
Abstract
Background
The Burkholderia cepacia complex (BCC) is comprised of at least seventeen Gram-negative species that cause infections in cystic fibrosis patients. Because BCC bacteria are broadly antibiotic resistant, phage therapy is currently being investigated as a possible alternative treatment for these infections. The purpose of our study was to sequence and characterize three novel BCC-specific phages: KS5 (vB_BceM-KS5 or vB_BmuZ-ATCC 17616), KS14 (vB_BceM-KS14) and KL3 (vB_BamM-KL3 or vB_BceZ-CEP511).
Results
KS5, KS14 and KL3 are myoviruses with the A1 morphotype. The genomes of these phages are between 32317 and 40555 base pairs in length and are predicted to encode between 44 and 52 proteins. These phages have over 50% of their proteins in common with enterobacteria phage P2 and so can be classified as members of the Peduovirinae subfamily and the "P2-like viruses" genus. The BCC phage proteins similar to those encoded by P2 are predominantly structural components involved in virion morphogenesis. As prophages, KS5 and KL3 integrate into an AMP nucleosidase gene and a threonine tRNA gene, respectively. Unlike other P2-like viruses, the KS14 prophage is maintained as a plasmid. The P2 E+E' translational frameshift site is conserved among these three phages and so they are predicted to use frameshifting for expression of two of their tail proteins. The lysBC genes of KS14 and KL3 are similar to those of P2, but in KS5 the organization of these genes suggests that they may have been acquired via horizontal transfer from a phage similar to λ. KS5 contains two sequence elements that are unique among these three phages: an ISBmu 2-like insertion sequence and a reverse transcriptase gene. KL3 encodes an EcoRII-C endonuclease/methylase pair and Vsr endonuclease that are predicted to function during the lytic cycle to cleave non-self DNA, protect the phage genome and repair methylation-induced mutations.
Conclusions
KS5, KS14 and KL3 are the first BCC-specific phages to be identified as P2-like. As KS14 has previously been shown to be active against Burkholderia cenocepacia in vivo, genomic characterization of these phages is a crucial first step in the development of these and similar phages for clinical use against the BCC.
Similar content being viewed by others

Background
The Burkholderia cepacia complex (BCC) is a group of at least seventeen species of Gram-negative opportunistic pathogens. Although these organisms can infect patients with a broad range of chronic conditions, the majority of infections occur in those with cystic fibrosis (CF) [1, 2]. Because the lungs of these individuals contain thick mucus that cannot be cleared by the mucociliary escalator, they are susceptible to pulmonary infections by microorganisms such as Pseudomonas, Staphylococcus, Haemophilus and Burkholderia[3, 4]. The prevalence of BCC infection in American CF patients was 3.1% in 2005 [5]. Although this prevalence is low compared to that of Pseudomonas aeruginosa (56.1% in 2005), there are three reasons why the BCC is a serious problem for the CF population [5]. First, BCC bacteria cause severe and potentially fatal respiratory infections. When compared to patients infected with Pseudomonas, those with BCC infections have reduced lung function and, depending on the species present, increased mortality [6, 7]. In approximately 20% of cases, these individuals develop a rapidly fatal condition called 'cepacia syndrome,' which is characterized by lung abscesses and septicemia [2, 8]. Second, BCC bacteria can spread from person-to-person. It has been shown that at least five BCC species can be transmitted in this manner: Burkholderia cepacia, Burkholderia multivorans, Burkholderia cenocepacia, Burkholderia dolosa and Burkholderia contaminans[9–11]. Because of the potential for these organisms to spread among a susceptible population, BCC culture-positive patients are isolated from other individuals with CF, a measure that has serious social and psychological implications [12, 13]. Finally, BCC bacteria are resistant to most antibiotics. These species have a variety of resistance mechanisms including β-lactamases, efflux pumps and biofilm formation [14–16]. The most effective anti-BCC antibiotics - ceftazidime, meropenem and minocycline - only inhibit between 23-38% of clinical isolates [17].
Because conventional antibiotics are largely ineffective against the BCC, phage therapy is being explored as a possible alternative treatment. Phage therapy is the clinical administration of bacteriophages (or phages) to prevent and/or to treat bacterial infections [18]. Although phages have been used therapeutically for almost a century, this treatment fell out of favor in North America and Western Europe when penicillin and other chemical antibiotics became widely available in the 1940s [18]. However, there has been renewed interest in this field following the emergence of multidrug resistant bacteria such as those of the BCC [18]. Three recent studies have shown that phages are active against the BCC in vivo. Seed and Dennis showed that treatment of B. cenocepacia-infected Galleria mellonella larvae with phages KS14, KS4-M or KS12 increased survival 48 hours post-infection, even when treatment with the latter two phages was delayed for 6 to 12 hours [19]. Carmody et al. showed that intraperitoneal administration of phage BcepIL02 to B. cenocepacia-infected mice decreased bacterial density in the lungs and led to decreased expression of the pro-inflammatory cytokines MIP-2 and TNF-α [20]. Lynch et al. published the first description of an engineered BCC phage and showed that this mutant (a repressor knockout of phage KS9) was able to increase survival of B. cenocepacia-infected G. mellonella 48 hours post-infection [21].
Before a phage can be safely used clinically, its complete genome sequence must be determined to assess whether the phage is obligately lytic or temperate, and to determine by homology whether the phage genome encodes any putative virulence factors. This report describes the genome sequence of three novel BCC phages and their relatedness to enterobacteria phage P2. P2 is a temperate myovirus that was isolated from E. coli strain Li by Bertani in 1951 [22]. P2 has recently been classified as part of a novel subfamily, placing it in the order Caudovirales, family Myoviridae, subfamily Peduovirinae and genus "P2-like viruses" [23]. This genus includes phages P2, Wϕ, 186 and PsP3 of enterobacteria, L-413C of Yersinia, Fels-2 and SopEϕ of Salmonella, ϕ-MhaA1-PHL101 of Mannheimia, ϕCTX of Pseudomonas, RSA1 of Ralstonia, ϕE202 of Burkholderia thailandensis and ϕ52237 and ϕE12-2 of Burkholderia pseudomallei[23]. Based on sequence analysis, it is proposed that the BCC-specific phages KS5 (vB_BceM-KS5 or vB_BmuZ-ATCC 17616), KS14 (vB_BceM-KS14) and KL3 (vB_BamM-KL3 or vB_BceZ-CEP511) should also be classified as part of this genus [24].
Results and Discussion
Isolation, host range and morphology
Seed and Dennis isolated KS5 from an extract of onion soil plated on B. cenocepacia K56-2 [25]. This phage produces clear plaques on K56-2 with a diameter of 0.5-1.0 mm [25]. Previously, KS5 was tested for its ability to form plaques on K56-2 LPS mutants and it was determined that it could efficiently lyse wildtype K56-2 (EOP = 1), XOA7 (waaL::pGPΩTp, EOP = 0.8), XOA15 (wabR::pGPΩTp, EOP = 1.3), XOA17 (wabS::pGPΩTp, EOP = 1.1) and RSF19 (wbxE::pGPΩTp, EOP = 0.5), but not XOA8 (wabO::pGPΩTp) or CCB1 (waaC::pGPΩTp) [21, 26, 27]. Based on these results, it was predicted that KS5 uses the K56-2 LPS as a receptor and that it binds within the core region [21]. KS5 has a relatively wide host range compared to many BCC phages, infecting B. multivorans C5393 and B. cenocepacia 715J, J2315, K56-2, C6433 and C5424 [25].
KS14 was isolated from an extract of Dracaena sp. soil plated on B. multivorans C5393 [19]. Both BCC phages and bacteria have been recovered from the Dracaena rhizosphere [19, 28]. On C5393, KS14 forms small clear plaques 0.5-1.0 mm in diameter, similar to the morphology of KS5 on K56-2. The host range of KS14 includes B. multivorans C5393 and C5274, B. cenocepacia 715J, C6433, C5424 and PC184, B. dolosa LMG 21443 and Burkholderia ambifaria LMG 17828 [19].
KL3 was isolated from a single plaque on a lawn of B. cenocepacia CEP511, an Australian CF epidemic isolate [29]. Phage induction from CEP511 was stochastic, as treatment with inducing agents such as UV or mitomycin C was not necessary. On LMG 17828, KL3 forms small turbid plaques 0.5-1.0 mm in diameter. KL3 has a narrow host range, infecting B. ambifaria LMG 17828.
Electron microscopy of KS5, KS14 and KL3 indicates that these phages belong to the family Myoviridae (Figure 1). These three phages exhibit the A1 morphotype, with icosahedral capsids and contractile tails [30]. KS5, KS14 and KL3 have similarly sized capsids, each 65 nm in diameter (Figure 1). In contrast, their tails vary in length: 140 nm for KS14, 150 nm for KS5 and 160 nm for KL3 (Figure 1). These sizes correspond to the length of the tail tape measure protein for each of these three phages: KS14 gp12 is 842 amino acids (aa) in length, KS5 gp15 is 920 aa and KL3 gp17 is 1075 aa (Tables 1, 2 and 3).

Transmission electron micrographs of KS5 (A), KS14 (B) and KL3 (C). Phages were stained with 2% phosphotungstic acid and viewed at 140,000-fold magnification. Scale bars represent 50 nm.
Genome characterization
KS5
The KS5 genome is 37236 base pairs (bp) in length and encodes 46 proteins (including the transposase of a predicted insertion sequence, discussed below) (Table 1). This genome has a 63.71% G+C content. Forty-three of the start codons are ATG, 2 are GTG and 1 is TTG (Table 1). As KS5 was isolated from an environmental sample, it was predicted that this phage might be obligately lytic [25]. However, KS5 encodes an integrase and a repressor and is found as a prophage in chromosome 2 of the fully sequenced B. multivorans strain ATCC 17616 (GenBank:NC_010805.1; BMULJ_03640-BMULJ_03684, bp 477496-514731) (Table 1). Because of this similarity, the possibility exists that KS5 originated from ATCC 17616 or a closely related strain found in the soil enrichment. Excluding the ATCC 17616 prophage, KS5 is most similar to a putative prophage element in Burkholderia multivorans CGD1. Twenty-three of 46 KS5 proteins are most closely related to a protein from CGD1, with percent identities ranging from 72-99% (Table 1).
KS14
The KS14 genome is 32317 bp in length and encodes 44 proteins (Table 2). This genome has a 62.28% G+C content. Forty-one of the start codons are ATG, 2 are GTG and 1 is TTG (Table 2). All predicted KS14 proteins show similarity to at least one protein in the database (as determined by a BLASTP search) except for gp38 and gp42. The protein with the most similarity to others in the database is the terminase large subunit, gp35, which has 75% identity with a protein of unknown function DUF264 of Burkholderia sp. CCGE1001. Aside from gp38 and gp42, the least similar protein is the hypothetical protein gp39, which has 29% identity with the flagellar hook-associated protein FlgK of Acidovorax ebreus TPSY (Table 2).
KL3
The KL3 genome is 40555 bp in length and encodes 52 proteins (Table 3). This genome has a 63.23% G+C content. Fifty-one of the start codons are ATG and 1 is GTG (Table 3). Similarly to KS14, all predicted KL3 proteins show similarity to at least one protein in the database except for gp43. The proteins with the most similarity to others in the database are the terminase large subunit (gp41) and the portal protein (gp42) that have 99% identity with Burkholderia glumae BGR1 proteins and the hypothetical protein gp50 which has 99% identity with a B. ambifaria MEX-5 protein. Aside from gp43, the least similar protein is the hypothetical protein gp14, which has 50% identity with the hypothetical protein BuboB_27112 of Burkholderia ubonensis Bu (Table 3).
Modular organization
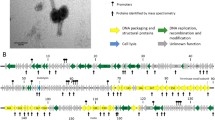
The genome maps of KS5, KS14 and KL3 are shown in Figure 2. Each of these phages has a modular organization, with genes for tail formation (shown in purple), lysis (shown in light blue) and head formation (shown in dark blue) clustered in each phage (Figure 2). In KS5, genes 13-19, 22-27, 29-30 and 36 encode tail proteins, genes 31-35 encode lysis proteins and genes 38-43 encode capsid proteins (Table 1, Figure 2). In KS14, genes 10-24 and 30 encode tail proteins, genes 25-29 encode lysis proteins and genes 31-36 encode capsid proteins (Table 2, Figure 2). In KL3, genes 15-27, 29-30 and 36 encode tail proteins, genes 31-35 encode lysis proteins and genes 37-42 encode capsid proteins (Table 3, Figure 2). In addition, KL3 has a three-gene DNA modification module (discussed below): gene 45, encoding an EcoRII-C restriction endonuclease, gene 46, encoding a Vsr endonuclease and gene 47, encoding an EcoRII methylase (Table 3, Figure 2).

Genome maps of KS5, KS14 and KL3. Genes transcribed from the plus strand are shown above and genes transcribed from the minus strand are shown below. The scale (in kbp) is shown on the bottom. The prophage gene order is shown for KS5 and KL3. The gene order for KS14 (which is maintained as a plasmid prophage) was chosen based on alignment with the other two sequences. Legend: orange, recombinase; yellow, transcriptional or translational regulation; black, insertion sequence; purple, tail morphogenesis; red, DNA modification; light blue, lysis; dark blue, capsid morphogenesis and DNA packaging; green, reverse transcription; brown, replication and partitioning; gray, unknown function.
Similarity to P2
KS5, KS14 and KL3 all show similarity to enterobacteria phage P2 [GenBank:NC_001895.1]. A four-way comparison of the P2, KS5, KS14 and KL3 genomes prepared using PROmer/MUMmer/Circos is shown in Figure 3. In this comparison, regions of similarity on the same strand are shown in green, while regions of similarity on the opposite strand are shown in red. The majority of similar regions among these phages are on the same strand, except for a short conserved region in KS5 and KL3 containing DNA methylase genes (KS5 20 and KL3 28, discussed below) on the minus strand in KS5 and on the plus strand in KL3 (Tables 1 and 3). KS5, KS14 and KL3 all encode proteins similar to phage P2 D, U, T, E, E+E', FII, FI, I, J, W, V, S, R and X (involved in tail formation) and L, M, N, O, P and Q (involved in capsid formation) (Table 4). In addition, KS5 gp8 and KL3 gp9 are similar to Ogr (transcriptional activator), KS5 gp28 is similar to Old (phage immunity protein), KS14 gp17 is similar to G (tail fiber assembly protein) and KS14 gp26/gp25 and KL3 gp32/gp31 are similar to LysBC (Rz/Rz1-like lysis proteins, discussed below) (Table 4). The percent identity of the similar proteins ranges from 25-64% in KS5, 24-64% in KS14 and 31-62% in KL3 (Table 4).

PROmer/MUMmer/Circos comparison of the KS5, KS14, KL3 and P2 prophages. Regions of similarity on the same strand are shown in green and regions of similarity on the opposite strand are shown in red. The scale (in kbp) is shown on the outside. The sequence start site for the KS14 prophage (which is maintained as a plasmid) was chosen based on alignment with the other three sequences. PROmer parameters: breaklen = 60, maxgap = 30, mincluster = 20, minmatch = 6.
The genes in common between P2 and the P2-like BCC phages are almost exclusively limited to structural genes involved in virion formation (Table 4). Other P2 genes, such as those involved in DNA replication, phage immunity, lysogeny and lysis are dissimilar among these phages. A similar pattern is observed (with some exceptions) following CoreGenes analysis of the P2-like phages ϕE202 of B. thailandensis and ϕ52237 and ϕE12-2 of B. pseudomallei (data not shown) [23]. A likely explanation for this pattern is that, while phage structural components predominantly interact with each other, components from other phage systems may interact with host-specific proteins (such as those involved in transcription and DNA replication) [31, 32]. KS5, KS14 and KL3 appear to have retained P2 modules for the closely interacting capsid and tail proteins, while acquiring new modules for carrying out Burkholderia host-specific processes. These genes replace P2 genes at the right end of the P2 genome (the TO-region), P2 Z/fun (the Z-region) and P2 orf30 (Table 4) [33]. As it is very common for genes not found in P2 to be identified in these three regions in other P2-like phages, it is predicted that these loci contain genes that have been acquired via horizontal transfer [33].
Although a phage may show relatedness to a well-characterized phage such as P2, specific guidelines must be used to determine both the degree of relatedness of two phages and if the novel phage can be classified as a "P2-like virus" in a strict taxonomic sense. Lavigne et al. proposed the use of the comparison program CoreGenes to aid in phage taxonomic analysis [34]. This program can be used to compare the proteomes of two or more phages [34]. If a phage shares at least 40% of its proteins (those with a BLASTP score ≥ 75) with a reference phage such as P2, then these two phages can be considered as part of the same genus, while if it shares 20-39% of its proteins with a reference phage, then they can be considered as part of the same subfamily [34]. When KS5, KS14 and KL3 were analyzed with CoreGenes using P2 as a reference genome, the percentage of proteins in common with respect to P2 were 51.16%, 53.49% and 53.49%, respectively. These are similar to the percentages for ϕE202 (55.81%), ϕ52237 (51.16%) and ϕE12-2 (48.84%) [23]. Based on these results, KS5, KS14 and KL3 can be classified as members of the Peduovirinae subfamily and "P2-like viruses" genus [23].
Integration site characterization
In E. coli, P2 is able to integrate at over 10 different loci, but certain sites may be used more commonly than others [35]. None of the three P2-like BCC phages characterized here were found to integrate into a locus similar to that of P2. Only KL3 was found to have a previously characterized integration site. Following PCR amplification and sequencing from the B. cenocepacia CEP511 chromosome (where KL3 is carried as a prophage), it was determined that, like many phages, KL3 integrates into a tRNA gene. Specifically, it integrates into the middle of a threonine tRNA gene: bp 1 of the KL3 prophage corresponds to bp 32 of the tRNA based on comparison with a 76 bp threonine tRNA gene of B. cenocepacia HI2424 chromosome 1 (Bcen2424_R0015, bp 491047-491122). Other phages that integrate into threonine tRNA genes include enterobacteria phage P22, Shigella flexneri phage SfV and Salmonella enterica serovar Typhimurium phage ST104 [36–38]. KL3 integration should not affect threonine tRNA synthesis as bp 1-45 of KL3 has an identical sequence to bp 32-76 of the tRNA gene.
In both B. multivorans ATCC 17616 and B. cenocepacia C6433, KS5 integrates into the 3' end of an AMP nucleosidase gene. AMP nucleosidases convert AMP into adenine and ribose 5-phosphate [39]. This gene has not been previously identified as a phage integration site. KS5 bases 1-815 (including the integration site and the integrase gene sequence) show similarity to sequences encoding pairs of adjacent AMP nucleosidase and integrase genes in several Burkholderia genomes. For example, in B. pseudomallei K96243 chromosome 2, the AMP nucleosidase (BPSS1777) and integrase (BPSS1776) genes are adjacent to genes annotated as encoding a putative phage capsid related protein (fragment) (BPSS1775) and putative phage-related tail protein (fragment) (BPSS1774A). Similarly, in B. pseudomallei 1106a chromosome 2, the AMP nucleosidase (BURPS1106A_A2416) and integrase (BURPS1106A_A2415) genes are adjacent to genes annotated as encoding a phage portal domain protein (BURPS1106A_A2414) and phage tail completion protein (BURPS1106A_A2413). The identification of phage related genes at this site in other Burkholderia genomes suggests that the AMP nucleosidase gene may be a conserved integration site among some Burkholderia-specific temperate phages.
KS14 is different from other P2-like phages in that it does not encode a tyrosine integrase. Most temperate phages use a tyrosine recombinase (or, in rare cases, a serine recombinase) to facilitate recombination between the phage attP site and the host attB site [40]. KS14 encodes a serine recombinase (gp6), but this protein is unlikely to mediate prophage integration for three reasons. First, gp6 is more closely related to invertases such as Mu Gin (49% identity, E-value: 8e-44) and P1 Cin (49% identity, E-value: 7e-43) than to integrases such as those from Streptomyces lividans phage ϕC31 (29% identity, E-value: 1.2) and Mycobacterium smegmatis phage Bxb1 (29% identity, E-value: 3e-4) [41–44]. Second, gp6 lacks the conserved cysteine-rich and leucine/isoleucine/valine/methionine-rich regions found in other serine integrases [45]. Third, gp6 is only 225 aa in length, which is substantially smaller than the serine integrases that are typically between 450-600 aa in length [45]. We did not believe KS14 to be obligately lytic because it encodes a putative repressor protein (gp5) and because previously collected KS14-resistant C6433 isolates were predicted to be lysogenized based on PCR-positivity with KS14-specific primers (Figure 4) [19].

Detection of lysogeny in KS14-resistant B. cenocepacia C6433 isolates [19]. Bacterial genomic DNA was amplified using KS14-specific primers. Lane 1: 1 Kb Plus DNA ladder (Invitrogen), lane 2: DNA-free control, lane 3: C6433 control, lane 4: KS14-resistant C6433 isolate I, lane 5: KS14-resistant C6433 isolate II, lane 6: KS14-resistant C6433 isolate III, lane 7: KS14-resistant C6433 isolate IV, lane 8: KS14-resistant C6433 isolate V. The size of the markers (in kbp) is shown on the left.
Phages such as P1, P7 and N15 of enterobacteria, ϕ20 of Bacillus anthracis, ϕBB-1 of Borrelia burgdorferi, LE1 of Leptospira biflexa, pGIL01 of Bacillus thuringiensis and pKO2 of Klebsiella oxytoca lysogenize their hosts as plasmids [46–53]. Because KS14 gene 39 encodes a putative ParA protein (involved in partitioning in other plasmid prophages), we predicted that the KS14 prophage might exist as a plasmid [54, 55]. To test this hypothesis, we used a standard protocol for the QIAprep Spin Miniprep plasmid isolation kit with cells of C6433 (a KS14 host), ATCC 17616 (a KS5 lysogen), CEP511 (a KL3 lysogen), K56-2 (a lysogen of KS10, a previously characterized BCC-specific phage) and five putatively lysogenized KS14-resistant C6433 isolates [19, 56]. These preparations were then treated with EcoRI and the resulting fragments were separated using agarose gel electrophoresis. For each of the four control strains, no distinct bands were observed (Figure 5, left). In contrast, preparations from each of the five putatively lysogenized strains contained identical distinct bands (Figure 5, right). Furthermore, these bands were the same size as those predicted and observed for an EcoRI digest of KS14 DNA (with predictions based on a circular genome sequence) (Figure 5, far right) and sequences from selected bands matched the KS14 genome sequence. Based on these results, we predict that KS14 is a temperate phage that, in contrast to other P2-like phages, lysogenizes host strains as a plasmid.

Isolation of the KS14 plasmid prophage. DNA was isolated using a QIAprep Spin Miniprep plasmid isolation kit (Qiagen) and digested with EcoRI (Invitrogen). Lane 1: 1 Kb Plus DNA ladder (Invitrogen), lane 2: B. cenocepacia C6433, lane 3: B. multivorans ATCC 17616, lane 4: B. cenocepacia CEP511 lane 5: B. cenocepacia K56-2, lane 6: blank, lane 7: 1 Kb Plus DNA ladder, lane 8: KS14-resistant C6433 isolate I, lane 9: KS14-resistant C6433 isolate II, lane 10: KS14-resistant C6433 isolate III, lane 11: KS14-resistant C6433 isolate IV, lane 12: KS14-resistant C6433 isolate V. The size of the markers (in kbp) is shown on the left. A KS14 EcoRI DNA digest and the size of the bands predicted for this digest (> 1 kbp in size) are shown on the far right.
It is important to note that, although one of these phages has been shown to be active in vivo, temperate phages are generally considered to be suboptimal for use in a phage therapy protocol [19, 21]. In contrast to obligately lytic phages, temperate phages are associated with superinfection immunity, lysogenic conversion and specialized transduction [reviewed in] [21]. In a previous study, we have shown that temperate BCC-specific phages can be engineered to their lytic form by inactivating the repressor gene [21]. This strategy could potentially be used with the three phages described here, thus making them more appropriate candidates for clinical use.
Morphogenesis genes
As discussed above, the KS5, KS14 and KL3 structural genes are related to those from P2 and function to construct a P2-like myovirus with a contractile tail. The only virion morphogenesis genes of P2 that these phages lack are G (encoding the tail fiber assembly protein, missing in KS5 and KL3) and H (encoding the tail fiber protein) (Table 4). Because the tail fibers are involved in host recognition, it is expected that these proteins would be dissimilar in phages infecting E. coli and those infecting the BCC.
A commonly identified characteristic in tailed phages is the expression of two tail proteins from a single start codon via a translational frameshift [57]. These proteins (encoded in a region between the genes for the tail tape measure and the major tail protein) share the same N-terminus but have different C-termini due to stop codon readthrough in the -1 frame [57]. In P2, this -1 frameshift occurs at a TTTTTTG sequence and produces the 91 aa protein E and the 142 aa protein E+E' from the same translational start site (Figure 6) [57, 58]. KS5, KS14 and KL3 all encode proteins similar to both E and E+E' with percent identities ranging from 49-59% (Table 4). Despite the relatively low degree of similarity, the P2 frameshift site appears to be conserved amongst these phages, suggesting that they likely use a similar frameshifting mechanism (Figure 6). In rare cases, RNA secondary structure can be identified downstream of the phage frameshift sequence [21, 57]. When the KS5, KS14 and KL3 E+E' sequences 60 bp downstream of the TTTTTTG sequence were screened for secondary structure, no predicted hairpins were identified (data not shown). This result was anticipated based upon the absence of these structures in the P2 phage E+E' gene [57].

Conservation of the P2 E / E+E' frameshift sequence in KS5, KS14 and KL3. For each phage, the DNA sequence is shown in the first line, the translation in the original frame is shown in the second line, the translation in the -1 frame is shown in the third line and the amino acid sequence of the frameshifted protein is shown in the fourth line. The conserved TTTTTTG frameshift sequence is underlined. The frameshift is predicted to occur after the terminal G in this sequence.
Lysis genes
In P2, the lysis module consists of five genes: Y (holin), K (endolysin), lysA (antiholin), lysB (Rz) and lysC (Rz1) [59, 60]. The P2-like BCC phages are predicted to encode endolysins, holins and antiholins that are dissimilar to those of P2 (Table 4). KS5 gp33, KS14 gp27 and KL3 gp33 are putative endolysins as they all have the conserved domain pfam01471 (PG_binding_1, putative peptidoglycan binding domain; E-values: 3e-11, 3e-10 and 9e-10, respectively) and show similarity to other phage endolysins. P2 Y is a type I holin with three transmembrane domains [61]. Although KS5 34, KS14 28 and KL3 34 are dissimilar to P2 Y, it is predicted that these three genes encode holins because they are each immediately upstream of a putative endolysin gene and they each encode proteins that a) have three transmembrane domains based on OCTOPUS analysis and b) show similarity to other phage holins.
Antiholins such as P2 LysA inhibit holin activity and delay lysis of infected cells in order to optimize the phage burst size [59, 62]. Although some phages such as λ express antiholins from a second translational start site two codons upstream of the holin start codon, phages such as P2 and ϕO1205 of Streptococcus thermophilus encode an antiholin from a separate gene [63, 59, 64]. The location of the putative antiholin genes KS5 35, KS14 29 and KL3 35 is similar to that in ϕO1205, in which the holin and antiholin genes are adjacent immediately upstream of the endolysin gene (as opposed to P2, in which gene K separates Y and lysA) [64, 59]. Based on OCTOPUS analysis, KS5 gp35 has three transmembrane domains, while KS14 gp29, KL3 gp35 and P2 LysA have four. Based on gene organization and protein transmembrane structure, it is predicted that the P2-like BCC phages have separate antiholin genes in their lysis modules.
P2 encodes two proteins, LysB and LysC, that are predicted to function similarly to λ Rz and Rz1 [60]. Rz is an inner membrane protein with an N-terminal transmembrane domain and Rz1 is a proline-rich outer membrane lipoprotein [65]. Rz/Rz1 pairs fuse the inner and outer membranes following holin and endolysin activity and facilitate phage release [65]. The P2 lysC start codon is in the +1 frame within the lysB gene, while the lysC stop codon is out of frame in the downstream tail gene R[66]. In contrast, the Rz1 gene in λ is entirely contained within the Rz gene [67]. KS14 and KL3 LysBC pairs (gp26/gp25 and gp32/gp31, respectively) are similar to that of P2 (Table 4). In KS14 and KL3, the lysC genes start approximately 160 bp upstream from the lysB stop codon and extend into the first 8 bp of R (gene 24 in KS14 and 30 in KL3) (Figure 7). Both KS14 and KL3 LysC proteins are predicted to have a signal peptidase II cleavage site between positions 20 (alanine) and 21 (cysteine). Signal peptidase II cleavage would produce a 72 aa lipoprotein with 7 prolines (9.7% proline) for KS14 LysC and a 74 amino acid lipoprotein with 7 prolines (9.5% proline) for KL3 LysC.

Organization of the lysBC genes in KS5, KS14 and KL3. R genes (KS5 30, KS14 24 and KL3 30) are shown in light gray, lysB genes (KS5 32, KS14 26 and KL3 32) are shown in dark gray and lysC genes (KS5 31, KS14 25 and KL3 31) are shown in white. The scale (in bp) is shown below.
In contrast to the P2-like lysBC gene organization found in KS14 and KL3, the KS5 genes 32/31 have a similar organization to λ Rz/Rz1. KS5 Rz1 is encoded in the +1 frame within the Rz gene (Figure 7). It is predicted to have a signal peptidase II cleavage site between positions 18 (alanine) and 19 (cysteine), which would produce a 46 amino acid lipoprotein with 12 prolines (26.1%). The differences in both gene organization and proline content between the P2-like KS14 and KL3 LysC proteins and the λ-like KS5 Rz1 protein suggest that KS5 may have acquired genes 31 and 32 - and potentially the entire lysis module - through horizontal transfer from a phage similar to λ.
Sequence elements unique to KS5 and/or KL3
Insertion sequences
Insertion sequences (ISs) are short genetic elements that can insert into nonhomologous regions of DNA [68]. These elements, comprised of a transposase gene and inverted repeats, create flanking direct repeats following insertion [68]. Many mutants of well-characterized phages have been found to carry ISs, including λ and Mu [69, 70]. However, it is relatively rare for wildtype phages to carry ISs because they can interfere with gene expression [71]. Sakaguchi et al. determined the genome sequence of the Clostridium botulinum phage c-st and determined that it carries 12 ISs (5 of which are incomplete) [71]. Of the 284 genomes sequenced at the time, one IS was found in each of eight phages: Burkholderia phages ϕE125 and Bcep22, enterobacteria phages P1 and HK022, Lactobacillus phages ϕAT3 and LP65, Rhodothermus phage RM378 and Shigella phage Sf6 [71].
A novel insertion sequence (named ISBmu 23 in vB_BmuZ-ATCC 17616) is found in the KS5 genome between gene 12, encoding a membrane protein and gene 13, encoding the tail protein D (Table 1). This IS does not appear to disrupt any putative ORFs and so may not have any significant effect on phage gene expression. ISBmu 23 is 1210 bp in length and contains two imperfect 16 bp inverted repeats (Table 1, Figure 8). In KS5, it is flanked by two copies of a 5 bp direct repeat, CCTAA. ISBmu 23 encodes a 330 aa transposase that has the conserved domain COG3039 (transposase and inactivated derivatives, IS5 family; E-value: 8e-29). This protein is most similar to the transposase of ISBmu 2 (85% identity), an IS5-like IS present in nine copies in ATCC 17616 [72]. ISBmu 2 and ISBmu 23 are very similar as they a) are present in the same genome, b) are both 1210 bp in length, c) encode similar 330 aa transposases, d) have similar 16 bp inverted repeats (the right inverted repeats of ISBmu 2 and ISBmu 23 are identical, while the left repeats differ by 3 bp) and e) preferentially integrate into CTAA sequences (Figure 8). Ohtsubo et al. found that the transposition of ISs in ATCC 17616 increased when the cells were grown at high temperatures [72]. Because these temperatures are similar to what the cell may encounter during infection of an animal or human, it is suggested that this change may provide a selective advantage to ATCC 17616 by modifying its genome under in vivo conditions [72]. Further experiments are required to determine if ISBmu 23 transposition is affected by temperature and if this IS may provide a selective advantage to KS5 lysogens in vivo.

Comparison of the IS Bmu 23 and IS Bmu 2 insertion sequences. A) Structure of ISBmu 23. IR, inverted repeats. Relative positions of the inverted repeats and transposase gene (in bp) are shown below. B) Alignment of the ISBmu 2 and ISBmu 23 inverted repeats. Non-consensus bases are underlined. ISBmu 2 sequences are from Ohtsubo et al. [72]. L, left repeat; Ri, right repeat inverted.
Reverse transcriptases
Reverse transcriptases (RTs) are RNA-dependent DNA polymerases most commonly associated with retroviruses and retrotransposons [73]. RTs have also been identified in several phage genomes, including those of P2-like phages [74–76]. One function of these proteins was extensively characterized in Bordetella bronchiseptica phage BPP-1. This phage has the ability to change its host range by making amino acid substitutions in its tail fiber protein, Mtd (major tropism determinant) [77]. This switch requires the phage-encoded RT Brt (Bordetella RT) that synthesizes a DNA copy of a 134 bp locus (the template repeat, TR) that has 90% identity with a 134 bp region of the mtd gene (the variable repeat, VR) [77, 74]. Adenines in the reverse transcribed copy of TR are mutagenized and the altered DNA integrates or recombines at VR by an unknown mechanism, generating a tail fiber gene with multiple base substitutions [74, 75].
A second function associated with phage RTs is phage exclusion. In Lactococcus lactis, expression of the putative RT AbiK lowers the efficiency of plating of infecting phages by an unknown mechanism (potentially involving single-strand annealing recombinases) [78]. Expression of Orf570, an RT identified in the P2-like enterobacteria prophage P2-EC30, was found to inhibit T5 infection of E. coli[76]. When a region of Orf570 containing an RT conserved motif was deleted, T5 infection was no longer inhibited [76].
KS5 encodes a putative RT, gp44. This protein has the conserved domain cd03487 (RT_Bac_retron_II, reverse transcriptases in bacterial retrotransposons or retrons; E-value: 2e-45). It is unlikely that gp44 and Brt have the same function: the two proteins show minimal similarity (21% identity, E-value: 7e-4), gene 44 is located distal to the tail fiber gene (in contrast to brt and mtd), neither nucleotide substitutions in the tail fiber gene nor variations in KS5 tropism were observed and no repeated sequences were identified in the KS5 genome longer than 28 bp [77]. When compared to Orf570, gp44 shows almost no relatedness (41% over 12/546 amino acids; E-value: 2.7) but is found at the same locus (in the prophage, both orf570 and 44 would be located proximal to the portal protein gene Q). Further experiments are required to determine if the KS5 RT is involved in tropism modification, phage exclusion or some uncharacterized function.
DNA methylation, restriction and repair
DNA methylase and endonuclease genes are commonly found in phage genomes. Methylases modify the DNA such that it becomes resistant to bacterial restriction systems [79]. Although P2 does not encode any putative methylases, such proteins are encoded by both KS5 and KL3 (KS5 gp20 and KL3 gp28 and gp47) (Tables 1 and 3). All three methylases are predicted to belong to the AdoMet_MTase superfamily (cl12011; S-adenosylmethionine-dependent methyltransferases). KS5 gp20 is most similar to a DNA methylase N-4/N-6 domain protein of B. ambifaria MEX-5 (89% identity). KL3 gp28 is most similar to a site-specific DNA methyltransferase of B. pseudomallei K96243 (78% identity). Both of these proteins have the conserved domain pfam01555 (N6_N4_Mtase, DNA methylase; KS5 gp20 E-value: 5e-22, KL3 gp28 E-value: 4e-25). Because this domain is associated with both N-4 cytosine and N-6 adenine methylases, these proteins may have either cytosine or adenine methylase activity [80]. KL3 gp47 shows similarity to a modification methylase EcoRII from several bacterial species, with E-values as low as 4e-114. This protein has the conserved domain cd00315 (Cyt_C5_DNA_methylase, Cytosine-C5 specific DNA methylases; E-value: 6e-68) and so can be classified as a cytosine-C5 methylase. KS5 gp20 and KL3 gp28 are likely involved in protecting the phage DNA from BCC restriction systems. As discussed below, the function of KL3 gp47 is likely to protect the phage DNA from a phage-encoded restriction enzyme.
Phage nucleases have a number of functions, including degradation of the bacterial DNA (to both inhibit the host and provide nucleotides for the phage), phage exclusion and DNA processing [81]. KL3 encodes two endonucleases, gp45 and gp46. Gp45 is most similar to a type II restriction endonuclease, EcoRII-C domain protein of Candidatus Hamiltonella defensa 5AT (Acyrthosiphon pisum) (77% identity). This protein has the conserved domain pfam09019 (EcoRII-C, EcoRII C terminal; E-value: 6e-65). Gp46 is most similar to a DNA mismatch endonuclease Vsr of Burkholderia graminis C4D1M (77% identity). This protein has the conserved domain cd00221 (Vsr, Very Short Patch Repair [Vsr] endonuclease; E-value: 9e-38).
The organization of genes 45-47 (encoding an EcoRII-C endonuclease, Vsr endonuclease and EcoRII methylase, respectively) in a single module suggests that the proteins that they encode are functionally related. The EcoRII-C endonuclease (which has a CCWGG recognition sequence where W = A or T) is likely to degrade either bacterial DNA to inhibit the host during the KL3 lytic cycle or superinfecting phage DNA [81, 82]. KL3 DNA would be protected from this cleavage by EcoRII methylation at the second position in the EcoRII-C recognition sequence (forming CCmWGG where Cm = 5-methylcytosine) [83]. Expression of the Dcm methylase, which has an identical recognition sequence and methylation site as EcoRII methylase, is mutagenic in E. coli because 5-methylcytosines are deaminated to thymines, causing T/G mismatches [84, 85]. EcoRII methylase expression would presumably cause mismatched sites in KL3 with the sequence C(T/G)WGG. In E. coli, these mismatches are repaired by very short patch (VSP) repair which starts with the recognition and nicking of the sequence C(T/G)WGG by a Vsr endonuclease [86]. As KL3 expresses a Vsr endonuclease, it could repair post-methylation T/G mismatches using the same mechanism.
The proposed model for methylase and endonuclease interaction during the KL3 lytic cycle is as follows. Unmethylated host DNA (or, alternatively, superinfecting phage DNA) is degraded by gp45. KL3 DNA is protected from gp45 degradation by gp47-mediated conversion of cytosine to 5-methylcytosine. These 5-methylcytosine bases are deaminated to thymine, but the resulting T/G mismatches are cleaved by gp46 and fixed using VSP repair. Although further experiments are required to test the validity of this model, KL3 appears to encode an elegant system for degradation of bacterial or superinfecting phage DNA, protection of the phage genome and repair of resulting mutations.
Conclusions
This study is the first to identify and characterize P2-like phages of the BCC. Like other previously characterized P2-like Burkholderia phages, KS5, KS14 and KL3 share structural genes with P2 but encode dissimilar accessory proteins. KS5, a 37236 bp prophage of B. multivorans ATCC 17616, integrates into an AMP nucleosidase gene, has a λ-like Rz/Rz1 cassette, carries an ISBmu 2-like insertion sequence and encodes a reverse transcriptase. KS14, a 32317 bp phage previously shown to be active against B. cenocepacia in vivo, encodes a serine recombinase but is maintained as a plasmid prophage [19]. KL3, a 40555 bp prophage of B. cenocepacia CEP511, integrates into a threonine tRNA gene and encodes a series of proteins capable of degrading bacterial or superinfecting phage DNA, methylating the phage genome and repairing methylation-induced mismatches. As KS14 has already been shown to be active in vivo, characterization of these three related phages is an important preliminary step in the development of a phage therapy protocol for the BCC.
Methods
Bacterial strains and growth conditions
BCC strains used for phage isolation and propagation were obtained from Belgium Coordinated Collection of Microorganisms LMG Bacteria Collection (Ghent, Belgium) and the Canadian Burkholderia cepacia complex Research and Referral Repository (Vancouver, BC). Many of the strains used are from the Burkholderia cepacia complex experimental strain panel and updated experimental strain panel [29, 87]. Strains were grown aerobically overnight at 30°C on half-strength Luria-Bertani (LB) solid medium or in half-strength LB broth with shaking. Transformations were performed with chemically-competent DH5α (Invitrogen, Carlsbad, CA), plated on LB solid medium containing 100 μg/ml ampicillin and grown aerobically overnight at 37°C. Strains were stored in LB broth containing 20% glycerol at -80°C.
Electron microscopy
To prepare samples for transmission electron microscopy, phage lysates were filter sterilized using a Millex-HA 0.45 μm syringe driven filter unit (Millipore, Billerica, MA), incubated on a carbon-coated copper grid 5 minutes at room temperature and stained with 2% phosphotungstic acid for 2 minutes. Micrographs were taken with the assistance of the University of Alberta Department of Biological Sciences Advanced Microscopy Facility using a Philips/FEI (Morgagni) transmission electron microscope with charge-coupled device camera at 140,000-fold magnification.
Phage isolation, propagation and DNA isolation
Isolation of KS5 from onion soil and KS14 from Dracaena sp. soil has been described previously [25, 19]. KL3 was isolated from a single plaque on a lawn of B. cenocepacia CEP511. The plaque was isolated using a sterile Pasteur pipette, suspended in 1 ml of suspension medium (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 10 mM MgSO4, 0.01% gelatin solution) with 20 μl CHCl3 and incubated 1 hour at room temperature to generate a KL3 stock. KL3 was propagated on B. ambifaria LMG 17828 in soft agar overlays: 100 μl of phage stock and 100 μl of liquid culture were incubated 20 minutes at room temperature and 3 ml of soft nutrient agar was added to this mixture, poured onto half-strength LB solid medium and incubated overnight at 30°C.
Phage genomic DNA was isolated using a modified version of a λ proteinase K/SDS lysis protocol [88]. Half-strength LB agarose plates (prepared with soft nutrient agarose) showing confluent phage lysis were overlaid with 3 ml of suspension media and incubated for 6 hours at 4°C on a platform rocker. The lysate was pelleted by centrifugation at 10 000 × g for 2 minutes and filter-sterilized using a 0.45 μm filter. 10 ml of lysate was treated with 10 μl DNase I/10 μl DNase I buffer and 6 μl RNase I (Fermentas, Burlington, ON) and incubated 1 hour at 37°C. Following addition of 0.5 M EDTA (pH 8.0) to 20 mM, proteinase K to 50 μg/ml and SDS to 0.5%, the solution was mixed and incubated 1 hour at 37°C. Standard phenol:chloroform extraction and ethanol precipitation were then used to purify the phage DNA. Samples were resuspended in TE (pH 8.0) and quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA).
KS14 plasmid prophage DNA was isolated from five putatively lysogenized KS14-resistant C6433 isolates [19] using a QIAprep Spin Miniprep kit (Qiagen, Hilden, Germany). Lysogeny was predicted using PCR with KS14-specifc primers (KS14F: GCAGCTAACCGAGTCGCACG, KS14R: CTCTGAAAAGGTGGGCGGTGG) (Sigma-Genosys, Oakville, ON) and TopTaq DNA polymerase and buffers (Qiagen). B. multivorans ATCC 17616 and B. cenocepacia C6433, CEP511 and K56-2 were used as negative controls. 2 ml aliquots of 16 hour overnight cultures (OD600: 2.0-2.2) were pelleted, washed 3× with sterile H2O to remove exogenous phages and treated using the standard kit protocol. For each sample, 2 20 μl EcoRI (Invitrogen) reactions each containing 17 μl of plasmid DNA were digested overnight, pooled and separated on 0.8% (wt/vol) agarose gels in 1× TAE (pH 8.0).
Sequencing and bioinformatics analysis
Preliminary sequence analysis was performed using a shotgun cloning protocol. Phage DNA was digested using EcoRI (Invitrogen), separated on 0.8% (wt/vol) agarose gels, purified using the GeneClean II kit (Qbiogene, Irvine, CA), ligated into pUC19 or pGEM-7Z and transformed into DH5α (Invitrogen). Following blue-white selection on LB solid medium containing 100 μg/ml ampicillin, constructs with phage DNA inserts were isolated using a QIAprep Spin Miniprep kit (Qiagen), digested using EcoRI and viewed using gel electrophoresis. Inserts were sequenced with the assistance of the University of Alberta Department of Biological Sciences Molecular Biology Service Unit using an ABI 3730 DNA analyzer (Applied Biosystems, Foster City, CA). Sequences were edited using EditView and aligned using AutoAssembler (Perkin-Elmer, Waltham, MA). For completion of the three genomes, DNA samples were submitted for pyrosequencing analysis (454 Life Sciences, Branford, CT). Gaps between the assembled sequences were filled following PCR amplification and cloning using primers (Sigma-Genosys) designed to amplify across the gaps, TopTaq DNA polymerase and buffers (Qiagen) and the CloneJET PCR cloning kit (Fermentas). The complete genome sequences of KS5, KS14 and KL3 were deposited in GenBank with the accession numbers GU911303, HM461982 and GU911304, respectively.
Annotation of the assembled sequences was performed using GeneMark.hmm-P http://exon.biology.gatech.edu[89]. For KS5, annotations were based on those of the ATCC 17616 chromosome 2 sequence (GenBank:NC_010805.1; BMULJ_03640 - BMULJ_03684, bp 477496-514731). Manual annotations were performed for the E+E' and lysC/Rz1 genes. Proteins were numbered based on the order of the genes in the prophage (i.e. the integrase gene was named 1 and the integrase was named gp1). Relatedness of the predicted proteins was assessed using BLASTP http://blast.ncbi.nlm.nih.gov[90]. Protein transmembrane domains, stem-loop structures and signal peptide cleavage sites were identified using OCTOPUS http://octopus.cbr.su.se, mfold http://mfold.rna.albany.edu and LipoP http://www.cbs.dtu.dk/services/LipoP, respectively [91–93]. Repeat sequences in the DNA were identified using REPuter http://bibiserv.techfak.uni-bielefeld.de/reputer[94]. Restriction sites were predicted using NEBcutter http://tools.neb.com/NEBcutter2[95]. Whole genome sequence comparisons were performed using CoreGenes with a stringency setting of 75 http://www.binf.gmu.edu/genometools.html[96, 34]. Comparison figures were constructed using PROmer/MUMmer http://mummer.sourceforge.net Circos http://mkweb.bcgsc.ca/circos[97, 98].
To identify the KS5 prophage insertion site in ATCC 17616, the assembled KS5 sequence was compared to the vB_BmuZ-ATCC 17616 sequence in a BLASTN search and the left prophage junction was determined. Primers designed to this region (KS5attL F: TGCACGGCGAGCTGAAACTG, KS5attL R: GAAGGCACGCGAGGTAGAACG) were used to amplify the C6433/KS5 prophage junction in C6433 lysogens. To identify the KL3 insertion site in CEP511, the region proximal to the KL3 integrase gene 1 was analyzed using BLASTN and found to be similar to a region containing a tRNA-Thr gene in several Burkholderia strains including B. ambifaria AMMD (Bamb_R0016; chromosome 1, bp 403358-403433). Primers designed to this region (KL3attL F: AGCTGCAGATGGGTAACGAGTGG, KL3attL R: CCACTCACGAAGGGCAAGCTG) were used to amplify the CEP511/KL3 prophage junction.
References
Estivariz CF, Bhatti LI, Pati R, Jensen B, Arduino MJ, Jernigan D, LiPuma JJ, Srinivasan A: An outbreak of Burkholderia cepacia associated with contamination of albuterol and nasal spray. Chest. 2006, 130: 1346-1353. 10.1378/chest.130.5.1346.
Isles A, Maclusky I, Corey M: Pseudomonas cepacia infection in cystic fibrosis: An emerging problem. J Pediatr. 1984, 104: 206-210. 10.1016/S0022-3476(84)80993-2.
Robinson M, Bye PTB: Mucociliary clearance in cystic fibrosis. Pediatr Pulmonol. 2002, 33: 293-306. 10.1002/ppul.10079.
Harrison F: Microbial ecology of the cystic fibrosis lung. Microbiology. 2007, 153: 917-923. 10.1099/mic.0.2006/004077-0.
Razvi S, Quittell L, Sewall A, Quinton H, Marshall B, Saiman L: Respiratory microbiology of patients with cystic fibrosis in the United States, 1995 to 2005. Chest. 2009, 136: 1554-1560. 10.1378/chest.09-0132.
Courtney JM, Dunbar KEA, McDowell A, Moore JE, Warke TJ, Stevenson M, Elborn JS: Clinical outcome of Burkholderia cepacia complex infection in cystic fibrosis adults. J Cyst Fibros. 2004, 3: 93-98. 10.1016/j.jcf.2004.01.005.
Jones AM, Dodd ME, Govan JRW, Barcus V, Doherty CJ, Morris J, Webb AK: Burkholderia cenocepacia and Burkholderia multivorans: Influence on survival in cystic fibrosis. Thorax. 2004, 59: 948-951. 10.1136/thx.2003.017210.
Coenye T, Vandamme P, Govan JRW, Lipuma JJ: Taxonomy and identification of the Burkholderia cepacia complex. J Clin Microbiol. 2001, 39: 3427-3436. 10.1128/JCM.39.10.3427-3436.2001.
Vandamme P, Holmes B, Vancanneyt M, Coenye T, Hoste B, Coopman R, Revets H, Lauwers S, Gillis M, Kersters K, Govan JRW: Occurrence of multiple genomovars of Burkholderia cepacia in cystic fibrosis patients and proposal of Burkholderia multivorans sp. nov. Int J Syst Evol Microbiol. 1997, 47: 1188-1200.
Biddick R, Spilker T, Martin A, LiPuma JJ: Evidence of transmission of Burkholderia cepacia, Burkholderia multivorans and Burkholderia dolosa among persons with cystic fibrosis. FEMS Microbiol Lett. 2003, 228: 57-62. 10.1016/S0378-1097(03)00724-9.
Vanlaere E, Baldwin A, Gevers D, Henry D, De Brandt E, LiPuma JJ, Mahenthiralingam E, Speert DP, Dowson C, Vandamme P: Taxon K, a complex within the Burkholderia cepacia complex, comprises at least two novel species, Burkholderia contaminans sp. nov. and Burkholderia lata sp. nov. Int J Syst Evol Microbiol. 2009, 59: 102-111. 10.1099/ijs.0.001123-0.
Zhou J, Garber E, Saiman L: Survey of infection control policies for patients with cystic fibrosis in the United States. Am J Infect Control. 2008, 36: 220-222. 10.1016/j.ajic.2007.05.009.
Duff AJA: Psychological consequences of segregation resulting from chronic Burkholderia cepacia infection in adults with CF. Thorax. 2002, 57: 756-758. 10.1136/thorax.57.9.756.
Hirai K, Iyobe S, Inoue M, Mitsuhashi S: Purification and properties of a new β-lactamase from Pseudomonas cepacia. Antimicrob Agents Chemother. 1980, 17: 355-358.
Burns JL, Wadsworth CD, Barry JJ, Goodall CP: Nucleotide sequence analysis of a gene from Burkholderia (Pseudomonas) cepacia encoding an outer membrane lipoprotein involved in multiple antibiotic resistance. Antimicrob Agents Chemother. 1996, 40: 307-313.
Conway B-D, Venu V, Speert DP: Biofilm formation and acyl homoserine lactone production in the Burkholderia cepacia complex. J Bacteriol. 2002, 184: 5678-5685. 10.1128/JB.184.20.5678-5685.2002.
Zhou J, Chen Y, Tabibi S, Alba L, Garber E, Saiman L: Antimicrobial susceptibility and synergy studies of Burkholderia cepacia complex isolated from patients with cystic fibrosis. Antimicrob Agents Chemother. 2007, 51: 1085-1088. 10.1128/AAC.00954-06.
Merril CR, Scholl D, Adhya SL: The prospect for bacteriophage therapy in Western medicine. Nat Rev Drug Discov. 2003, 2: 489-497. 10.1038/nrd1111.
Seed KD, Dennis JJ: Experimental bacteriophage therapy increases survival of Galleria mellonella larvae infected with clinically relevant strains of the Burkholderia cepacia complex. Antimicrob Agents Chemother. 2009, 53: 2205-2208. 10.1128/AAC.01166-08.
Carmody LA, Gill JJ, Summer EJ, Sajjan US, Gonzalez CF, Young RF, Lipuma JJ: Efficacy of bacteriophage therapy in a model of Burkholderia cenocepacia pulmonary infection. J Infect Dis. 2010, 201: 264-271. 10.1086/649227.
Lynch KH, Seed KD, Stothard P, Dennis JJ: Inactivation of Burkholderia cepacia complex phage KS9 gp41 identifies the phage repressor and generates lytic virions. J Virol. 2010, 84: 1276-1288. 10.1128/JVI.01843-09.
Bertani G: Studies of lysogeny. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol. 1951, 62: 293-300.
Lavigne R, Darius P, Summer EJ, Seto D, Mahadevan P, Nilsson AS, Ackermann H-W, Kropinski AM: Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 2009, 9-
Kropinski AM, Prangishvili D, Lavigne R: Position paper: The creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ Microbiol. 2009, 11: 2775-2777. 10.1111/j.1462-2920.2009.01970.x.
Seed KD, Dennis JJ: Isolation and characterization of bacteriophages of the Burkholderia cepacia complex. FEMS Microbiol Lett. 2005, 251: 273-280. 10.1016/j.femsle.2005.08.011.
Loutet SA, Flannagan RS, Kooi C, Sokol PA, Valvano MA: A complete lipopolysaccharide inner core oligosaccharide is required for resistance of Burkholderia cenocepacia to antimicrobial peptides and bacterial survival in vivo. J Bacteriol. 2006, 188: 2073-2080. 10.1128/JB.188.6.2073-2080.2006.
Ortega X, Silipo A, Saldfas MS, Bates CC, Molinaro A, Valvano MA: Biosynthesis and structure of the Burkholderia cenocepacia K56-2 lipopolysaccharide core oligosaccharide: Truncation of the core oligosaccharide leads to increased binding and sensitvity to polymyxin B. J Biol Chem. 2009, 284: 21738-21751. 10.1074/jbc.M109.008532.
Vandamme P, Henry D, Coenye T, Nzula S, Vancanneyt M, LiPuma JJ, Speert DP, Govan JRW, Mahenthiralingam E: Burkholderia anthina sp. nov. and Burkholderia pyrrocinia, two additional Burkholderia cepacia complex bacteria, may confound results of new molecular diagnostic tools. FEMS Immunol Med Microbiol. 2002, 33: 143-149. 10.1111/j.1574-695X.2002.tb00584.x.
Mahenthiralingam E, Coenye T, Chung JW, Speert DP, Govan JRW, Taylor P, Vandamme P: Diagnostically and experimentally useful panel of strains from the Burkholderia cepacia complex. J Clin Microbiol. 2000, 38: 910-913.
Ackermann H-W: Frequency of morphological phage descriptions in the year 2000. Arch Virol. 2001, 146: 843-857. 10.1007/s007050170120.
Minakhin L, Semenova E, Liu J, Vailov A, Severinova E, Gabisonia T, Inman R, Mushegian A, Severinov K: Genome sequence and gene expression of Bacillus anthracis bacteriophage Fah. J Mol Biol. 2005, 354: 1-15. 10.1016/j.jmb.2005.09.052.
Datta I, Sau S, Sil AK, Mandal NC: The bacteriophage λ DNA replication protein P inhibits the oriC DNA- and ATP-binding functions of the DNA replication initiator protein DnaA of Escherichia coli. J Biochem Mol Biol. 2005, 38: 97-103.
Nilsson AS, Haggård-Ljungquist E: Evolution of P2-like phages and their impact on bacterial evolution. Res Microbiol. 2007, 158: 311-317. 10.1016/j.resmic.2007.02.004.
Lavigne R, Seto D, Mahadevan P, Ackermann H-W, Kropinski AM: Unifying classical and molecular taxonomic classification: analysis of the Podoviridae using BLASTP-based tools. Res Microbiol. 2008, 159: 406-414. 10.1016/j.resmic.2008.03.005.
Barreiro V, Haggård-Ljungquist E: Attachment sites for bacteriophage P2 on the Escherichia coli chromosome: DNA sequences, localization on the physical map, and detection of a P2-like remnant in E. coli K-12 derivatives. J Bacteriol. 1992, 174: 4086-4093.
Lindsey DF, Martinez C, Walker JR: Physical map location of the Escherichia coli attachment site for the P22 prophage (attP22). J Bacteriol. 1992, 174: 3834-3835.
Guan S, Verma NK: Serotype conversion of a Shigella flexneri candidate vaccine strain via a novel site-specific chromosome-integration system. FEMS Microbiol Lett. 1998, 166: 79-87. 10.1111/j.1574-6968.1998.tb13186.x.
Hermans APHM, Beuling AM, van Hoek AHAM, Aarts HJM, Abee T, Zwietering MH: Distribution of prophages and SGI-1 antibiotic-resistance genes among different Salmonella enterica serovar Typhimurium isolates. Microbiology. 2006, 152: 2137-2147. 10.1099/mic.0.28850-0.
Zhang Y, Cottet SE, Ealick SE: Structure of Escherichia coli AMP nucleosidase reveals similarity to nucleoside phosphorylases. Structure. 2004, 12: 1383-1394. 10.1016/j.str.2004.05.015.
Groth AC, Calos MP: Phage integrases: Biology and applications. J Mol Biol. 2004, 335: 667-678. 10.1016/j.jmb.2003.09.082.
Plasterk RHA, Ilmer TAM, Van de Putte P: Site-specific recombination by Gin of bacteriophage Mu: Inversions and deletions. Virology. 1983, 127: 24-36. 10.1016/0042-6822(83)90367-7.
Hiestand-Nauer R, Iida S: Sequence of the site-specific recombinase gene cin and of its substrates serving in the inversion of the C segment of bacteriophage P1. EMBO J. 1983, 2: 1733-1740.
Kuhstoss S, Rao RN: Analysis of the integration function of the Streptomycete bacteriophage ϕC31. J Mol Biol. 1991, 222: 897-908. 10.1016/0022-2836(91)90584-S.
Kim AI, Ghosh P, Aaron MA, Bibb LA, Jain S, Hatfull GF: Mycobacteriophage Bxb1 integrates into the Mycobacterium smegmatis groEL1 gene. Mol Microbiol. 2003, 50: 463-473. 10.1046/j.1365-2958.2003.03723.x.
Smith MCM, Thorpe HM: Diversity in the serine recombinases. Mol Microbiol. 2002, 44: 299-307. 10.1046/j.1365-2958.2002.02891.x.
Ikeda H, Tomizawa J: Prophage P1, an extrachromosomal replication unit. Cold Spring Harb Symp Quant Biol. 1968, 33: 791-798.
Yun T, Vapnek D: Electron microscopic analysis of bacteriophages P1, P1Cm, and P7. Determination of genome sizes, sequence homology, and location of antibiotic resistance determinants. Virology. 1977, 77: 376-385. 10.1016/0042-6822(77)90434-2.
Ravin VK, Shulga MG: Evidence for extrachromosomal location of prophage N15. Virology. 1970, 40: 800-807. 10.1016/0042-6822(70)90125-X.
Inal JM, Karunakaran KV: φ20, a temperate bacteriophage isolated from Bacillus anthracis exists as a plasmidial prophage. Curr Microbiol. 1996, 32: 171-175. 10.1007/s002849900030.
Eggers CH, Casjens S, Hayes SF, Garon CF, Damman CJ, Oliver DB, Samuels DS: Bacteriophages of Spirochetes. J Mol Microbiol Biotechnol. 2000, 2: 365-373.
Saint Girons I, Bourhy P, Ottone C, Picardeau M, Yelton D, Hendrix RW, Glaser P, Charon N: The LE1 bacteriophage replicates as a plasmid within Leptospira biflexa: construction of an L. biflexa-Escherichia coli shuttle vector. J Bacteriol. 2000, 182: 5700-5705. 10.1128/JB.182.20.5700-5705.2000.
Verheust C, Jensen G, Mahillon J: pGIL01, a linear tectiviral plasmid prophage originating from Bacillus thuringiensis serovar israelensis. Microbiology. 2003, 149: 2083-2092. 10.1099/mic.0.26307-0.
Casjens SR, Gilcrease EB, Huang WM, Bunny KL, Pedulla ML, Ford ME, Houtz JM, Hatfull GF, Hendrix RW: The pKO2 linear plasmid prophage of Klebsiella oxytoca. J Bacteriol. 2004, 186: 1818-1832. 10.1128/JB.186.6.1818-1832.2004.
Abeles AL, Friedman SA, Austin SJ: Partition of unit-copy miniplasmids to daughter cells III. The DNA sequence and functional organization of the P1 partition region. J Mol Biol. 1985, 185: 261-272. 10.1016/0022-2836(85)90402-4.
Ludtke DN, Eichorn BG, Austin SJ: Plasmid-partition functions of the P7 prophage. J Mol Biol. 1989, 209: 393-406. 10.1016/0022-2836(89)90005-3.
Goudie AD, Lynch KH, Seed KD, Stothard P, Shrivastava S, Wishart DS, Dennis JJ: Genomic sequence and activity of KS10, a transposable phage of the Burkholderia cepacia complex. BMC Genomics. 2008, 9-
Xu J, Hendrix RW, Duda RL: Conserved translational frameshift in dsDNA bacteriophage tail assembly genes. Mol Cell. 2004, 16: 11-21. 10.1016/j.molcel.2004.09.006.
Christie GE, Temple LM, Bartlett BA, Goodwin TS: Programmed translational frameshift in the bacteriophage P2 FETUD tail gene operon. J Bacteriol. 2002, 184: 6522-6531. 10.1128/JB.184.23.6522-6531.2002.
Ziermann R, Bartlett B, Calendar R, Christie GE: Functions involved in bacteriophage P2-induced host cell lysis and identification of a new tail gene. J Bacteriol. 1994, 176: 4974-4984.
Summer EJ, Berry J, Tran TAT, Niu L, Struck DK, Young R: Rz/Rz1 lysis gene equivalents in phages of Gram-negative hosts. J Mol Biol. 2007, 373: 1098-1112. 10.1016/j.jmb.2007.08.045.
Wang I-N, Smith DL, Young R: Holins: The protein clocks of bacteriophage infections. Annu Rev Microbiol. 2000, 54: 799-825. 10.1146/annurev.micro.54.1.799.
Kutter E, Raya R, Carlson K: Molecular mechanisms of phage infection. Bacteriophages: Biology and Applications. Edited by: Kutter E, Sulakvelidze, A. 2005, Boca Raton: CRC Press, 165-222.
Graschopf A, Bläsi U: Molecular function of the dual-start motif in the λ S holin. Mol Microbiol. 1999, 33: 569-582. 10.1046/j.1365-2958.1999.01501.x.
Sheehan MM, Stanley E, Fitzgerald GF, Van Sinderen D: Identification and characterization of a lysis module present in a large proportion of bacteriophages infecting Streptococcus thermophilus. Appl Environ Microbiol. 1999, 65: 569-577.
Berry J, Summer EJ, Struck DK, Young R: The final step in the phage infection cycle: The Rz and Rz1 lysis proteins link the inner and outer membranes. Mol Microbiol. 2008, 70: 341-351. 10.1111/j.1365-2958.2008.06408.x.
Markov D, Christie GE, Sauer B, Calendar R, Park T, Young R, Severinov K: P2 growth restriction on an rpoC mutant is suppressed by alleles of the Rz1 homolog lysC. J Bacteriol. 2004, 186: 4628-4637. 10.1128/JB.186.14.4628-4637.2004.
Hanych B, Kedzierska S, Walderich B, Uznanski B, Taylor A: Expression of the Rz gene and the overlapping Rz1 reading frame present at the right end of the bacteriophage lambda genome. Gene. 1993, 129: 1-8. 10.1016/0378-1119(93)90689-Z.
Mahillon J, Chandler M: Insertion sequences. Microbiol Mol Biol Rev. 1998, 62: 725-774.
Pilacinski W, Mosharrafa E, Edmundson R: Insertion sequence IS2 associated with int-constitutive mutants of bacteriophage lambda. Gene. 1977, 2: 61-74. 10.1016/0378-1119(77)90073-7.
Chow LT, Broker TR: Adjacent insertion sequences IS2 and IS5 in bacteriophage Mu mutants and an IS5 in a lambda darg bacteriophage. J Bacteriol. 1978, 133: 1427-1436.
Sakaguchi Y, Hayashi T, Kurokawa K, Nakayama K, Oshima K, Fujinaga Y, Ohnishi M, Ohtsubo E, Hattori M, Oguma K: The genome sequence of Clostridium botulinum type C neurotoxin-converting phage and the molecular mechanisms of unstable lysogeny. Proc Natl Acad Sci USA. 2005, 102: 17472-17477. 10.1073/pnas.0505503102.
Ohtsubo Y, Genka H, Komatsu H, Nagata Y, Tsuda M: High-temperature-induced transposition of insertion elements in Burkholderia multivorans ATCC 17616. Appl Environ Microbiol. 2005, 71: 1822-1828. 10.1128/AEM.71.4.1822-1828.2005.
Finnegan DJ: Transposable elements: How non-LTR retrotransposons do it. Curr Biol. 1997, 7: R245-R248. 10.1016/S0960-9822(06)00112-6.
Doulatov S, Hodes A, Dal L, Mandhana N, Liu M, Deora R, Simons RW, Zimmerly S, Miller JF: Tropism switching in Bordetella bacteriophage defines a family of diversity-generating retroelements. Nature. 2004, 431: 476-481. 10.1038/nature02833.
Medhekar B, Miller JF: Diversity-generating retroelements. Curr Opin Microbiol. 2007, 10: 388-395. 10.1016/j.mib.2007.06.004.
Odegrip R, Nilsson AS, Haggård-Ljungquist E: Identification of a gene encoding a functional reverse transcriptase within a highly variable locus in the P2-like coliphages. J Bacteriol. 2006, 188: 1643-1647. 10.1128/JB.188.4.1643-1647.2006.
Liu M, Deora R, Doulatov SR, Gingery M, Eiserling FA, Preston A, Maskell DJ, Simons RW, Cotter PA, Parkhill J, Miller JF: Reverse transcriptase-mediated tropism switching in Bordetella bacteriophage. Science. 2002, 295: 2091-2094. 10.1126/science.1067467.
Fortier L-C, Bouchard JD, Moineau S: Expression and site-directed mutagenesis of the lactococcal abortive phage infection protein AbiK. J Bacteriol. 2005, 187: 3721-3730. 10.1128/JB.187.11.3721-3730.2005.
Snyder L, Champness W: Molecular Genetics of Bacteria. 2003, Washington DC: ASM Press, 2
InterPro: IPR002941 DNA methylase N-4/N-6. [http://www.ebi.ac.uk/interpro/IEntry?ac=IPR002941]
Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Ruger W: Bacteriophage T4 genome. Microbiol Mol Biol Rev. 2003, 67: 86-156. 10.1128/MMBR.67.1.86-156.2003.
Mücke M, Grelle G, Behlke J, Kraft R, Krüger DH, Reuter M: EcoRII: A restriction enzyme evolving recombination functions?. EMBO J. 2002, 21: 5262-5268. 10.1093/emboj/cdf514.
Schroeder SG, Samudzi CT: Structural studies of EcoRII methylase: Exploring similarities among methylases. Protein Eng. 1997, 10: 1385-1393. 10.1093/protein/10.12.1385.
Lieb M: Spontaneous mutation at a 5-methylcytosine hotspot is prevented by very short patch (VSP) mismatch repair. Genetics. 1991, 128: 23-27.
Coulondre C, Miller JH, Farabaugh PJ, Gilbert W: Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978, 274: 775-780. 10.1038/274775a0.
Hennecke F, Kolmar H, Brundl K, Fritz H-J: The vsr gene product of E. coli K-12 is a strand- and sequence-specific DNA mismatch endonuclease. Nature. 1991, 353: 776-778. 10.1038/353776a0.
Coenye T, Vandamme P, LiPuma JJ, Govan JRW, Mahenthiralingam E: Updated version of the Burkholderia cepacia complex experimental strain panel. J Clin Microbiol. 2003, 41: 2797-2798. 10.1128/JCM.41.6.2797-2798.2003.
Sambrook J, Russell DW: Extraction of bacteriophage λ DNA from large-scale cultures using proteinase K and SDS. Molecular Cloning: A Laboratory Manual. Edited by: Sambrook J, Russell DW. 2001, Cold Spring Harbor: Cold Spring Harbor Laboratory Press, 2.56-2.58. 3
Lukashin AV, Borodovsky M: GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 1998, 26: 1107-1115. 10.1093/nar/26.4.1107.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Viklund H, Elofsson A: OCTOPUS: Improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics. 2008, 24: 1662-1668. 10.1093/bioinformatics/btn221.
Zuker M: Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31: 3406-3415. 10.1093/nar/gkg595.
Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A: Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003, 12: 1652-1662. 10.1110/ps.0303703.
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R: REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29: 4633-4642. 10.1093/nar/29.22.4633.
Vincze T, Posfai J, Roberts RJ: NEBcutter: A program to cleave DNA with restriction enzymes. Nucleic Acids Res. 2003, 31: 3688-3691. 10.1093/nar/gkg526.
Zafar N, Mazumder R, Seto D: CoreGenes: A computational tool for identifying and cataloging "core" genes in a set of small genomes. BMC Bioinformatics. 2002, 3: 12-10.1186/1471-2105-3-12.
Delcher AL, Kasif S, Fleischmann RD, Peterson J, White O, Salzberg SL: Alignment of whole genomes. Nucleic Acids Res. 1999, 27: 2369-2376. 10.1093/nar/27.11.2369.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA: Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19: 1639-1645. 10.1101/gr.092759.109.
Acknowledgements
JJD gratefully acknowledges funding from the Canadian Cystic Fibrosis Foundation and the Canadian Institutes of Health Research to the CIHR Team on Aerosol Phage Therapy. KHL is indebted to the Canadian Cystic Fibrosis Foundation, the Alberta Heritage Foundation for Medical Research, the Killam Trusts and the Natural Sciences and Engineering Research Council of Canada for financial support.
The authors thank Randy Mandryk in the University of Alberta Department of Biological Sciences Advanced Microscopy Facility for assistance with electron micrographs, the University of Alberta Department of Biological Sciences Molecular Biology Service Unit for sequence data collection, Miles Peterson for assistance with figure construction and Kimberley Seed for preliminary sequence collection.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
KHL isolated KL3, took electron micrographs, isolated, sequenced, annotated and analyzed the three genomes and drafted the manuscript. PS prepared the PROmer/MUMmer/Circos genome comparison. JJD devised the study, assisted with experimental design and interpretation of data, performed sequence data collection and edited the manuscript. All authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article
Cite this article
Lynch, K.H., Stothard, P. & Dennis, J.J. Genomic analysis and relatedness of P2-like phages of the Burkholderia cepacia complex. BMC Genomics 11, 599 (2010). https://doi.org/10.1186/1471-2164-11-599
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-11-599