
Abstract
Background
Gummosis and root rot caused by Phytophthora are among the most economically important diseases in citrus. Four F1 resistant hybrids (Pool R), and four F1 susceptible hybrids (Pool S) to P. parasitica, were selected from a cross between susceptible Citrus sunki and resistant Poncirus trifoliata cv. Rubidoux. We investigated gene expression in pools of four resistant and four susceptible hybrids in comparison with their parents 48 hours after P. parasitica inoculation. We proposed that genes differentially expressed between resistant and susceptible parents and between their resistant and susceptible hybrids provide promising candidates for identifying transcripts involved in disease resistance. A microarray containing 62,876 UniGene transcripts selected from the CitEST database and prepared by NimbleGen Systems was used for analyzing global gene expression 48 hours after infection with P. parasitica.
Results
Three pairs of data comparisons (P. trifoliata/C. sunki, Pool R/C. sunki and Pool R/Pool S) were performed. With a filter of false-discovery rate less than 0.05 and fold change greater than 3.0, 21 UniGene transcripts common to the three pairwise comparative were found to be up-regulated, and 3 UniGene transcripts were down-regulated. Among them, our results indicated that the selected transcripts were probably involved in the whole process of plant defense responses to pathogen attack, including transcriptional regulation, signaling, activation of defense genes participating in HR, single dominant genes (R gene) such as TIR-NBS-LRR and RPS4 and switch of defense-related metabolism pathway. Differentially expressed genes were validated by RT-qPCR in susceptible and resistant plants and between inoculated and uninoculated control plants
Conclusions
Twenty four UniGene transcripts were identified as candidate genes for Citrus response to P. parasitica. UniGene transcripts were likely to be involved in disease resistance, such as genes potentially involved in secondary metabolite synthesis, intracellular osmotic adjustment, signal transduction pathways of cell death, oxidative burst and defense gene expression. Furthermore, our microarray data suggest another type of resistance in Citrus-Phytophthora interaction conferred by single dominant genes (R gene) since we encountered two previously reported R genes (TIR-NBS-LRR and RPS4) upregulated in the resistant genotypes relative to susceptible. We identified 7 transcripts with homology in other plants but yet unclear functional characterization which are an interesting pool for further analyses and 3 transcripts where no significant similarity was found. This is the first microarray study addressing an evaluation of transcriptional changes in response to P. parasitica in Citrus.
Similar content being viewed by others


Background
Phytophthora nicotianae Breda de Haan (Phytophthora parasitica Dastur) and Phytophthora citrophthora (Smith & Smith) have caused severe damage in Citrus nurseries and orchards worldwide. In Brazil, P. parasitica is the predominant species associated with the disease, found in more than 95% of groves and nurseries [1]. These pathogens infect the main scaffold branches of the tree, inducing the formation of cankers with gum exudation and the expansion of the lesions upwards affects secondary branches, while downward expansion affects the trunk and roots [2]. Infected trees usually lack vigor and may die prematurely [3]. P. parasitica is an oomycete, belonging to the kingdom Stremenopiles, which comprises a diverse group of organisms that has been consolidated as a result of analysis of mitochondrial and ribosomal DNA sequences [4]. These pathogens establish intimate relations with their hosts by forming haustoria during the infection, which are structures used for obtaining nutrients from the plant, redirecting host metabolism and suppressing host defence in biotrophy [5]. P. parasitica is considered a hemibiotroph, and therefore it first establishes itself in host tissues as a biotroph but then switches to a more necrotrophic type of growth, rapidly invading and killing host cells.
Selection and breeding for resistance to Phytophthora in citrus species is considered the most efficient approach to control the disease, since there are varying degrees of resistance within the genera Citrus and its relatives. In this context, Poncirus trifoliata is an important genotype because of its agronomic valuable characteristics including resistance to Phytophthora, Citrus Tristeza Virus (CTV) and citrus nematode (Tylenchulus semipenetrans) [6].
In response to pathogen attack, a series of plant defense responses leading to the hypersensitive reaction, cell wall modifications, production of reactive oxygen species (ROS), accumulation of phytoalexins, and synthesis of pathogenesis related (PR) proteins may be activated [7]. In general, the salicylic acid (SA)-dependent signaling leads to expression of PR proteins, the production of ROS, and localized cell death. This leads to a defense that is effective against biotrophs because it restricts pathogen growth via hypersensitive death of the infected cells. The same reaction allows growth of a necrotrophic pathogen. Against necrotrophic pathogens, jasmonic acid (JA) and ethylene (ET)-dependent defenses are successfully employed in plants activating a different set of PR proteins [8]. But there is evidence for extensive cross-talk between signaling pathways involving antagonistic and synergistic interactions [9]. In these aspects, it is interesting to investigate the plant response mechanisms in the presence of hemibiotrophic pathogens like P. parasitica, since they comprise both lifestyles during their development.
Biotechnology tools associated with conventional citrus breeding programs have facilitated the development of cultivars with desirable characteristics [10]. Microarray technology has been used to identify gene expression changes in several responses to stress, enabling comparison of transcript levels for thousands of genes simultaneously, such as response of sweet orange (Citrus sinensis) to 'Candidatus Liberibacter asiaticus' infection [7] and [11]. In citrus, the first transcript profiling data was reported by Shimada et al., [12] who constructed a cDNA microarray to monitor expression of mRNA during fruit development. Subsequently, several citrus DNA microarray platforms were developed, such as The Spanish Citrus Genomic Consortium. Affymetrix developed and released a citrus GeneChip containing 960,444 total 25- mer oligos in an 11 micron format (http://www.affymetrix.com/analysis/index.affx), based on the NCBI citrus EST collection [6].
A new approach to study hybrid vigor at the molecular scale is to survey gene expression as a phenotype in F1 hybrids and their parental [13]. Genes in hybrids are inherited from the parents; thus, variation in regulation of the genes often leads to variation in the level of gene expression in hybrids, which in turn may alter the phenotype of these hybrids [14]. Due to the sensitivity of microarrays, plant-to-plant variation of gene expression could be reduced by bulk harvesting of resistant and susceptible hybrids. According to Wenger et al., [15] pooling segregants based on their phenotype allows the region of the genome responsible for the phenotype to be detected because DNA polymorphisms in regions unlinked to the responsible locus will segregate randomly and be ''evened'' out, while sequences or polymorphisms either directly responsible for the trait, or very closely linked to it, will be present in all positive segregants and absent in all negative segregants. According to Meng et al., [16] the majority of DNA microarrays in use today are created from single genomes that do not reflect the genetic diversity of a heterogeneous group. One alternative approach is to incorporate genetic information from several species within a single microarray slide. Mixed-DNA microarrays can be used to quickly assess the distribution of genetic diversity across multiple species.
In previous studies, four resistant and four susceptible F1 hybrids were selected from the population derived from a cross between Citrus sunki Hort. ex. Tan. and P. trifoliata (L.) Raf cv. Rubidoux, which are respectively, susceptible and resistant to P. parasitica[17, 18]. We proposed that genes differentially expressed between resistant and susceptible parents and between their resistant and susceptible hybrids provide promising candidates for identifying transcripts involved in disease resistance. In the present study, we investigated gene expression in resistant and susceptible hybrids in comparison with their parents 48 hours after P. parasitica infection using a microarray chip containing 62,876 UniGene transcripts selected from P. trifoliata, C. sinensis and C. reticulata libraries. Five differentially expressed genes were validated by reverse transcription quantitative real-time PCR (RT-qPCR) in susceptible and resistant plants and used to detect differences in expression between inoculated and uninoculated control plants.
Methods
Plant material and inoculation
In a previous study, four resistant and four susceptible five-year-old hybrids (F1 population of Citrus sunki Hort. ex. Tan. × Poncirus trifoliata (L.) Raf cv. Rubidoux) were selected from 314 field-grown plants. The selected hybrids were the most resistant and susceptible to P. parasitica infection according to Boava [17] and Siviero et al. [18]. Three buds from each hybrid and parental line were collected and grafted onto 6-month-old Rangpur lime rootstocks. After six months, plants were inoculated by a mycelia disc; a mycelial block was placed onto the center of a cut made in the stem and covered with Parafilm. After 48 hours, leaves were harvested individually, separately flash-frozen in liquid nitrogen, and then stored at 80°C prior to RNA isolation. The selection of this harvesting time was based in previous studies on the time course of the type local and systemic defense followed by activation of many genes involved in the interaction Citrus-Phytophthora, detailed by Teixeira [19]. All inoculated plants were kept in a greenhouse at an average temperature of 25°C and 90% (± 0.5%) relative humidity until the final evaluation, measurement of the lesion size, at 40 days postinoculation. Experimental design was completely randomized including three biological replicates for each parent and three for each individual hybrid.
In addition, in another experiment with compatible plant-pathogen interactions, we used the P. trifoliata genotypes to detect differences in gene expression level between inoculated and uninoculated control plants. The plants for these experiments were grown and inoculated independently from microarray experiments. In this new experiment, we collected leaves of P. trifoliata for RNA isolations at 48 hours after P. parasitica inoculation and uninoculated control plants Experimental design was completely randomized including three biological replicates.
Isolation of total RNA and sample labeling
Total RNA was isolated with RNeasy Plant Mini Kit (Qiagen) and treated with RNase-free DNase (Qiagen) according to the manufacturer's instructions. The concentration of total RNA was determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies). RNA integrity was verified using a Bioanalyzer 1000 (Agilent). Following RNA isolation, the samples were pooled into resistant (Pool R) and susceptible hybrids (Pool S) to minimize variation between individual RNA samples. Three biological replicates of each parent and each resistant and susceptible pooled hybrids were used for hybridization with a cDNA microarray. RNA samples were sent to Roche NimbleGen Systems, where cDNA synthesis and Cy3 labeling were performed. Equal amounts of total RNA for each sample were converted to double-stranded cDNA using the SuperScript II cDNA Conversion Kit (Invitrogen). Because this method uses an oligo (dT) primer, RNA strands lacking poly(A) tails are likely to be underrepresented. Cy3 labeling, hybridization and data acquisition were performed at the NimbleGen facility following the manufacturer's procedures.
Microarray data analysis
A total of 62,876 UniGene transcripts (31,583 of C. sinensis, 18,712 of C. reticulata and 12,581 of P. trifoliata) selected from the CitEST database, assembled from the ESTs submitted to NCBI (GenBank accession numbers EY649559 to EY842485) were used to construct oligonucleotide microarray chips by Roche NimbleGen Systems using a multi-step approach to select probes with optimal predicted hybridization characteristics. Three probes were selected per UniGene, comprising a probe set, and each probe set is represented on the final array by two replicates. All probes were designed as perfect match oligonucleotides. The experimental design and all microarray data have been deposited in the NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo accession number GSE20412). Arrays were hybridized and processed by Roche NimbleGen Systems as previously described [20]. For each resistant and susceptible hybrid pool and each parent sample, hybridization was performed on independent microarrays. Arrays were scanned by Roche NimbleGen using a GenePix 4000B microarray scanner (Molecular Devices, Sunnyvale, CA) and the data were extracted using NimbleScan software. For each probe set, an expression measure was calculated using the robust multiarray average (RMA) [21], consisting of three preprocessing steps: convolution background correction, quantile normalization [22], and a summarization based on a robust multiarray model fit using the median polish algorithm. Probe set data with these normalized expression values, provided by Roche NimbleGen Systems in RMA calls files, were imported to ArrayStar software 3.0 version (DNASTAR Inc., Madison, WI), where statistical analysis was performed.
Three pairs of data comparisons that might reveal an association with the resistance of citrus to P. parasitica, were performed: (i) P. trifoliata Rubidoux versus C. sunki; (ii) resistant hybrids (Pool R) versus C. sunki; and (iii) resistant hybrids (Pool R) versus susceptible hybrids (Pool S). For each comparison, a moderated t-test value was calculated, and p-values were adjusted for multiple comparisons by the false-discovery rate correction [23]. Afterwards, UniGene transcripts that were consistently differentially expressed (greater than 3.0-fold up or less than 0.33-fold down; p-value ≤ 0.05) between replicate samples were identified as involved in disease resistance. In order to eliminate UniGene transcripts selected as a consequence of genotype differences but not directly related to disease resistance, Venn diagrams were used to identify the intersection among these three sets of informative transcripts. These transcripts were then rechecked by BLASTX searches against the GenBank database, and further classified into categories according to the Munich Information Center for Protein Sequences classification system (MIPS; http://www.helmholtz-muenchen.de/mips).
Real time-qPCR
Reverse transcription quantitative real-time PCR (RT-qPCR) was performed with five selected genes to validate the microarray experiments (Table 1). Our candidate gene analysis focused on upregulated UniGene transcripts in all comparison: P. trifoliata relative to C. sunki, in the resistant pool relative to C. sunki, and in the resistant pool relative to the susceptible pool. These candidate genes were judged to be biologically interesting on the basis of their predicted function retrieved from CitEST. These selected genes were also used to detect differences in expression between inoculated and uninoculated control plants.
In order to find a reference gene to normalize the RT-qPCR results, the stability of five endogenous control genes in Citrus was analyzed to confirm their stability according to geNorm software [24] and to ensure the existence of gene expression variation due to the experimental conditions. Oligonucleotides primers were designed using Primer Express 2.0 software (Applied Biosystems). From the RNA isolated as described above, cDNAs were synthesized from 3.0 μg of total RNA using Superscript III (200 U/μL) (Invitrogen) with an oligo (dT) primer (dT12-18, Invitrogen) according to the manufacturer's instructions. cDNA was treated with RNAse H (1 μl) for 20 min at 37°C to remove any contaminating RNA. RT-qPCR was performed using Power SYBR® Green PCR Master Mix reagent (Applied Biosystems). The reaction consisted of 2.0 μL of cDNA and 120 nM of each gene-specific primer in a final volume of 15 μL. Amplification was carried out for three technical replicates for each sample, including negative controls. An ABI PRISM 7500 SDS (Applied Biosystems) was used for the following thermal cycles: 50°C for 2 min, 95°C for 10 min; 40 cycles of 95°C for 15 s, and 60°C for 1 min. Expression levels were assessed based on the number of amplification cycles needed to reach a common fixed threshold (cycle threshold - Ct) in the exponential phase of PCR. Ct data were analyzed using the GenEx version 4.3.6 software (http://www.multid.se/). For relative quantification, the 2-ΔΔCTmethod between conditions in RT-qPCR was applied [25].
Results and Discussion
Response to infection by P. parasitica
Resistant and susceptible F1 hybrids of a population consisting of recombinants derived from a cross between C. sunki and P. trifoliata Rubidoux, respectively susceptible and resistant to P. parasitica, were evaluated in response to infection by P. parasitica. The F1 population was initially developed for mapping genetic loci for resistance against the citrus tristeza virus [26] and against Phytophthora gummosis [18]. This population was a good choice for mapping resistance to Phytophthora because the Poncirus genera possesses genes conferring many agriculturally important traits not found in Citrus, including genes responsible for resistance to Phytophthora[27]. In the present study, four selected resistant hybrids (H70, H73, H142, H150) and four selected susceptible hybrids (H19, H47, H105, H148) and their parents lines were inoculated by the disc method. The parents represented the extremes in lesion lengths, 13.5 mm for P. trifoliata and 33.75 mm for C. sunki, 40 days after P. parasitica inoculation. The F1 hybrids showed substantial differences in lesion lengths between resistant hybrids and susceptible hybrids (Figure 1). The four resistant hybrids and four susceptible ones showed respectively means of lesion size of 15.18 and 33.06 mm. These results were in accordance with previous studies carried out by [17, 18]

Mean longitudinal length of the lesion (in mm) caused by P. parasitica , in resistant (H70, H73, H142, H150) and susceptible (H19, H47, H105, H148) F 1 hybrids, and in their parents Citrus sunki and Poncirus trifoliata Rubidoux 48 hours after inoculation. Vertical bars represent standard deviations of the means of three replicates.
Microarray analysis
We proposed that genes differentially expressed between resistant and susceptible parents and between their resistant and susceptible hybrids provide promising candidates for identifying transcripts involved in citrus resistance against Phytophthora. Oligonucleotide microarray chips prepared by Roche NimbleGen Systems were used to profile the expression patterns. Expression was detected in tissues for many of the UniGene transcripts (a total of 62,876) present on the array selected from the CitEST database. The data were analyzed based on the RMA-processed expression values. Three possible comparisons (P trifoliata/C. sunki, Pool R/C. sunki and Pool R/Pool S) were analyzed individually using the log (base 2)-transformed normalized expression values as input data. Based on the criterion for differentially expressed, the transcripts were considered up- or down-regulated if the log2 ratio of resistant genotypes inoculated to susceptible genotypes results were greater (positive) than or less than (negative) 3.0-fold, respectively and p-value less than or equal to the level of significance α = 0.05. Thus, 6,735 UniGene transcripts (10.71%) were selected as differentially expressed in P. trifoliata relative to C. sunki (Figure 2A; Table 2), while 1,296 (2.06%) UniGene transcripts were selected as differentially expressed in resistant pool relative to C. sunki (Figure 2B; Table 2). When comparing the resistant with the susceptible hybrids pools, 564 (0,90%) transcripts were selected as differentially expressed (Figure 2C; Table 2).

Analysis of differential gene expression in P. trifoliata 'Rubidoux', C. sunki , resistant (Pool R) and susceptible (Pool S) hybrids. Signal correlation plots were used to examine comparisons between: (A) The average signal derived from the three biological replicates of resistant parent (Rubidoux) (Y-axis) and susceptible parent (Sunki) (X-axis) graphed on a logarithmic (base 2) scale. Note the prevalence of genes showing distinct expression patterns in the two genotypes; (B) A similar graph was made to compare expression in the resistant hybrids pool (Pool R) (Y-axis) and the susceptible parent (Sunki) (X-axis); (C) A similar graph of expression data of the resistant hybrids pool (Y-axis) plotted against the expression data of the susceptible hybrids pool (X-axis).
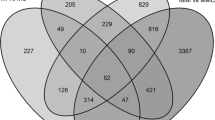
In addition, we observed differences in the expression levels in the resistant and susceptible parental genotypes and associated those with the differences in the expression levels in the resistant (pool R) and susceptible (pool S) hybrids. This association was accomplished by restricting the sets of differentially expressed UniGene transcripts to those identified as up- and downregulated in all three pairwise comparisons, Venn diagrams were used to identify the intersection among these three sets of informative transcripts, which resulted in the selection of 24 UniGene transcripts common to the three pairwise comparative analyses (Figure 3A, Table 3) of which 21 were up-regulated (Figure 3B, Table 3) and 3 were down-regulated (Figure 3C, Table 3) during P. parasitica infection, which were further classified into 7 categories according to the MIPS classification scheme (Table 3). The experimental design was in fact enriched with transcripts involved in pathogen response belonging to the categories disease/defense. We identified 11 UniGene transcripts that were likely to be involved in disease resistance; 7 transcripts with homology in other plants but yet unclear functional characterization which are an interesting pool for further analyses and 3 transcripts where no significant similarity was found.

Venn diagrams showing the differentially expressed UniGene transcripts in three different comparisons between resistant (Pool R) and susceptible (Pool S) F 1 hybrids and their parents, Citrus sunki (Sunki) and Poncirus trifoliata Rubidoux (Rub), respectively susceptible and resistant to P. parasitica , 48 hours after inoculation. Transcripts profile greater (positive) than or less than (negative) 3.0-fold, respectively and p-value less than or equal to the level of significance α = 0.05 were considered differentially expressed. A) differentially expressed UniGene; B) upregulated; C) downregulated
The 24 UniGene transcripts common to the three pairwise comparative analyses were distributed among the 3 different libraries used for built of the our microarray chip from the CitEST database, 12 UniGene transcripts were identified in the UniGene set of C. sinensis, 10 in P. trifoliata, and 2 in C. reticulate. According to Wan et al.[28], the use of mixed-DNA microarrays is advantageous because much more information is incorporated into the analysis. In the present work, there were 18,712 UniGene transcripts from P. trifoliata libraries of the CitEST database and we decided to incorporate UniGene sequences from C. sinensis and C. reticulata to be represented in the array and consequently enrich the analyses. The Spanish Citrus Genomic Consortium also developed a mixed-DNA microarrays composed of 24,000- element cDNA array containing 20,000 unigenes, based on nearly 90,000 high-quality sequences generated from 52 different cDNA libraries [6].
Real time-qPCR validation
RT-qPCR is currently the most sensitive method to compare gene expression at both low and high levels. To avoid distortions, RT-qPCR requires a reference gene (or a few genes) with the most stable expression over the experiment, i.e., a gene presenting expression levels that is little influenced by the experimental or environmental conditions, to correct for sample-to-sample variation in RT-qPCR efficiency and errors in sample quantification. Several reference genes tested presented varied expression due to the experimental conditions in different genotypes exposed to biotic and abiotic stimuli [29]. To support the choice of the best reference genes, i.e., the most stable genes following pathogen inoculation in the different genotypes, the GeNorm algorithm [24], implemented in GenEx version 4.3.6. software (http://www.multid.se/) was used. In the present work, among the five reference genes tested (Table 1), ubiquitin (UBQ) and cyclophilin (CYP) had the lowest expression stability mean values (M-value = 0.0257), i.e., these genes had more stable expression than other evaluated genes (Figure 4), and the data were normalized by the normalization factor calculated using these most stable reference genes.

Expression stability mean values (M-values) of 5 endogenous control genes, in tissue samples from citrus genotypes 48 hours after P. parasitica inoculation after analysis using geNorm software.
Five UniGene transcripts that were differentially expressed in the microarray analysis were selected for RT-qPCR. Our candidate gene analysis focused on upregulated genes in the resistant genotypes that were judged to be biologically interesting on the basis of their predicted function retrieved from CitEST. We selected genes that are likely involved in disease resistance, such as one gene potentially involved in secondary metabolite synthesis and protein activity regulation (miraculina) and genes potentially involved in cell rescue, defense and virulence (TIR-NBS-LRR and RPS4) and one gene homolog in other plant but yet unclear functional characterization which are an interesting for further analyses. Comparison of the results from RT-qPCR with those from microarray analyses revealed roughly similar patterns or tendencies of expression in the three comparative pairwise analyses (Figure 5). Notably, using high stringency in the analysis of the microarray, data showed only twenty one genes were upregulated in common among the three comparative pairwise analyses. The validation with RT-qPCR showed that all the selected genes were in fact upregulated to similar levels in all comparison (≥ 3.0-fold, p-value ≤ 0.05). All five transcripts were upregulated in P. trifoliata relative to C. sunki, in the resistant pool relative to C. sunki, and in the resistant pool relative to the susceptible pool. These results revealed similar patterns of expression with those from microarray analyses.

Validation of microarray data by quantitative real-time. RT-qPCR fold-changes are shown for five genes upregulated in all microarray pairwise comparisons (Rub/Sun, Pool R/Sun, and Pool R/Pool S) 48 hours after P. parasitica inoculation, and compared with fold-changes obtained by microarray analysis. RT-qPCR data were normalized to the two most stable endogenous control genes (UBQ and CYP).
In addition, besides of estimating and confirm the fold change at each comparative pairwise analyses on microarray, we include in our RT-qPCR analysis, five selected genes in inoculated and uninoculated plants of P. trifoliata. (Figure 6). Thus, this confirmation offers an opportunity to determine transcription patterns of the systemic response of Citrus to infection with the pathogen P. parasitica providing information on different defense and metabolic pathways. With this analysis the differences in gene expression probably are involved in the response to infection to P. parasitica and maybe some of them can be related to the resistance mechanism and the changes found are not only due to inherent constitutive gene expression differences among the genotypes"

Validation of microarray data by RT-qPCR using P. trifoliata parents uninoculated control and inoculated with P. parasitica after 48 hours. RT-qPCR data were normalized to the two most stable candidate endogenous control genes (UBQ and CYP).
Functional analyses of differentially-expressed genes
Upon recognition of P. parasitica by its host Citrus, a series of signaling pathways are switched on, which lead to the metabolic reprogramming of the host plant. Phytophthora species are usually biotrophic or hemibiotrophic pathogens that keep host cells alive (at least initially) to enable nutrient uptake from the plant cells [30]. A more extensive cell death response could cause opposite effects on the outcome of biotrophic versus necrotrophic plant-pathogen interactions. Hemibiotrophic pathogens adopt a two-step infection style. According to Kanneganti et al.[31], during the phase of infection that follows penetration of host tissue, they require living cells, much like biotrophic pathogens. In contrast, in a later phase of the disease, they cause extensive necrosis of host tissue, resulting in profuse colonization and sporulation. This infection cycle suggests that host cell death may impact the disease differently depending on its timing.
The time-point of infection was selected for the isolation of defense-related transcripts because, in compatible interactions, transcripts involved in pathogen response were observed at 48 hours (e.g., [19]) and this is reflected by the low number of transcripts of secondary metabolism isolated in this study. According to Grenville-Briggs and van West [32]Phytophthora species are hemibiotrophs being biotrophic for the initial stage of up to 36 h after inoculation. Molecular studies carried out with hemibiotrophic pathogens infecting plants helped the identification of several putative genes that are expressed at the stage biotrophy and are involved in membrane or cell wall biosynthesis, amino acid metabolism, osmoregulation, phosphorylation, protein secretion and energy consumption [33].
In our study, genes potentially involved in secondary metabolite synthesis were identified, including leucoanthocyanidin dioxygenase, which is the key enzyme leading to the synthesis of anthocyanins. Anthocyanins are known to be potent agents acting against oxidative stress [34]. The exact role of anthocyanins in defense is not clear but it could be that these compounds neutralize damaging reactive oxygen species (ROS) [35]. A late embryogenesis-abundant LEA5 protein was also found upregulated in the resistant genotypes. LEA proteins have been considered to play roles in maintaining membrane structures, binding of water, and acting as molecular chaperones [36]. According to Galau [37], these genes are believed to be induced in response to the expansion and maintenance of giant cells, and may play a role in intracellular osmotic adjustment. These results demonstrate that a transcriptional reprogramming has occurred within the first 48 hours after inoculation, which might correspond to the P. parasitica biotrophic phase.
Cell death triggered by ROS at the interface of necrotrophic fungus-plant interactions has been shown to be required for disease susceptibility [38]. Therefore, ROS in conjunction with P. parasitica-induced ET, may play a role in development of visible necrotrophic symptoms, observed 40 days after inoculation. Several results suggest that P. parasitica may produce a toxin-like virulence factor similar to necrosis and ET-inducing peptides characterized in other fungal pathogens such as Phytophthora spp. [39], Botrytis spp. [40] and Pythium aphanidermatum, during compatible-necrotrophic interactions. According to Attard et al. [41] all Phytophthora species abundantly secrete 10 kDa proteins, which form a superfamily called elicitins. The EST sequencing project for P. parasitica led to the identification of 10 different elicitin classes [42, 43]. Elicitins have been found to be up regulated lately when the pathogen entered the necrotrophic growth stage [44].
Elicitins can induce a hypersensitive reaction (HR) and further defense mechanisms in the host [45]. In our study, we found two Unigenes transcripts encoding serine-threonine protein kinase upregulated in the resistant genotypes. According to Sasabe et al. [46] protein phosphorylation is an indispensable process for signal transduction pathways of cell death, oxidative burst and defense gene expression. One or more proteins whose activity is controlled by Ser/Thr protein kinases and phosphatases might be activated by elicitin-treatment, or kinase proteins which are sensitive to phosphatases might play an important role in cell death, oxidative burst and defense gene expression at the upstream of common signal transduction pathway and/or by independent pathways.
Sasabe et al. [46] report that Phytophthora citricola elicitin molecule induces apoptotic cell death in tobacco and they isolated several EST involved in plant oxidative or respiratory burst like superoxide dismutases and NADPH oxidase. Here, we identified one superoxide dismutase downregulated in the resistant genotypes, which were consequently upregulated in susceptible plants relative to resistant. Several enzymes, such as superoxide dismutase are responsible for the removal of production of signaling compounds such as reactive oxygen intermediates (ROIs) [47]. According to Mitler et al., [48] cell death of plant tissues resulted from attack by pathogens does not only occur in HR, resistant responses, but also in susceptible reactions such as necrotic symptom, for example which is caused by toxin. The late induction of elicitins observed in hemibiotrophic pathogen interaction [44] is therefore probably a component of Phytophthora's pathogenic strategy under positive selection. This explains the late but extended formation of necrosis observed in this study in susceptible genotypes.
Our microarray data, which identified different defense and metabolic pathways, is congruent with the results from previous studies by Siviero et al [18]. These authors, studying of the mode of inheritance associated to the resistance against Phytophthora gummosis in Poncirus trifoliata, identified three quantitative trait loci associated to the resistance against Phytophthora gummosis in citrus, indicating the quantitative pattern of the disease. Because this resistance is controlled by multiple genes, the pathogen has to undergo multiple mutations to adapt to partial resistance which is more difficult than a single locus mutation. In this case, plants may exhibit resistance to most isolates to a certain degree by reducing pathogen reproduction, infection efficiency and colonization. This resistance is more durable than that mediated by resistance genes (R gene), but is difficult to move into cultivated varieties by crossing and phenotypic selection.
However, our microarray data suggest another type of resistance in Citrus-Phytophthora interaction, which resistance may be inherited qualitatively conferred by single dominant genes (R gene). We encountered two previously reported R genes (TIR-NBS-LRR and RPS4) upregulated in the resistant genotypes relative to susceptible. The largest class of R genes is characterized by the presence of nucleotide-binding site (NBS) sequence followed by leucine-rich repeats (LRRs) [49]. NBS-LRR class R genes, such as RPS4 in Arabidopsis can also be characterized by the presence of a leading sequence homologous to the TIR (Toll-Interleukin-1 receptor) that is responsible for cytoplasmic signaling in animals [49]. Previous data suggest that the TIR domain of RPS4 is important for induction of cell death when RPS4 is transiently expressed in tobacco leaves [50]. NBS-LRR-mediated resistance has been identified against numerous types of biotrophic or hemibiotrophic pathogens, including fungi, oomycetes, viruses and bacteria, and these types of resistance genes have been identified across a wide range of plants [51].
Qualitative resistance is mediated by R genes that lead to a race-specific hypersensitive response. These R genes only provide short-lived resistance in the field as new virulent races of the pathogen rapidly overcome the resistance encoded by single race-specific resistance genes [52]. In contrast, quantitative resistance is controlled by many interacting genes that do not prevent infection, but slow down the development of the pathogen at individual infection sites on the plant, and hence, lasts longer [53].
Although the presence of R gene does not contradict the statement that the Poncirus genera used in our experiments possess quantitative pattern of resistance to P. parasitica , these genes showed strong induction with an alteration of more than 4-fold in resistant genotypes relative to susceptible (Table 3). This finding further support the previous point of view that the HR was one of the main defense responses. R genes are also thought to encode specific receptors that recognize elicitors and initiate signal transduction cascades resulting in the HR [54]. Although a major feature of the HR is a rapid and local cell death, many defense-related genes that are not involved in cell death are also activated and may play a more important role in resistance to pathogen. Therefore, it is further inferred that the HR in resistant genotypes may result from a broad-spectrum recognition of the pathogen by the product of unknown R gene(s) or R gene analogues, or that a same or similar defense system, especially the downstream signaling components such as kinases and other defense genes, may exist specific resistances.
Conclusion
This is the first global gene expression study in Citrus × Phytophthora gummosis interaction. We identified 24 Phytophthora gummosis responsive transcripts. The functional classification of these genes and their expression profile at 48 h after inoculation provide useful information on resistance of citrus to Phytophthora. The selected transcripts were validated by RT-qPCR in susceptible and resistant plants and between inoculated and uninoculated control plants. Our results indicated that the selected transcripts were really upregulated in the resistant genotypes and probably involved in the whole process of plant defense responses to pathogen attack, including transcriptional regulation, signaling, activation of defense genes participating in HR, such as TIR-NBS-LRR and RPS4, switch of defense-related metabolism pathway.
As we have a linkage map for P. trifoliata and three QTL for Phytophthora gummosis resistance localized on this map, the integration of genomics and genetic mapping using genetic genomics approaches will provide new insights into resistance to this disease and help with the development of improved disease management strategies. The genes that we have identified as upregulated across the resistant genotypes will be valuable for ongoing work in eQTL mapping.
References
Medina Filho HP, Bordignon R, Siqueira W, Feichtenberger E, Carvalho MRT: Tolerância de híbridos e de clones de porta-enxertos de citros à infecção de raízes por Phytophthora nicotianae. Fitopatologia Brasileira. 2004, 29: 169-178. 10.1590/S0100-41582004000200008.
Alvarez LA, Gramaje D, Abad-Campos P, García JJ: Seasonal susceptibility of citrus scions to Phytophthora citrophthora and P. nicotianae and the influence of environmental and host-linked factors on infection development. Eur J Plant Pathol. 2009, 124: 621-635. 10.1007/s10658-009-9449-8.
Queiroz BPV, Melo IS: Antagonism of Serratia marcescens towards Phytophthora parasitica and its effects in promoting the growth of citrus. Braz J Microbiol. 2006, 37: 448-450. 10.1590/S1517-83822006000400008.
Alexopoulos CJ, Mims CW, Blackwell M: Phylum Oomycota. Introductory Mycology. Edited by: Alexopoulos CJ, Mims CW, Blackwell M. 1996, John Wiley & Sons Press, 683-737.
O'Connell RJ, Panstruga R: Tete a tete inside a plant cell. Establishing compatibility between plants and biotrophic fungi and oomycetes. New Phytologist. 2006, 171: 699-718.
Talon M, Gmitter FG: Citrus genomics. Int J Pl Genomics. 2008, Article ID 528361, 17 pages
Albrecht U, Bowman KD: Gene expression in Citrus sinensis (L.) Osbeck following infection with the bacterial pathogen Candidatus Liberibacter asiaticus causing Huanglongbing in Florida. Plant Sci. 2008, 175: 291-306. 10.1016/j.plantsci.2008.05.001.
Cordelier S, de Ruffray P, Fritig B, Kauffmann S: Biological and molecular comparison between localized and systemic acquired resistance induced in tobacco by a Phytophthora megasperma glycoprotein elicitin. Plant Mol Biol. 2003, 51: 109-118. 10.1023/A:1020722102871.
De Vos M, Van Oosten VR, Van Poecke RM, Van Pelt JA, Pozo MJ, Mueller MJ, Buchala AJ, Me'traux JP, Van Loon LC, Dicke M, Pieterse CM: Signal signature and transcriptome.
Mourão Filho FAA, Pio R, Mendes BMJ, Azevedo FA, Schinor EH, Entelmann FA, Alves ASR, Cantuarias-Avilés TE: Evaluation of citrus somatic hybrids for tolerance to Phytophthora nicotianae and citrus tristeza virus. Sci Hortic. 2008, 115: 301-308.
Kim JS, Sagaram US, Burns JK, Li JL, Wang N: Response of sweet orange (Citrus sinensis) to 'Candidatus Liberibacter asiaticus' infection: microscopy and microarray analyses. Phytopathology. 2009, 99: 50-57. 10.1094/PHYTO-99-1-0050.
Shimada T, Fujii H, Endo T, Yazaki J, Kishimoto N, Shimbo K, Kikuchi S, Omura M: Toward comprehensive expression profiling by microarray analysis in Citrus: monitoring the expression profiles of 2213 genes during fruit development. Plant Sci. 2005, 168: 1383-1385. 10.1016/j.plantsci.2005.01.013.
Holland JB: Genetic architecture of complex traits in plants. Curr Opin Plant Biol. 2007, 10: 156-161. 10.1016/j.pbi.2007.01.003.
Doebley J, Lukens L: Transcriptional regulators and the evolution of plant form. Plant Cell. 1998, 10: 1075-1082. 10.1105/tpc.10.7.1075.
Wenger JW, Schwartz K, Sherlock G: Bulk Segregant Analysis by High-Through. put Sequencing Reveals a Novel Xylose Utilization Gene from Saccharomyces cerevisiae. PLoS Genet. 2010, 6 (5): e1000942-
Meng D, Broschat SL: Call DR: A Java-based tool for the design of classification microarra ys. BMC Bioinformatics. 2008, 9: 328-10.1186/1471-2105-9-328.
Boava LP: Estabilidade de QTLs ligados à resistência dos citros a gomose, causada por Phytophthora parasitica. PhD thesis. 2004, Universidade Estadual Paulista
Siviero A, Cristofani M, Furtado EL, Garcia AAF, Coelho ASG, Machado MA: Identification of QTLs associated with citrus resistance to Phytophthora gummosis. J Appl Genet. 2006, 47: 23-28. 10.1007/BF03194595.
Teixeira JEC: Genes de defesa de Citrus sunki e Poncirus trifoliata: expressão constitutiva e induzida por Phytophthora parasítica. PhD thesis. 2005, Universidade Federal de Lavras
Ulijasz AT, Andes DR, Glasner JD, Weisblum B: Regulation of iron transport in Streptococcus pneumoniae by RitR, an orphan response regulator. J Bacteriol. 2004, 186: 8123-8136. 10.1128/JB.186.23.8123-8136.2004.
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP: Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003, 4: 249-264. 10.1093/biostatistics/4.2.249.
Bolstad BM, Irizarry RA, Astrand M, Speed TP: A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003, 19: 185-193. 10.1093/bioinformatics/19.2.185.
Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995, 57: 289-300.
Vandesompele J, De Preter K, Pattyn F: Accurate normalisation of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3: 0034.1-0034. 10.1186/gb-2002-3-7-research0034.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔC Tmethod. Methods. 2001, 25: 402-408. 10.1006/meth.2001.1262.
Cristofani M, Machado MA, Grattapaglia D: Genetic linkage maps of Citrus sunki Hort. ex.Tan. and Poncirus trifoliata (L.) Raf. and mapping of citrus tristeza virus resistance gene. Euphytica. 1999, 109: 25-32. 10.1023/A:1003637116745.
Chen C, Bowman KD, Choi Ya, Dang PM, Rao MN, Huang S, Soneji JR, McCollum TG, Gmitter FG: EST-SSR genetic maps for Citrus sinensis and Poncirus trifoliate. Tree Genet Genomes. 2008, 4: 1-10. 10.1007/s11295-007-0083-3.
Wan Y, Broschat SL, Call DR: Validation of mixed-genome microarrays as a method for genetic discrimination. Appl Environ Microbiol. 2007, 73: 1425-1432. 10.1128/AEM.01647-06.
Boava LP, Laia ML, Jacob TR, Dabbas KM, Gonçalves JF, Ferro JA, Ferro MI, Furtado EL: Selection of endogenous genes for gene expression studies in Eucalyptus under biotic (Puccinia psidii) and abiotic (acibenzolar-S-methyl) stresses using RT-qPCR. BMC Res Notes. 2010, 24: 3:43-
Takemoto D, Hardham AR, Jones DA: Differences in cell death induction by Phytophthora elicitins are determined by signal components downstream of MAP Kinase kinase in different species of Nicotiana and cultivars of Brassica rapa and Raphanus sativus. Plant Physiol. 2005, 138: 1491-1504. 10.1104/pp.104.058388.
Kanneganti TD, Huitema E, Cakir C, Kamoun S: Synergistic interaction of the plant cell death pathway induced by Phytophthora infestans Nep1-like protein piNPP1.1 and INF1 elicitin. Mol Plant Microbe Interact. 2006, 19: 854-863. 10.1094/MPMI-19-0854.
Grenville-Briggs LJ, van West P: The biotrophic stages of oomycete-plant interactions. Advances in Applied. Microbiology. 2005, 57: 217-243.
Tör M: Tapping into molecular conversation between oomycete plant pathogens and their hosts. Eur J Plant Pathol. 2008, 122: 57-69.
Stintzing FC, Stintzing AS, Carle R, Frei B, Wrolstad RE: Color and antioxidant properties of cyanidin-based anthocyanin pigments. J Agric Food Chem. 2002, 50: 6172-6181. 10.1021/jf0204811.
Calla B, Vuong T, Radwan O, Hartman GL, Clough SJ: Gene Expression Profiling Soybean Stem Tissue Early Response to Sclerotinia sclerotiorum and in Silico Mapping in Relation to Resistance Markers. The Plant Genome. 2009, 2: 149-166. 10.3835/plantgenome2008.02.0008.
Wise M, Tunnaciliffe A: POPP the question: What do LEA proteins do?. Trends Plant Sci. 2004, 9: 13-17. 10.1016/j.tplants.2003.10.012.
Galau GA, Wang HYC, Hughes DW: Cotton Lea 5 and Lea 14 encode atypical late embryogenesis-abundant proteins. Plant Physiol. 1993, 101: 695-696. 10.1104/pp.101.2.695.
Okubara PA, Paulitz TC: Root defenses to fungal pathogens: A molecular prospective. Plant Soil. 2005, 274: 215-226. 10.1007/s11104-004-7328-9.
Qutob D, Kemmerling B, Brunner F, Küfner I, Engelhardt S, Gust AA, Luberacki B, Seitz HU, Stahl D, Rauhut T, Glawischnig E, Schween G, Lacombe B, Watanabe N, Lam E, Schlichting R, Scheel D, Nau K, Dodt G, Hubert D, Gijzen M, Nürnberger T: Phytotoxicity and innate immune responses induced by Nep1-like proteins. Plant Cell. 2006, 18: 3721-3744. 10.1105/tpc.106.044180.
Staats M, van Baarlen P, Schouten A, van Kan JA, Bakker FT: Positive selection in phytotoxic protein-encoding genes of Botrytis species. Fungal Genet Bio. 2007, 44: 52-63. 10.1016/j.fgb.2006.07.003.
Attard A, Gourgues M, Galiana E, Panabières F, Ponchet M, et al: Strategies of attack and defense in plant-oomycete interactions, accentuated for Phytophthora parasitica Dastur (syn. P. Nicotianae Breda de Haan). J Plant Physiol. 2008, 165: 83-94. 10.1016/j.jplph.2007.06.011.
Panabieres F, Amselem J, Galiana E, Le Berre JY: Gene identification in the oomycete pathogen Phytophthora parasitica during in vitro vegetative growth through expressed sequence tags. Fungal Genet Biol. 2005, 42: 611-23. 10.1016/j.fgb.2005.03.002.
Le Berre JY, Engler G, Panabieres F: Exploration of the late stages of the tomato-Phytophthora parasitica interactions through histological analysis and generation of expressed sequence tags. New Phytol. 2008, 177 (2): 480-492.
Moy P, Qutob D, Chapman BP, Atkinson I, Gijzen M: Patterns of gene expression upon infection of soybean plants by Phytophthora sojae. Mol Plant Microbe Interact. 2004, 17: 1051-1062. 10.1094/MPMI.2004.17.10.1051.
Kamoun S: A catalogue of the effector secretome of plant pathogenic oomycetes. Annu Rev Phytopathol. 2006, 44: 41-60. 10.1146/annurev.phyto.44.070505.143436.
Sasabe M, Takeuchi K, Kamoun S, Ichinose Y, Govers F, Toyoda K, Shiraishi T, Yamada T: Independent pathways leading to apoptotic cell death, oxidative burst and defense gene expression in response to elicitin in tobacco cell suspension culture. Eur J Biochem. 2000, 267: 5005-5013. 10.1046/j.1432-1327.2000.01553.x.
Möller IM: Plant mitochondria and oxidative stress: electron transport, NADPH turnover, and metabolism of reactive oxygen species. Annu Rev Plant Physiol Plant Mol Biol. 2001, 52: 561-59.
Mittler R, Shulaev V, Seskar M, Lam E: Inhibition of programmed cell death in tobacco plants during a pathogen-induced hypersensitive response at low oxygen pressure. Plant Cell. 1996, 8: 1991-2001. 10.1105/tpc.8.11.1991.
Hammond-Kosack KE, Jones JDG: Plant disease resistance genes. Annu Rev Plant Physiol Plant Mol Biol. 1997, 48: 575-607. 10.1146/annurev.arplant.48.1.575.
Zhang Y, Dorey S, Swiderski M, Jones JDG: Expression of RPS4 in tobacco induces an AvrRps4-independent HR that requires EDS1, SGT1 and HSP90. Plant J. 2004, 40: 213-224. 10.1111/j.1365-313X.2004.02201.x.
Jones JDG, Dangl JL: The plant immune system. Nature. 2006, 444: 323-329. 10.1038/nature05286.
Colon LT, Turkensteen LJ, Prummel W, Budding DJ, Hoogendoorn J: Durable resistance to late blight (Phytophthora infestans) in old potato cultivars. Eur J Plant Pathol. 1995, 101: 387-397. 10.1007/BF01874852.
Bormann CA, Rickert AM, Ruiz RAC, Paal J, Strahwald J, Buhr K, Gebhardt C: Tagging quantitative trait loci formaturity-corrected late blight resistance in tetraploid potato with PCR-based candidate gene markers. Mol Plant Microbe Interact. 2004, 17: 1126-1138. 10.1094/MPMI.2004.17.10.1126.
Baker B, Zambryski P, Staskawicz B, Dineshkumar SP: Signaling in plant microbe interactions. Science. 1997, 276: 726-733. 10.1126/science.276.5313.726.
Acknowledgements
The present study was conducted with the financial support of Fundação de Amparo a Pesquisa do estado de São Paulo (FAPESP) (Process n. 2007/08435-5) and Instituto Nacional de Ciência e Tecnologia (INCT) de Genômica para Melhoramento de Citros (Process n. 573848/2008-4) which provided a scholarship to LPB (process n. 150721/2009-9) from the Conselho Nacional de Pesquisa de Desenvolvimento (CNPq). The authors thank Marta Tanrikulu of http://ScienceDocs.com for editing of an earlier version of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
MCY and MAM planned and supervised the study. LPB, MCY, contributed to the design and execution of the experiments detailed. LPB and MCY carried out plant growth and inoculation. LPB, VSM and KK participated in extraction of plant RNA. LPB and MRA performed microarray analysis. LPB, VSM and KK contributed to the primer design and RT-qPCR validation. KTL performed annotations of the UniGene transcripts. LPB and MCY drafted the manuscript. MCY, MAT and MAM provided intellectual input. MAM, MRA, VSM, KK and MAT revised the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Boava, L.P., Cristofani-Yaly, M., Mafra, V.S. et al. Global gene expression of Poncirus trifoliata, Citrus sunki and their hybrids under infection of Phytophthora parasitica. BMC Genomics 12, 39 (2011). https://doi.org/10.1186/1471-2164-12-39
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-12-39