
Abstract
Background
Infectious laryngotracheitis virus (ILTV) is an alphaherpesvirus that causes acute respiratory disease in chickens worldwide. To date, only one complete genomic sequence of ILTV has been reported. This sequence was generated by concatenating partial sequences from six different ILTV strains. Thus, the full genomic sequence of a single (individual) strain of ILTV has not been determined previously. This study aimed to use high throughput sequencing technology to determine the complete genomic sequence of a live attenuated vaccine strain of ILTV.
Results
The complete genomic sequence of the Serva vaccine strain of ILTV was determined, annotated and compared to the concatenated ILTV reference sequence. The genome size of the Serva strain was 152,628 bp, with a G + C content of 48%. A total of 80 predicted open reading frames were identified. The Serva strain had 96.5% DNA sequence identity with the concatenated ILTV sequence. Notably, the concatenated ILTV sequence was found to lack four large regions of sequence, including 528 bp and 594 bp of sequence in the UL29 and UL36 genes, respectively, and two copies of a 1,563 bp sequence in the repeat regions. Considerable differences in the size of the predicted translation products of 4 other genes (UL54, UL30, UL37 and UL38) were also identified. More than 530 single-nucleotide polymorphisms (SNPs) were identified. Most SNPs were located within three genomic regions, corresponding to sequence from the SA-2 ILTV vaccine strain in the concatenated ILTV sequence.
Conclusions
This is the first complete genomic sequence of an individual ILTV strain. This sequence will facilitate future comparative genomic studies of ILTV by providing an appropriate reference sequence for the sequence analysis of other ILTV strains.
Similar content being viewed by others

Background
Infectious laryngotracheitis virus (ILTV) is an alphaherpesvirus that causes acute respiratory disease in chickens. This disease causes economic loss in poultry industries worldwide and is a significant concern for animal health and welfare [1]. The virus contains a linear, double-stranded DNA genome in a herpesvirus type D arrangement. This genome arrangement consists of a unique long region and a unique short region flanked by identical internal and terminal repeat sequences [2, 3], with the short region able to invert with respect to the long region. In previous studies, several regions of the ILTV genome of different strains have been sequenced and annotated [3–12]. Recently, a full genomic sequence of ILTV was assembled by concatenating partial sequences of six different ILTV strains [13]. However, the whole genomic sequence of a single strain of ILTV has not been reported. In this study, the whole genome sequence of a commercial live attenuated vaccine strain of ILTV was examined using high-throughput sequencing technology.
Results and Discussion
Sequencing and coverage
Genomic sequencing using the SOLiD™ system generated 526.69 Mb of sequence and 12,046,726 reads. A total of 230 contigs were mapped to the virus genome after de novo assembly. A consensus sequence of 137,693 bp (without the terminal repeat region) was generated after assembly of contigs using the concatenated reference sequence. The depth of coverage against the concatenated ILTV sequence was greater than 150-fold. This depth of coverage exceeds the level required for precision whole-genome sequencing and demonstrates the suitability of the massively parallel, ligation-mediated sequencing method for herpesvirus genome sequencing [14]. Although the depth of coverage was sufficient to generate long contigs able to cover the whole genome, a total of 185 gaps or regions of ambiguous sequence were detected after de novo assembly. Most gaps were less than 100 bp in size. While sonication theoretically produces random fragment libraries, some sequences, such as A-T rich regions, have been found to be more susceptible to breakage [15]. Such weak regions may have been cleaved more frequently during our fragment library preparation, resulting in an absence of high-throughput sequencing data across these regions.
Overview of the Serva ILTV genome
The general features of the Serva ILTV genome sequence were examined and were consistent with those described in previous genome studies of ILTV [3–12]. The genome size of the Serva ILTV strain was 152,630 bp, with a G + C content of 48%. The unique long and unique short sequences were 113,930 bp and 13,094 bp in length, respectively, with the unique short sequence flanked by internal and terminal repeat sequences (each 12,803 bp in length). A total of 80 predicted open reading frames (ORFs) were identified. Sixty-five ORFs were located within the long unique region, nine within the unique short region, and three pairs of ORFs within the repeat regions of the genome. An annotated genome map of the Serva ILTV strain is shown in Figure 1.

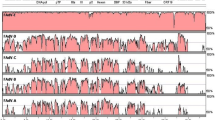
Nucleotide alignment of the Serva ILTV sequence and the concatenated ILTV reference sequence. Single vertical black lines indicate single nucleotide differences. Wider black boxes indicate larger regions of sequence differences, including areas where there are large gaps. Dashes indicate single base gaps in sequence alignments. The alignment was performed using Clustal W version 2.0. The locations and sizes of predicted ORFs and origins of DNA replication were annotated using the Geneious software package. The solid lines below the residue numbers indicate the locations of the internal and terminal repeat sequences.
Comparison of the Serva ILTV genome with the concatenated ILTV reference sequence
Comparative analyses showed that the Serva strain had the same gene arrangement as that of the concatenated ILTV genomic reference sequence (Figure 1). In complete genomic alignment analysis, the Serva strain had 96.5% DNA sequence identity with the concatenated sequence. Differences between the two sequences were mostly located within the left terminal region of the genome (extending over 18,169 bp), the middle of the unique long region of the genome (extending over 31,332 bp) and within the repeat regions (extending over 8,364 bp) (Figure 1). In the concatenated reference sequence, all these regions were obtained from SA-2 ILTV, a commercial vaccine strain of ILTV produced from an Australian field isolate [13]. Australian strains of ILTV may contain different genetic features compared with other ILTV strains, due to their evolution in a geographically isolated environment [16]. Excluding SA-2 sequence regions, DNA sequence identity between the Serva strain sequenced in this study and the concatenated sequence was 99.9%, containing 41 single-nucleotide polymorphisms and 13 nucleotide insertions or deletions. This high level of identity is consistent with the stable genome and low mutation rates observed in herpesviruses [17, 18]. All nucleotide differences between the concatenated reference and the Serva strain of ILTV are listed in Additional File 1.
Four large regions of sequence were absent in the concatenated ILTV sequence compared with the Serva strain. These included 528 bp and 594 bp of sequence missing in the UL29 and UL36 genes, respectively. Additionally, in each of the repeat regions the concatenated sequence lacked 1,563 bp of sequence compared to the Serva strain. To verify these findings, PCR was used to amplify the relevant regions of the UL29 and UL36 genes, and the repeat region of the Serva genome. The PCR products were sequenced using Sanger sequencing. The results obtained from Sanger sequencing were identical to those obtained by the high-throughput sequencing. The predicted translation products of the UL29 and UL36 genes of the Serva strain more closely resembled, and had more conserved domains when compared with, the UL29 and UL36 homologues from other alphaherpesviruses than the predicted products derived from the concatenated genome sequence (Figure 2). These results support the conclusion that the newly determined additional sequences are genomic sequence of ILTV, not sequencing errors. Within the left terminal region of the genome, the Serva sequence had a 189 bp deletion compared to the concatenated sequence (Figure 1).

Amino acid alignment of predicted translation products of homologous genes from the Serva ILTV strain, the concatenated ILTV reference sequence and other alphaherpesviruses. (A) UL29 gene product. (B) C terminal part of the UL36 gene product. Black boxes indicate 100% identity, dark grey boxes indicate 80-99% identity and pale grey boxes indicate 60-79% identity. Dashes indicate single amino acid gaps in sequence alignments. Alignments were performed using Clustal W version 2.0. The Blosum62 matrix was used to score sequence alignments.
There were differences in size of more than 50 amino acids between the predicted translation products of the UL54, UL29, UL30, UL36, UL37 and UL38 genes in the concatenated reference sequence and the Serva sequence. Smaller differences in the predicted sizes of other gene products were also identified (Table 1). All amino acid differences between predicted translation products in the concatenated reference sequence and the Serva sequence are listed in Additional File 2.
Given the conserved nature of the ILTV genome, future studies examining the genomic variation between different strains of ILTV may represent a strategic approach to examining the molecular pathogenesis of this virus. In particular it would be useful to compare sequence differences between virulent and attenuated strains of ILTV, especially within genes already known to be associated with ILTV virulence including gC, UL0, gG, gJ and TK genes [19–23].
Conclusions
This is the first complete genomic sequence of an individual ILTV strain. A number of differences between this strain and the concatenated reference ILTV sequence were identified. Significantly, four large missing regions were identified in the published concatenated reference sequence. Missing regions of genomic sequence considerably hamper genomic assembly, so the Serva sequence assembled in this study represents a much improved reference sequence for future high throughput ILTV sequencing studies and comparative genomic analyses.
Methods
Virus strain
This study utilised the chicken embryo origin live attenuated Serva vaccine strain of ILTV (Nobilis® ILT, Intervet), which has been recently introduced into Australia [16]. Virions were purified by Ficoll gradient centrifugation directly from commercial vaccine vials. Pelleted virions were washed and resuspended in TNE buffer (0.01 M Tris, 0.2 M NaCl, 1 mM EDTA, pH 7.4).
High-throughput sequencing
Following purification, total viral genomic DNA was extracted using the High Pure PCR Template Preparation Kit (Roche). Sequencing was performed using parallel, ligation-mediated sequencing technology (SOLiD™ 3 system, Applied Biosystems) following the manufacturer's standard procedures. Briefly, 1 μg of ILTV DNA was sheared and P1 and P2 adaptors were ligated to the fragments. Ligated DNA fragments were size-selected to an average length of 170 bp and amplified for 10 cycles. Approximately 10 pg of this ILTV fragment library/μl, as well as 50 pg of a similarly generated library of an unrelated bacterial genome of around one megabase pairs/μl, were added to an emulsion with 80 million beads. The libraries were sequenced in parallel using a flow cell divided into 8 segments. The resulting reads were unambiguously mapped to either the viral or the bacterial genome, allowing up to two mismatches for each read. The software package Velvet version 0.7.55 was used to perform de novo assembly of all reads [24] and the resulting contigs for the virus were identified bioinformatically and then aligned to the complete concatenated ILTV genomic sequence, with the exception of the terminal repeat region (identical to the internal repeat region), which was excluded from the analysis.
ILTV DNA sequence analysis
The software package Geneious [25] was used to manually curate the alignments of the contigs and to produce a consensus sequence with reference to the original mapped reads. Any sequence gaps or ambiguous regions of sequence were amplified and sequenced by Sanger sequencing methods using BDT version 3.1 (Applied Biosystems). Nucleotide and amino acid sequence alignments were performed using ClustalW version 2.0 [26], ORFs were then annotated using the Geneious software package. Open reading frames containing more than 50 amino acids were predicted using the ORF finder function of Geneious, based on the complete concatenated ILTV genomic sequence.
Nucleotide sequence accession numbers
The complete genome sequence of Serva ILTV has been deposited in the NCBI GenBank database under accession HQ_630064. The concatenated ILTV genomic sequence is available in the GenBank database under accession NC_006623. Nucleotide sequences for psittacid herpesvirus-1 (PsHV1), equid herpesvirus-1 (EHV1) and herpes simplex virus-1 (HSV1) were also utilized in this study. The translated amino acid sequences for the UL29 genes of these viruses are available in the GenBank database under accessions NP_944402, YP_053076 and NP_044631, respectively. The translated amino acid sequences for the UL36 genes of these viruses are available in the GenBank database under accessions NP_944409, YP_053069 and ABI63498, respectively.
References
Bagust TJ, Jones RC, Guy JS: Avian infectious laryngotracheitis. Rev Sci Tech. 2000, 19 (2): 483-492.
Roizmann B, Desrosiers RC, Fleckenstein B, Lopez C, Minson AC, Studdert MJ: The family Herpesviridae: an update. The Herpesvirus Study Group of the International Committee on Taxonomy of Viruses. Arch Virol. 1992, 123 (3-4): 425-449. 10.1007/BF01317276.
Ziemann K, Mettenleiter TC, Fuchs W: Gene arrangement within the unique long genome region of infectious laryngotracheitis virus is distinct from that of other alphaherpesviruses. J Virol. 1998, 72 (1): 847-852.
Fuchs W, Mettenleiter TC: DNA sequence and transcriptional analysis of the UL1 to UL5 gene cluster of infectious laryngotracheitis virus. J Gen Virol. 1996, 77 (9): 2221-2229. 10.1099/0022-1317-77-9-2221.
Fuchs W, Mettenleiter TC: DNA sequence of the UL6 to UL20 genes of infectious laryngotracheitis virus and characterization of the UL10 gene product as a nonglycosylated and nonessential virion protein. J Gen Virol. 1999, 80 (8): 2173-2182.
Griffin AM, Boursnell ME: Analysis of the nucleotide sequence of DNA from the region of the thymidine kinase gene of infectious laryngotracheitis virus; potential evolutionary relationships between the herpesvirus subfamilies. J Gen Virol. 1990, 71 (4): 841-850. 10.1099/0022-1317-71-4-841.
Johnson MA, Prideaux CT, Kongsuwan K, Tyack SG, Sheppard M: ICP27 immediate early gene, glycoprotein K (gK) and DNA helicase homologues of infectious laryngotracheitis virus (gallid herpesvirus 1) SA-2 strain. Arch Virol. 1995, 140 (4): 623-634. 10.1007/BF01309954.
Johnson MA, Tyack SG, Prideaux C, Kongsuwan K, Sheppard M: Nucleotide sequence of infectious laryngotracheitis virus (gallid herpesvirus 1) ICP4 gene. Virus Res. 1995, 35 (2): 193-204. 10.1016/0168-1702(94)00096-U.
Johnson MA, Tyack SG, Prideaux CT, Kongsuwan K, Sheppard M: Nucleotide sequence of the left-terminus of infectious laryngotracheitis virus (Gallid herpesvirus 1) SA-2 strain. Arch Virol. 1997, 142 (9): 1903-1910. 10.1007/s007050050209.
Poulsen DJ, Burton CR, O'Brian JJ, Rabin SJ, Keeler CL: Identification of the infectious laryngotracheitis virus glycoprotein gB gene by the polymerase chain reaction. Virus Genes. 1991, 5 (4): 335-347. 10.1007/BF00271532.
Wild MA, Cook S, Cochran M: A genomic map of infectious laryngotracheitis virus and the sequence and organization of genes present in the unique short and flanking regions. Virus Genes. 1996, 12 (2): 107-116. 10.1007/BF00572949.
Ziemann K, Mettenleiter TC, Fuchs W: Infectious laryngotracheitis herpesvirus expresses a related pair of unique nuclear proteins which are encoded by split genes located at the right end of the UL genome region. J Virol. 1998, 72 (8): 6867-6874.
Thureen DR, Keeler CL: Psittacid herpesvirus 1 and infectious laryngotracheitis virus: Comparative genome sequence analysis of two avian alphaherpesviruses. J Virol. 2006, 80 (16): 7863-7872. 10.1128/JVI.00134-06.
Smith DR, Quinlan AR, Peckham HE, Makowsky K, Tao W, Woolf B, Shen L, Donahue WF, Tusneem N, Stromberg MP, et al: Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res. 2008, 18 (10): 1638-1642. 10.1101/gr.077776.108.
Deininger PL: Random subcloning of sonicated DNA: application to shotgun DNA sequence analysis. Anal Biochem. 1983, 129 (1): 216-223. 10.1016/0003-2697(83)90072-6.
Blacker H, Kirkpatrick N, Rubite A, O'Rourke D, Noormohammadi A: Epidemiology of recent outbreaks of infectious laryngotracheitis in poultry in Australia. Aust Vet J. 2011, 89 (3): 89-94. 10.1111/j.1751-0813.2010.00665.x.
Cheng TP, Valentine MC, Gao J, Pingel JT, Yokoyama WM: Stability of murine cytomegalovirus genome after in vitro and in vivo passage. J Virol. 2010, 84 (5): 2623-2628. 10.1128/JVI.02142-09.
Sakaoka H, Kurita K, Iida Y, Takada S, Umene K, Kim YT, Ren CS, Nahmias AJ: Quantitative analysis of genomic polymorphism of herpes simplex virus type 1 strains from six countries: studies of molecular evolution and molecular epidemiology of the virus. J Gen Virol. 1994, 75 (3): 513-527. 10.1099/0022-1317-75-3-513.
Devlin JM, Browning GF, Hartley CA, Kirkpatrick NC, Mahmoudian A, Noormohammadi AH, Gilkerson JR: Glycoprotein G is a virulence factor in infectious laryngotracheitis virus. J Gen Virol. 2006, 87 (10): 2839-2847. 10.1099/vir.0.82194-0.
Fuchs W, Wiesner D, Veits J, Teifke JP, Mettenleiter TC: In vitro and in vivo relevance of infectious laryngotracheitis virus gJ proteins that are expressed from spliced and nonspliced mRNAs. J Virol. 2005, 79 (2): 705-716. 10.1128/JVI.79.2.705-716.2005.
Han MG, Kweon CH, Mo IP, Kim SJ: Pathogenicity and vaccine efficacy of a thymidine kinase gene deleted infectious laryngotracheitis virus expressing the green fluorescent protein gene. Arch Virol. 2002, 147 (5): 1017-1031. 10.1007/s00705-001-0794-y.
Pavlova SP, Veits J, Blohm U, Maresch C, Mettenleiter TC, Fuchs W: In vitro and in vivo characterization of glycoprotein C-deleted infectious laryngotracheitis virus. J Gen Virol. 2010, 91 (4): 847-857. 10.1099/vir.0.016634-0.
Veits J, Luschow D, Kindermann K, Werner O, Teifke JP, Mettenleiter TC, Fuchs W: Deletion of the non-essential UL0 gene of infectious laryngotracheitis (ILT) virus leads to attenuation in chickens, and UL0 mutants expressing influenza virus haemagglutinin (H7) protect against ILT and fowl plague. J Gen Virol. 2003, 84 (12): 3343-3352. 10.1099/vir.0.19570-0.
Zerbino DR, Birney E: Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18 (5): 821-829. 10.1101/gr.074492.107.
Drummond AJ, Ashton B, Cheung M, Heled J, Kearse M, Moir R, Stones-Havas S, Sturrock S, Thierer T, Wilson A: Geneious v5.0. 2010, [http://www.geneious.com/]
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al: Clustal W and Clustal X version 2.0. Bioinformatics. 2007, 23 (21): 2947-2948. 10.1093/bioinformatics/btm404.
Acknowledgements
The Rural Industries Research and Development Corporation, Australia, funded this study. JMD is supported by a fellowship from the Australian Research Council. NICTA is funded by the Australian Government as represented by the Department of Broadband, Communications and the Digital Economy and the Australian Research Council through the ICT Centre of Excellence program. The authors thank Kelly Ewen-White (Lifetech) for her expert advice and assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
S-WL carried out the sequencing studies, sequence analyses, sequence alignments and drafted the manuscript. NPF performed preparation of viral DNA. IP performed sequencing. JFM performed sequence analyses. CAH performed preparation of viral DNA, participated in sequence analyses and helped to draft the manuscript. JMD, PFM, GFB and AHN conceived of the study, designed and coordinated the study, participated in preparation of viral DNA, sequencing studies, sequence analyses, and helped to draft the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2010_10090_MOESM1_ESM.XLS
Additional file 1:Summary of nucleotide differences. Nucleotide differences detected between the concatenated ILTV reference sequence and the Serva ILTV sequence after whole genome alignment. (XLS 355 KB)
12864_2010_10090_MOESM2_ESM.XLS
Additional file 2:Summary of all amino acid differences. Amino acid differences detected between the predicted translation products of the concatenated ILTV reference sequence and the Serva ILTV sequence. (XLS 135 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lee, SW., Markham, P.F., Markham, J.F. et al. First complete genome sequence of infectious laryngotracheitis virus. BMC Genomics 12, 197 (2011). https://doi.org/10.1186/1471-2164-12-197
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-12-197