: 1-45.
Ceyssens P-J, Glonti T, Kropinski AM, Lavigne R, Chanishvili N, Kulakov L, Lashkhi N, Tediashvili M, Merabishvili M: Phenotypic and genotypic variations within a single bacteriophage species. Virol J. 2011, 8: 134-10.1186/1743-422X-8-134.
Mahenthiralingam E, Coenye T, Chung JW, Speert DP, Govan JRW, Taylor P, Vandamme P: Diagnostically and experimentally useful panel of strains from the Burkholderia cepacia complex. J Clin Microbiol. 2000, 38: 910-913.
Coenye T, Vandamme P, LiPuma JJ, Govan JRW, Mahenthiralingam E: Updated version of the Burkholderia cepacia complex experimental strain panel. J Clin Microbiol. 2003, 41: 2797-2798. 10.1128/JCM.41.6.2797-2798.2003.
Lukashin AV, Borodovsky M: GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 1998, 26: 1107-1115. 10.1093/nar/26.4.1107.
Söding J, Biegert A, Lupas AN: The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33: W244-W248. 10.1093/nar/gki408.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Marchler-Bauer A, Bryant SH: CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32: W327-W331. 10.1093/nar/gkh454.
Moon S, Byun Y, Kim H-J, Jeong S, Han K: Predicting genes expressed via −1 and +1 frameshifts. Nucleic Acids Res. 2004, 32: 4884-4892. 10.1093/nar/gkh829.
Zuker M: Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31: 3406-3415. 10.1093/nar/gkg595.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA: Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19: 1639-1645. 10.1101/gr.092759.109.
Delcher AL, Phillippy A, Carlton J, Salzberg SL: Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002, 30: 2478-2483. 10.1093/nar/30.11.2478.
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL: Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol. 2001, 305: 567-580. 10.1006/jmbi.2000.4315.
Juncker AS, Willenbrock H, von Heijne G, Brunak S, Nielsen H, Krogh A: Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003, 12: 1652-1662. 10.1110/ps.0303703.
Acknowledgements
The authors would like to thank Amberlie Heaman for isolation and preliminary host range analysis of AH2, Miles Peterson for assistance with figure construction, Arlene Oatway (University of Alberta Department of Biological Sciences Advanced Microscopy Facility) for assistance with electron microscopy, and Miguel Valvano (University of Western Ontario) for providing K56-2 LPS mutants.
JJD gratefully acknowledges funding from the Canadian Institutes of Health Research (CIHR grant 200705XNE-170954), and Cystic Fibrosis Canada (CFC grant RES0001467), for operating grant support. KHL gratefully acknowledges funding from Cystic Fibrosis Canada, NSERC, the Killam Trusts, and Alberta Innovates – Health Solutions for scholarship support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KHL isolated KL1, performed electron microscopy, sequenced, annotated, and analyzed the genomes, and drafted the manuscript. PS constructed Figure 4 and performed FSFinder and preliminary HHpred analysis. JJD devised the study and assisted with experimental design, data analysis, and the writing of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2011_4321_MOESM3_ESM.pptx
Additional file 3: Figure S3. Stem-loop structures predicted by mfold analysis of the KL1 (left) and AH2 (right) frameshift regions (including the putative frameshift sites and 35 downstream bases). (PPTX 214 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lynch, K.H., Stothard, P. & Dennis, J.J. Comparative analysis of two phenotypically-similar but genomically-distinct Burkholderia cenocepacia-specific bacteriophages. BMC Genomics 13, 223 (2012). https://doi.org/10.1186/1471-2164-13-223
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-223