
Abstract
Background
Acidithiobacillus ferrooxidans gains energy from the oxidation of ferrous iron and various reduced inorganic sulfur compounds at very acidic pH. Although an initial model for the electron pathways involved in iron oxidation has been developed, much less is known about the sulfur oxidation in this microorganism. In addition, what has been reported for both iron and sulfur oxidation has been derived from different A. ferrooxidans strains, some of which have not been phylogenetically characterized and some have been shown to be mixed cultures. It is necessary to provide models of iron and sulfur oxidation pathways within one strain of A. ferrooxidans in order to comprehend the full metabolic potential of the pangenome of the genus.
Results
Bioinformatic-based metabolic reconstruction supported by microarray transcript profiling and quantitative RT-PCR analysis predicts the involvement of a number of novel genes involved in iron and sulfur oxidation in A. ferrooxidans ATCC23270. These include for iron oxidation: cup (copper oxidase-like), ctaABT (heme biogenesis and insertion), nuoI and nuoK (NADH complex subunits), sdrA1 (a NADH complex accessory protein) and atpB and atpE (ATP synthetase F0 subunits). The following new genes are predicted to be involved in reduced inorganic sulfur compounds oxidation: a gene cluster (rhd, tusA, dsrE, hdrC, hdrB, hdrA, orf2, hdrC, hdrB) encoding three sulfurtransferases and a heterodisulfide reductase complex, sat potentially encoding an ATP sulfurylase and sdrA2 (an accessory NADH complex subunit). Two different regulatory components are predicted to be involved in the regulation of alternate electron transfer pathways: 1) a gene cluster (ctaRUS) that contains a predicted iron responsive regulator of the Rrf2 family that is hypothesized to regulate cytochrome aa3 oxidase biogenesis and 2) a two component sensor-regulator of the RegB-RegA family that may respond to the redox state of the quinone pool.
Conclusion
Bioinformatic analysis coupled with gene transcript profiling extends our understanding of the iron and reduced inorganic sulfur compounds oxidation pathways in A. ferrooxidans and suggests mechanisms for their regulation. The models provide unified and coherent descriptions of these processes within the type strain, eliminating previous ambiguity caused by models built from analyses of multiple and divergent strains of this microorganism.
Similar content being viewed by others

Background
Acidithiobacillus ferrooxidans is an acidophilic, chemolithoautotrophic γ-proteobacterium that uses energy and electrons derived from the oxidation of ferrous iron (Fe(II)) and r educed i norganic s ulfur c ompounds (RISCs) for carbon dioxide and nitrogen fixation and other anabolic processes. It is a member of a consortium of microorganisms used for industrial copper recovery (bioleaching or biomining) and gold recovery (biooxidation) and contributes to the geobiochemical recycling of metals and nutrients in pristine and contaminated acid-rich environments.
When A. ferrooxidans requires reducing power it is confronted by a particularly challenging bioenergetic problem because the standard reduction half-potential of the Fe(II)/Fe(III) couple (+0.77 V at pH 2, the pH of the medium) is much more positive than that of the NAD(P)/NAD(P)H couple (-0.32 V at the cytoplasmic pH 7). This means that electrons have to be "pushed uphill" from Fe(II) to NAD(P) against the redox potential gradient (uphill pathway). The energy to accomplish this was proposed to come from the proton motive force that naturally occurs across the periplasmic membrane of A. ferrooxidans (outside = pH 2, inside = pH 7). It was further suggested that protons, entering the cell through a membrane associated ATP synthetase, could be consumed during the reduction of oxygen to water (2e- + 1/2 O2 + 2H+ → H2O) in which electrons for this reaction come from the oxidation of Fe(II) in a pathway that is thermodynamically favorable (downhill pathway). These ideas were first promulgated based upon theoretical considerations by Ingledew [1] but the model has been extended more recently and major parts of it are now supported by experimental evidence (reviewed in [2]).
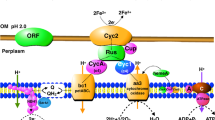
According to the model (see Figure 1), components involved in the downhill electron pathway from Fe(II) to oxygen include: an outer membrane high molecular-weight cytochrome c encoded by cyc2, a gene of unknown function (ORF1), a periplasmic soluble blue copper protein rusticyanin encoded by rus, a periplasmic membrane-bound dihemic cytochrome c4 encoded by cyc1 and a terminal cytochrome oxidase of the aa3-type encoded by the coxBACD gene cluster [3–6]. In A. ferrooxidans ATCC 33020 these genes are cotranscribed in an eight gene transcriptional unit, termed the rus operon that is upregulated in Fe(II)-grown cells [5, 7]. Gene context analysis of the recently annotated genomic sequence of A. ferrooxidans type strain ATCC 23270 demonstrates that the rus operon is similarly organized in this strain [8].

Model of Fe(II) oxidation in A. ferrooxidans ATCC 23270. The flow of electrons is shown from the oxidation of Fe+2 by Cyc2 to reduce oxygen via the aa3 complex (downhill pathway) or to reduce NAD+ via bc1/quinone pool/NADH complex (uphill pathway). The downhill pathway can consume protons entering via the ATPase complex helping to drive ATP synthesis or via the bc1/quinone pool/NADH complex that drives the flow of electrons in the uphill pathway. The switch point between the downhill and uphill flow is suggested to be at the level of rusticyanin (Rus). Abbreviations used can be found in the text.
The uphill components include: a cytochrome bc1 complex (complex III, ubiquinol-cytochrome c reductase), the quinone pool and a NADH1 dehydrogenase complex (hereafter abbreviated to NADH complex) functioning in reverse [9–11]. Genetic and biophysical evidence obtained for A. ferrooxidans ATCC 19859 and ATCC 33020 support this view [11–13]. In these two strains, the genes encoding the bc1 complex have been shown to be part of a five-gene operon, termed the petI operon, mainly transcribed in Fe(II) conditions and organized as following: a diheme cytochrome c4 (cycA1), a short-chain dehydrogenase (sdrA1) of unknown function, a cytochrome b (petA1), an iron-sulfur protein Rieske (petB1), and a cytochrome c1 (petC1) [2, 12, 13]. Based on the cotranscription of the diheme cytochrome c4 gene (cycA1) with those encoding the bc1 complex, it was proposed that electrons from the diheme cytochrome c4 take a thermodynamically uphill pathway via the bc1 complex to a NADH complex driven energetically by proton motive force [2, 12–14] (see Figure 1). Adjacent to the petI operon, is a three gene (resBC and a hypothetical gene) operon in which ResBC are predicted to be chaperones involved in heme insertion in c-type cytochromes, possibly those involved in the maturation of the c1 cytochrome of the bc1 complex [12]. The petI operon is similarly organized and transcribed in A. ferrooxidans ATCC 23270 [14] supporting this model.
The bifurcation in the flow of electrons from Fe(II) to reduce either NAD (uphill) or O2 (downhill) has been proposed to occur at the level of rusticyanin, a small periplasmic blue copper protein [2, 13, 14] (see Figure 1). By adjusting the flow of electrons at this branch point, it was suggested that A. ferrooxidans could balance its requirements for NADH and ATP [9].
The components involved in both the uphill and downhill electron flow have been shown to form a Fe-oxidizing/O2-reducing supercomplex spanning both inner and outer membranes in an unnamed strain of A. ferrooxidans [15] supporting previous models. The supercomplex has also been suggested to include a copper containing protein (ORF1) physically associated with the periplasmic Cyc1 that is proposed to be involved in downhill electron flow. In addition, in vitro reconstitution of the iron oxidation system of the A. ferrooxidans type strain with rusticyanin, aa3-type cytochrome oxidase, the cytochromes c Cyc2 and Cyc1 has been reported recently [16].
Despite progress in understanding iron oxidation in the Acidithiobacillus genus, several lacunae in our knowledge persist, such as the identification of components involved in the proposed connection between the bc1/quinone complex and the NADH complex, the identification of the chaperones used for cofactor insertion into the relevant redox proteins, and the mechanisms and components regulating the electron flux via the downhill pathway to O2 versus the uphill pathway to the NADH complex.
Reduced inorganic sulfur oxidation is widespread in prokaryotes (reviewed in [17, 18]. However, in contrast to iron that occurs in only two oxidation states, sulfur exists in multiple states from -2 to +6, complicating identification of intermediates and relevant enzymes in sulfur oxidation. Also, some sulfur compounds can be oxidized abiotically adding further difficulties in resolving enzymatic steps from chemical changes. Despite these difficulties, several biological pathways for RISCs oxidation have been identified including the phylogenetically widespread sulfur oxidizing (sox) gene pathway (reviewed in [17]) and the archaeal type sulfur oxygenase reductase (sor) gene system (reviewed in [19]). However, neither sox, nor sor have been detected in the genome of A. ferrooxidans [2, 8], raising the question as to how this organism oxidizes RISCs.
Much of our knowledge of RISCs oxidation in A. ferrooxidans comes from enzyme assays performed many years ago on different strains, some of which await phylogenetic characterization or, in some cases, have not yet even been isolated as pure cultures [2]. Those that have been characterized form genetically diverse clusters with at least three phylogenetic groups [20–23]. This raises the possibility that current models of RISCs oxidation reflect a patchwork assemblage of predicted pathways and activities that may not exist in toto in any one A. ferrooxidans strains and perhaps more accurately reflect the pangenomic capacity of the genus Acidithiobacillus for RISCs oxidation.
Thus the current investigation was prompted by the need to generate a more comprehensive picture of iron and sulfur bioenergetics by searching for missing steps and predicting novel enzymatic and electron transfer components and to provide a coherent picture of these processes in one strain of A. ferrooxidans (type strain, ATCC 23270), facilitating the recognition of species variation in bioenergetic pathways.
Results and Discussion
General Features of the Transcriptional Profiles
RNA, isolated from mid-log A. ferrooxidans grown in either sulfur (S0) or ferrous iron (Fe(II)) medium, was used to probe gene expression using microarrays displaying unique oligonucleotides representing about 3000 predicted genes of the A. ferrooxidans type strain genome. Using statistical criteria described previously [14], a 1.5 log ratio of median cut-off (corresponding to genes induced more than 2.8 fold) was selected as indicating differential gene expression in the two growth conditions (Table 1[7, 24, 25], Table 2[24–29] and Additional file 1[7, 24–26, 29]). The expression patterns observed with the microarrays were validated for some relevant genes by real-time quantitative PCR (Table 3[7, 24–26, 28, 29]). One hundred and ninety four genes presented a differential expression profile, of which 110 were upregulated (up to 38 fold) while 84 were downregulated (up to 10 fold) in iron compared to sulfur medium. Genes exhibiting differential expression were grouped by hierarchical clustering and were found to be mostly associated with unknown functions, energy metabolism, cell envelope and central intermediary processes (Additional file 1[7, 24–26, 29]).
Extending the current model of Fe(II) oxidation
The rus operon was induced in Fe(II)-grown cells (Table 1[7, 24, 25] and Table 3[7, 24–26, 28, 29]) supporting the current model of the involvement of Cyc2, rusticyanin, Cyc1 and cytochrome oxidase in the oxidation of Fe(II) and the downhill electron transfer chain terminating in the reduction of oxygen to water (Figure 1).
Embedded in the rus gene operon, is a hypothetical gene of unknown function (ORF1, AFE_3151). Its genetic linkage and congruent transcriptional activity suggest that it is involved in Fe(II) oxidation (Table 1[7, 24, 25] and Table 3[7, 24–26, 28, 29]). In agreement with these data, highest expression of ORF1 was detected in iron-compared to sulfur-grown ATCC 33020 cells [7]. ORF1 exhibits weak similarity to the putative type-3 multicopper oxidase from Halorubrum lacusprofundi (30% similarity, e-value: 8e-05) and to the predicted outer membrane type 3 multicopper oxidase protein Pan1 from Halobacterium sp. NRC-1 (32% similarity; e-value: 3e-04). It also exhibits weak similarity to rusticyanin including conservation of three out of its four critical copper binding residues. In addition, EPR analysis suggests that ORF1 contains copper [30]. We propose the name Cup (cup redoxin-like) for ORF1. A possible function for Cup is to deliver copper either to aa3 cytochrome oxidase or to rusticyanin. The well documented proteins Sco and Cox1 that are involved in copper delivery to copper-containing proteins in other organisms [31] have been not detected in A. ferrooxidans [8] and Cup may have assumed their role. Given its similarity to rusticyanin, an alternate hypothesis is that Cup is involved in electron transfer perhaps between Cyc2 and Cyc1 bypassing rusticyanin. Two arguments in favor of this hypothesis are: 1) Cup has been shown experimentally to be physically associated with Cyc1 and not with rusticyanin [15], and 2) Cup is bound to the outer membrane likely facing the periplasm [Amouric, Yarzabal and Bonnefoy, unpublished results]. Cup could provide an alternative route for electron flow during iron oxidation and an additional point for its regulation.
Immediately downstream of the rus operon is a cluster of six genes, upregulated in iron (Table 1[7, 24, 25] and Table 3[7, 24–26, 28, 29]), that are predicted to be involved in cytochrome aa3 oxidase biogenesis (ctaABT) and iron-responsive regulation of cytochrome aa3 oxidase biogenesis (ctaRUS): ctaA (AFE_3144) encoding an integral membrane protein with 7 out of 8 His located in transmembrane regions similar to heme A synthase CtaA ([32] and references therein), ctaB (AFE_3143) encoding a heme O synthase and ctaT (AFE_3142) encoding an integral membrane protein belonging to the major facilitator family transporter that could be involved in the exportation of heme A to cytochrome oxidase [33] (Figure 1); ctaR (AFE_3141) predicted to encode an iron responsive regulator [34] of the Rrf2 family that in T. denitrificans is clustered with cbb3 cytochrome oxidase biogenesis genes (data not shown), ctaU (AFE_3139) encoding a hypothetical protein of unknown function and ctaS (AFE_3138) encoding a predicted Fe(II)-dependent oxygenase superfamily member of unknown function. Immediately downstream but transcribed in the other direction, are two genes regBA (AFE_3136-3137), also upregulated in iron (Table 1[7, 24, 25] and Table 3[7, 24–26, 28, 29]), that are predicted to encode a sensor/regulator two-component signal transduction system of the RegB/RegA family with similarity to RegBA of Rhodobacter capsulatus. RegA directly controls synthesis of cytochrome cbb3 and ubiquinol oxidases that function as terminal electron acceptors in a branched respiratory chain [35]. Given the sequence similarity of the predicted A. ferrooxidans RegA with that of R. capsulatus, the conservation of the quinone binding site in the membrane spanning domain and of the redox active cysteine in the cytoplasmic transmitter domain of RegB, we propose that it is also involved in redox sensing. Because of the regBA juxtaposition to genes predicted to encode cytochrome aa3 oxidase biogenesis, this cytochrome oxidase is a likely candidate for the target of RegBA regulation in A. ferrooxidans. However, in R. capsulatus, RegBA also regulates other genes involved in respiratory electron components such as cytochromes c2, c(y) and the cytochrome bc1 complex, so that the actual RegBA target(s) in A. ferrooxidans requires experimental evaluation. RegBA of R. capsulatus have been shown to respond to the status of the aerobic respiratory chain, most likely the ubiquinone pool in the membrane [36] and, if this also proves to be the case in A. ferrooxidans, RegBA could help in making regulatory changes to balance electron equivalents between uphill and downhill electron flow, perhaps by adjusting the proportion of cytochrome Cyc1 and cytochrome oxidase aa3 (downhill electron flow) to the cytochrome CycA1 and the cytochrome bc1 complex (uphill electron flow). Alternately, RegBA could play a role in switching between iron and sulfur oxidation or between aerobic and anaerobic oxidation. It is clear that the discovery of the predicted iron and redox responsive regulators CtaRUS and RegAB will now allow the experimental biologist to focus on important regulatory switches that are most likely to be involved in cellular decisions related to energy metabolism.
The petI operon was also induced in Fe(II) medium (Table 1[7, 24, 25] and Table 3[7, 24–26, 28, 29]) supporting the current model for the role of the bc1 complex in the uphill flow of electrons during iron oxidation (Figure 1). Embedded within the petI operon is sdrA1 (AFE_3108) whose function remains unknown. SdrA1 has the characteristic NAD(P) binding site at its N-terminus (TG AGEG IG) of Ndu9 which is a subunit of the NADH complex involved in this complex assembly and stability in a variety of eukaryotes [37]. However, SdrA1 exhibits the conserved catalytic residues (N125, S147, Y166, K170) involved in electron input into the NADH complex or electron transfer within the complex, suggesting that this protein could function as an oxidoreductase [38, 39] (Additional file 2). SdrA1 has been predicted to be situated in the cytoplasm in A. ferrooxidans with a possible hydrophobic region embedded in the inner membrane [12] and we hypothesize that it transfers electrons from the quinone pool to the NADH complex (Figure 1).
Whereas most of the predicted genes encoding the subunits of the NADH complex (AFE_2630-2617) are equally expressed in Fe(II) and S0 growth conditions, nuoI (AFE_2622) and nuoK (AFE_2620) are upregulated in Fe(II) medium (Table 1[7, 24, 25]). The nuoI encodes a ferredoxin located in the cytoplasmic arm of the NADH complex and is involved in the intramolecular electron transfer between FMN and quinone, whereas nuoK encodes a membrane subunit thought to be involved in quinone reduction [40] and in proton translocation [41]. Given the predicted locations of NuoI and NuoK in the hinge region of the NADH complex [42] and the upregulation of nuoI, nuoK and sdrA1 in Fe(II) growth conditions, we suggest that they interact to facilitate uphill electron flow from quinone to the NADH complex (Figure 1).
Whereas most of the genes predicted to encode the ATP synthetase complex were similarly expressed in Fe(II) and in S0 grown cells, membrane embedded F0 subunits A and C, encoded by atpB (AFE_3209) and atpE (AFE_3208) respectively, were upregulated in Fe(II) growth conditions (Table 1[7, 24, 25]). These subunits are involved in proton translocation across the membrane [43] and their upregulation could allow more protons to pass through the ATP synthetase complex during Fe(II) oxidation, provided an increase in ATP synthesis. However, the resulting increase of intracellular protons requires a concomitant increase in intracellular electrons for their neutralization, so as not to compromise the internal pH of the cell. These electrons could come from the downhill pathway during Fe(II) oxidation as shown in Figure 1.
The organization and regulation of the components of Fe(II) oxidation in A. ferrooxidans appear to be unique to this organism, although Thiobacillus prosperus strain V6 possesses a transcriptional unit upregulated in iron conditions with some similarity to the rus operon [44]. However, the T. prosperus operon lacks the cyc1 and the rus genes encoding cytochrome c4 and rusticyanin, respectively. The latter is located downstream from the cluster, is monocistonic and is expressed in iron and sulfur growth conditions [44], suggesting different regulatory mechanisms compared to A. ferrooxidans. In Leptospirillum group II, the electron transfer chain involved in Fe(II) oxidation contains two cytochromes c and a cbb3 cytochrome oxidase [45–47] but no blue copper protein such as rusticyanin. In the archaea Ferroplasma acidarmanus a blue copper protein (sulfocyanin) has been suggested to transfer the electrons from Fe(II) to a cbb3-type terminal oxidase [48]. While the genes encoding four blue copper proteins (two sulfocyanin-like and two rusticyanin-like) have been identified in Metallosphaera sedula [49], none of them respond to the presence of iron in the medium [50]. However, foxA (cytochrome oxidase subunit I) and soxNL (cytochrome b and [2Fe-2S] Rieske) -cbsAB (cytochromes b) clusters have been predicted to be important for Fe(II) oxidation in M. sedula. Genes encoding cytochrome c oxidase subunits I and II (foxAB) and CbsA-like cytochrome b (foxC) have been also proposed to be involved in iron oxidation in Sulfolobus metallicus [51]. It appears therefore that different pathways for ferrous iron oxidation have evolved in prokaryotes.
Extending the current model of the oxidation of reduced inorganic sulfur compounds (RISCs)
Predicted genes, proteins or enzymatic activities previously identified in the oxidation of RISCs in different species of A. ferrooxidans include: sqr (AFE_1792) encoding sulfide quinone reductase from NASF-1 strain [26, 52], doxDA (AFE_0044) encoding thiosulfate quinone oxidoreductase from ATCC23270 [53] and from the CCM4253 strain [54], tetH (AFE_0029) encoding tetrathionate hydrolase from ATCC23270 [27], cydAB (AFE_0955-0954) encoding a bd oxidase and cyoABCD (AFE_0631-0634) encoding a bo3 oxidase from ATCC19859 [11] (See Figure 2). All these genes are upregulated in sulfur-containing media relatively to Fe(II) (Table 2[24–29] and Table 3[7, 24–26, 28, 29]). This is also the case for the petII operon (AFE_2727-2732) encoding a second bc1 complex, a cytochrome c4, SdrA2 and a high potential iron-sulfur protein, Hip, as already reported for ATCC19859 [11], ATCC33020 [13] and ATCC23270 [14] strains (Table 2[24–29] and Table 3[7, 24–26, 28, 29]). This second bc1 complex has been proposed to function directly transferring electrons from sulfur to oxygen (Figure 2), and possibly in the aerobic and anaerobic oxidation of sulfur and formate described by Pronk et al. [55]. In that case, the bc1 complex receives the electrons from the quinol pool and transfers them to the membrane-bound cytochrome c4 CycA2 and/or to the periplasmic high potential iron-sulfur protein Hip that subsequently gives the electrons to the terminal oxidase [2, 13, 14] (Figure 2). SdrA2, like SdrA1 (see above), may promote electron flow from the quinone pool to the NADH complex (Figure 2). These findings support earlier models of the branched electron transfer flow during S0 oxidation [2, 11, 13, 14] (Figure 2). However, this model is far from being complete and several outstanding questions remain unanswered, including the identification of the enzymes that oxidize sulfur and sulfite.

Model of sulfur oxidation in A. ferrooxidans ATCC 23270. Reduced inorganic sulfur compound (RISC) oxidation pathways are predicted to involve various enzymes, enzyme complexes and a number of electron carriers located in different cellular compartments: in the outer membrane facing the periplasm (tetrathionate reductase, TetH), in the periplasm (high potential iron-sulfur protein, HiPIP), attached to the cytoplasmic membrane on the periplasmic side (cytochrome c, CycA2), in the cytoplasmic membrane (sulfide quinone reductase (SQR), thiosulfate quinone reductase (TQR), bc1 complex, NADH complex I, bd and bo3 terminal oxidases) and in the cytoplasm (heterodisulfide reductase (HDR), and ATP sulfurylase (SAT)). Insoluble sulfur is first converted to sulfane sulfate (GSSH) which is then transferred to the heterodisulfide reductase (HDR) through a cascade of sulfur transferases (DsrE, TusA and Rhd). Electrons coming from sulfide (H2S), thiosulfate (S2O32-) or sulfane sulfate (GSSH) are transferred via the quinol pool (QH2) either (1) directly to terminal oxidases bd or bo3, or indirectly throught a bc1 complex and a cytochrome c(CycA2) or a high potential iron-sulfur protein (HiPIP) probably to the aa3 oxidase where O2 reduction takes place or (2) to NADH complex I to generate reducing power.
Sulfur
Seven genes, potentially encoding a heterodisulfide reductase complex HdrABC (AFE_2586 and AFE_2555-2550) were highly upregulated in cells grown in sulfur medium (Table 2[24–29] and Table 3[7, 24–26, 28, 29]). This complex catalyzes the reversible reduction of the disulfide bond X-S-S-X coupled with energy conservation in methanogenic archaea ([56] and references therein) and sulfate reducing archaea and bacteria [57]. This complex in A. ferrooxidans is predicted to have three different cytoplasmic HdrB subunits (AFE_2586, 2554 and 2550) with the cysteine-rich domain which binds the unusual type [4Fe-4S] cluster [58], involved in disulfide reduction [56, 59]. In addition, genes potentially encoding the ferredoxin-like and the flavoprotein subunits, HdrC (AFE_2555 and 2551) and HdrA (AFE_2553) are present in the same locus. The flavoprotein HdrA subunit exhibits a possible N-terminal membrane spanning region, a conserved FAD binding site (GXGXXGX16–19(D/E)), and the conserved four cysteine cluster (CXGXRDX6–8CSX2CC) that binds a Fe-S center, typical of the cytoplasmic iron-sulfur flavoprotein type enzyme.
Three genes encoding for sulfur metabolism accessory proteins, placed immediately upstream of the heterodisulfide reductase complex, are similarly upregulated (Table 2[24–29]). These encode a cytoplasmic rhodanase-related sulfurtransferase (AFE_2558, COG0607) [60], a cytoplasmic SirA-like disulfide bond formation regulator (AFE_2557, pfam01206, COG0425, IPR001455) and an inner membrane located peroxiredoxin of the DrsE superfamily (AFE_2556, COG2210 and 2044). The rhodanase AFE_2558 is 48% similar to the Sud protein from Wolinella succinogenes which binds and transfers polysulfide sulfur to the polysulfide reductase located in the cytoplasmic membrane [61, 62]. In turn, the SirA-like protein AFE_2557 is 54% similar to the TusA protein from Vibrio fischeri and other bacteria which belong to a complex sulfur-relay system that facilitates specific sulfur flow/trafficking from various pathways [63–65]. Finally, DsrE family proteins such as that predicted to be encoded by AFE_2556 are small proteins recently shown to be involved in sulfur transfer reactions during sulfur oxidation [66].
Significant sequence similarity and conserved gene organization of the A. ferrooxidans heterodisulfide reductase complex and accessory proteins is restricted only to Aquifex aeolicus and known acidophilic sulfur oxidizing microorganisms Hydrogenobaculum sp. Y04AAS1, Hydrogenivirga sp. 128-5-R1-1, Metallosphaera sedula [50], Sulfolobus acidocaldarius, S. tokodaii and S. solfataricus (Figure 3), associating the whole gene cluster with sulfur oxidation. In methanogenic and sulfate reducing archaea, HdrA receives the electrons from the hydrogenase and transfers them through HdrC to the heterodisulfide reductase catalytic site located in HdrB. Accompanying the reduction of heterodisulfide, protons are extruded across the membrane creating a proton motive force. We hypothesize that in A. ferrooxidans and the sulfur oxidizers referred above, the Hdr complex, driven by the naturally existing proton gradient, could be working in reverse and oxidizing disulfide intermediaries (from sulfur oxidation) to sulfite and delivering the collected electrons to the membrane quinol pool (Figure 2). In addition, the three accessory sulfurtransferases are likely involved in the transfer of the proposed sulfane sulfur [67, 68] to the heterodisulfide reductase (Figure 2).

Comparison of the hdr cluster between A. ferrooxidans ATCC 23270 and other sulfur oxidizers. Heterodisulfide reductase complex (HdrC1B1AOrf2HdrC2B2), accessory proteins (Rhd, TusA, DsrE) and ATP sulfurylase (Sat) in AF: A. ferrooxidans ATCC 23270 (NC_011206), AA: Aquifex aeolicus (NC_000918) and known acidophilic sulfur oxidizing microorganisms HB: Hydrogenobaculum sp. Y04AAS1 (NC_011126), HV: Hydrogenivirga sp. 128-5-R1-1 (NZ_ABHJ00000000), MS: Metallosphaera sedula (NC_009440), SA: Sulfolobus acidocaldarius (NC_007181), ST: S. tokodaii (NC_003106) and SS: S. solfataricus (NC_002754). Percentage of amino-acid similarity is indicated. Blue triangles represent inversion in the gene order.
The predicted A. ferrooxidans heterodisulfide reductase complex has additional features that are consistent with its proposed role in RISCs oxidation. Sulfur oxidation in a variety of different A. ferrooxidans strains has been shown to require glutathione [67, 68] and requires a neutral pH optimum [67], suggesting a cytoplasmic activity in agreement with the predicted localization of the heterodisulfide reductase catalytic site (Figure 2). Second, non-heme iron and labile sulfur have been shown to be present in a sulfur oxidizing enzyme preparation of A. ferrooxidans [67] and iron-sulfur clusters are predicted to be present in the A. ferrooxidans HdrB and C subunits. Third, sulfur oxidation has been shown to be inhibited by HQNO in A. ferrooxidans [69], in agreement with the proposal that the quinone pool is the physiological electron acceptor (Figure 2). Fourth, the actual substrate of the sulfur oxidizing enzyme in A. ferrooxidans is thought not to be elemental sulfur, which has poor water solubility and cannot enter the cell, but rather sulfane sulfur of GSnH species (n>1) and most likely GSSH [67, 68]. Sulfane sulfate would thus provide the necessary disulfide bond X-S-S-X to serve as a substrate for the predicted catalytic activity of the A. ferrooxidans heterodisulfide reductase (Figure 2). Conserved within the heterodisufide reductase gene cluster, between hdrA and hdrC2, is a putative gene of unknown function (AFE_2552) whose product is predicted to reside in the cytoplasm (Figure 2). Given its conserved gene context, we propose that it is also involved in RISCs oxidation and it is now pinpointed for experimental investigation.
Sulfite
Another step in the sulfur oxidation model that awaits genetic characterization is sulfite oxidation. Being metastable and considered short-lived in mine waste environments, one possibility is that sulfite rapidly oxidizes non-enzymically to sulfate, thiosulfate or glutathione S-sulfonate in the presence of Fe(III) [70, 71] or sulfur [72]. However, the involvement of a protein catalyzing this reaction is more likely since a sulfite oxidase activity was purified from three different strains of A. ferrooxidans, namely TM [73], ATCC13661 [74] and AP19-3 [75, 76]
Genes coding for known periplasmic enzymes involved in the direct oxidation of sulfite during sulfur dissimilatory metabolism (sorAB or soxCD [77]) have not been detected in the A. ferrooxidans genome. In our model (Figure 2), sulfite is hypothesized to be produced in the cytoplasm by heterodisulfide reductase. Therefore, subsequent oxidation of sulfite is likely to occur in this cellular location and therefore would not be expected to proceed via the classical periplasmic Sor or Sox. One possibility for the cytoplasmic activity is that sulfite is converted to adenosine-5'-phosphosulfate (APS) via the well characterized APS reductase complex encoded by aprBA [78–80]. However, the genome contains no candidates with significant similarity to aprBA, although it does have a predicted sat (AFE_0539) which, in other microorganisms, encodes an ATP sulfurylase responsible for the second step in this pathway. The A. ferrooxidans Sat shares 44% identity and 60% similarity with both domains of the bifunctional SAT/APS kinase from Aquifex aeolicus that catalyzes the production of ATP and sulfate from APS and pyrophosphate [81, 82]. If Sat is indeed catalyzing APS to sulfate (Figure 2), an enzyme catalyzing the oxidation of sulfite to APS is required. This missing function could be accomplished by the conserved hypothetical gene embedded in the hdr locus of sulfur oxidizers (Figure 3). The concordance of gene occurrence and organization between A. aeolicus, Hydrogenobaculum sp. Y04AAS1 the Sulfolobales and A. ferrooxidans including 1) the hdr locus with a gene of unknown function 2) sat and 3) a lack of aprBA, strongly suggests that these microorganisms have a novel sulfur oxidation pathway.
Our data agree with the model proposed for S0 oxidation in M. sedula, including a heterodisulfide reductase (hdr), a tetrathionate hydrolase (tetH), a terminal oxidase complex based on both quinol oxidase (soxCL) and aa3-type cytochrome oxidase (soxAB) components [50]. In S. metallicus, a gene (sor), encoding sulfur oxygenase reductase, which is absent in A. ferrooxidans, is the dominant transcript in sulfur-grown cells and is therefore proposed to be involved in sulfur oxidation [51]. In conclusion, the RISCs oxidation pathways of acidophiles are not only different from those of neutrophilic sulfur oxidizers [17, 18] but also appear to be different among the acidophilic sulfur-oxidizers including between members of the Acidithiobacillus genus [83]. In that sense, the conservation of the hdr locus in different acidophilic sulfur oxidizers, archaea and eubacteria, is noteworthy and merits further investigation.
Additional Discussion
Genomic and transcriptomic (microarrays and real-time quantitative PCR) studies of iron and sulfur energetic metabolism of A. ferrooxidans, not only confirm previous data, but elaborate on the complexity of these pathways. Both iron and RISCs respiratory chains are branched and redundant [2, 11, 13, 14] (Figures 1 and 2). This provides A. ferrooxidans with a flexible respiratory system that may allow it to adapt efficiently to environmental changes by modulating gene expression according to the growth conditions (substrate, oxygen concentration, growth phase, etc.). Another way to adapt efficiently to a change in the growth conditions is by modifying the association of complexes as suggested recently for iron respiration in A. ferrooxidans [15]. In the case of the cytochrome c oxidase complex of Dictyostelium discoideum, the oxygen concentration induces a switch between two interchangeable subunit isoforms of the cytochrome c oxidase [84–86]. This switch has been shown to be due to transcriptional regulation and also to different stabilities of the two subunits toward oxygen [86].
Both iron and RISCs oxidation pathways involve outer membrane, periplasmic and inner membrane components (Figures 1 and 2). According to our hypothesis, several super-complexes spanning the outer and/or the inner membranes are expected to conduct either the electrons to the oxygen, or the sulfane-sulfur to the catalytic side of the herodisulfide reductase, from pyrite (FeS2), which is a natural substrate of A. ferrooxidans. While such a super-complex has been isolated recently for iron oxidation [15], no biochemical data are available until now to substantiate the existence of a sulfane-sulfur transfer supramolecular structure. While a sulfurtransferase complex encoded by the hdr locus is likely to be involved in the transfer of the proposed sulfane sulfur to the heterodisulfide reductase from the inner membrane to the cytoplasm (Figure 2), expression data suggests no obvious upregulated outer membrane protein as proposed by Rohwerder and Sand [68] to allow it to cross the cell wall. We propose also the existence of a cytoplasmic super-complex catalyzing both the oxidation of sulfane-sulfur to sulfite and of sulfite to APS, preventing the accumulation of sulfite in the cytoplasm (Figure 2). While the existence of such a complex has not been demonstrated in A. ferrooxidans, a thiosulfate-oxidizing system oxidizing hydrogen sulfide, thiosulfate, sulfur and sulfite directly to sulfate without the presence of free intermediates has been evidenced in Paracoccus versutus and Paracoccus pantotrophus ([17, 87] and references therein). Moreover, the methanogenic and sulfate reducing archaea heterodisulfide reductase forms a tight complex with the hydrogenase, which catalyzes its reduction with H2 ([56, 57] and references therein). Such supramolecular structures will allow (1) stabilization of the different components (2) electron, or sulfane sulfur, channeling leading to more efficient transfer, and (3) diffusion avoidance preventing toxic compound leakage.
Conclusion
-
Bioinformatic analysis coupled with gene transcript profiling extends our understanding of the iron and reduced inorganic sulfur compounds oxidation pathways in A. ferrooxidans.
-
Novel genes predicted to be involved in iron oxidation (Figure 1) include those potentially encoding: i) heme biosynthesis and insertion into the terminal electron acceptor cytochrome aa3 oxidase used in the downhill flow of electrons from Fe(II) to reduce oxygen to water, ii) subunits of the Fo ATP synthetase that may be an adaptation to extremely low pHs encountered during iron oxidation, iii) a copper-containing protein that may be involved in electron transfer or in copper insertion, iv) two subunits and one predicted accessory protein of the NADH complex that may promote the flow of electrons from the quinone pool to the NADH complex during uphill electron flow and v) two potential regulators of alternate electron pathways predicted to respond to iron and the status of the quinone pool respectively.
-
Novel genes predicted to be involved in RISCs oxidation (Figure 2) include those potentially encoding: i) three sulfurtransferases, ii) a heterodisulfide reductase complex, iii) an ATP sulfurylase and iv) a NADH complex accessory protein that may promote uphill electron flow from the quinone pool to the NADH complex during RISCs oxidation.
-
The models (Figures 1 and 2) provide unified and coherent descriptions of iron and RISCs oxidation and suggests mechanisms for their regulation within the type strain, eliminating previous confusion caused by models built from analyses of multiple and divergent strains of this microorganism.
-
The identification of differentially expressed genes of unknown function predicted to be involved in iron and RISCs oxidation direct the experimental biologist to future research.
Methods
Strains and culture conditions
A. ferrooxidans type strain ATCC 23270 was obtained from the American Type Culture Collection. A. ferrooxidans was grown at 30°C under oxic conditions with 200 rpm agitation in (i) Fe(II)-medium (62 mM FeSO4-7H2O in basal salts solution ((NH4)2SO4: 0.4 g. L-1, K2HPO4: 0.4 g. L-1, MgSO4-7H2O: 0.4 g. L-1 adjusted to pH 1.6 with H2SO4 or (ii) S0-medium (1% (w/v) elemental sulfur in basal salts solution ((NH4)2SO4: 0.4 g. L-1, K2HPO4: 0.4 g. L-1, MgSO4-7H2O: 0.4 g. L-1 adjusted to pH 3.5 with H2SO4). When reaching mid-logarithmic phase cells were harvested by centrifugation at 4°C and washed with basal salt solution (pH adjusted). To remove iron traces or sulfur particles before further treatment several cycles of washing and 1 minute low speed spin-centrifugations were performed. Clean cell pellets were frozen in liquid nitrogen and stored at -80°C until used for RNA extraction.
RNA isolation
Total RNA was extracted using a modified acid-phenol extraction method [88], including a preliminary TRIZOL® reagent (Invitrogen) extraction step. This RNA was used directly for cDNA synthesis and labeling. For real-time PCR, RNAs were additionally purified with the High Pure RNA isolation kit (Roche Applied Biosystem) and treated twice with DNAse I (Roche Applied Biosystem). The lack of DNA contamination was checked by PCR on each RNA sample.
Microarray production
A. ferrooxidans ATCC 23270 oligonucleotide microarrays were designed and produced in-house. Internal 50-mers targeting each of the 3037 putative ORFs predicted in the genomic sequence of A. ferrooxidans ATCC 23270 (TIGR release September 2003; 2.98 Mb) were selected using the Oligoarray software (Version 1.0), synthesized by MWG Biotech and spotted in duplicate on Gamma Amino Propyl Silane Corning UltraGAPS slides, according to the manufacturer's instructions, with a ChipWriterProarrayer (Bio-Rad, 1000 Alfred Nobel Drive Hercules, CA) at the Marseille-Nice Genopole (France). A special set of control oligonucleotides (negative, positive, tiled and antisense sequences) was included to evaluate probe specificity and adjust the hybridization conditions. 50-mer oligonucleotides corresponding to the CDS of the gfp gene from Aequorea sp. (L29345), the rad9 gene form Saccharomyces cereviciae (M26049) and the idi2 gene from human (AK303669) were used as negative controls. The array design and spotting protocol were deposited in ArrayExpress database [89] under the accession codes A-MEXP-1478 and A-MEXP-1479.
Microarray experimental design
This experiment consisted of 8 independent hybridizations using total RNA obtained from 2 different iron- (FeIIA and FeIIB) and 2 different sulfur-replicate cultures (S°A and S°B). Each RNA sample was labeled once with each dye (e.g. S0 A_Cy®5 and S0 A_Cy®3) and hybridized against a reciprocally labeled cDNA sample (e.g. FeIIA_Cy®3 and FeIIA_Cy®5). The cDNAs obtained for each biological replicate (e.i. A or B) were cohybridized against two different types of microarrays obtained in different spotting campaigns (Oligoarray v.1 or Oligoarray v.2). More details of the experimental design are available in the ArrayExpress database under the accession code E-MEXP-1990.
Labeling and hybridization
The ChipShot™ Labeling Clean-Up System (Promega-Corning) was used to generate fluorescently labeled cDNA via direct incorporation of Cy®3 and Cy®5-labeled nucleotides (Amersham Biosciences). The reverse transcription reaction was performed in the presence of 5 μg total RNA and random hexamers according to manufacturer's recommendations. Two independent cDNA preparations were labeled once with each dye (reverse dye labeling) to account for sampling differences, biases in dye coupling or emission efficiency of Cy®-dyes. Labeled cDNA was purified from contaminating fluorescent dNTPs and degraded RNA using the ChipShot Labeling Clean-Up System (Promega-Corning). Dye incorporation efficiency was determined by absorbance readings at 260, 550 and 650 nm and the frequency of incorporation (FOI, pmol of dye incorporated per ng of cDNA) was calculated according to Promega's instructions. Optimally labeled samples were combined, vacuum-dried and resuspended to a final volume of 40–50 μL in Corning Pronto! Universal Hybridization Buffer. The combined denatured target cDNA samples (95°C for 5 min) were hybridized to the spotted slides for 14 h at 42°C in the Corning hybridization chamber. Following hybridization, slides were washed in serial dilutions of Corning Wash Buffers as recommended by the manufacturer and dried by centrifugation at 1200 × g for 2 min.
Image acquisition and data analysis
Microarrays were scanned for the Cy®3 and Cy®5 fluorescent signals using the ScanArray 5000 Microarray Analysis System (PerkinElmer Life Sciences, Inc.). Scans stored as 16-bit TIFF (Tagged Information File Format) image files and then analyzed with the image quantification software package GenePix Pro 6.0 (Axon Instruments, Inc.). Saturated and low-quality spots (spots smaller than 60 μm or bigger than 160 μm in diameter, sub-circular in shape and/or exhibiting uneven fluorescence distribution) were flagged and filtered out. Local background was subtracted from the recorded spot intensities and median values for each spot were Log transformed (log2) and normalized by average intensity of each slide to account for any difference in total intensity between the scanned images using the Acuity microarray analysis software (Version 4.0, Axon Instrument Inc.). The processed Fe(II) and S0 signal intensities for each spot were used for calculating the expression ratio Fe(II)/S0. Twenty four replicate ratio values per gene, resulting from direct and reverse labeling, replicate spots and replicate experiments, were used for further statistical analysis of the data. The statistical significance of differential expression in Fe(II) or S0 grown cells, was assessed using Acuity package. The raw data, as well as the processed (filtered and Median normalized) data, for all hybridizations was submitted to the ArrayExpress database and is available under the accession code E-MEXP-1990. A 1.5 fold deviations from the 1:1 hybridization ratio was taken as indicative of differential gene expression in the two growth conditions analyzed. Hierarchical cluster analysis (Pearson correlation, average linkage) was performed using Genesis software suit [90]. Functional annotations were retrieved from Valdés et al. [8].
Real-time quantitative PCR
The relative abundances of a set of differentially expressed genes and a set of invariant genes, according to the microarray results obtained, were determined in Fe(II)- and S0-grown cells by real-time PCR. Specific primers for the genes of interest amplifying average products of 300 bp with about 50 GC% and about 55°C Tm were designed (Additional file 3). Equal amounts of A. ferrooxidans DNAseI-treated total RNA were retro-transcribed from these primers with the Superscript II™ RNase (Invitrogen Life Technologies) at 42°C for 50 min, and then treated at 70°C for 15 min to inactivate the enzyme. The real-time PCR quantifications were performed on the cDNA obtained, using the Light-Cycler instrument and the LightCycler Fast Start DNA master (plus) SYBR Green I PCR kit (Roche Applied Biosystems) with external standards as described in Roche Molecular Biochemicals technical note no. LC 11/2000. The real-time PCR experiments were performed twice, with both independent total RNA and cDNA preparations by the comparative threshold cycle method. The calculated threshold cycle (Ct) for each gene was normalized to Ct of the rrs gene.
Bioinformatic analysis
The sequence and annotation of the complete A. ferrooxidans strain ATCC 23270 genome was retrieved from GenBank/EMBL/DDBJ (CP001219). Functional assignments for hypothetical and predicted genes of interest were performed manually on a gene-by-gene basis. Amino acid sequences for these genes were used to query the following databases: National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG and InterPro. Comparative genomic analyses were performed using the Comprehensive Microbial Resource [91], Microbesonline [92] and String [93].
References
Ingledew WJ: Thiobacillus ferrooxidans. The bioenergetics of an acidophilic chemolithotroph. Biochim Biophys Acta. 1982, 683: 89-117.
Holmes DS, Bonnefoy V: Genetic and bioinformatic insights into iron and sulfur oxidation mechanisms of bioleaching organisms. Biomining. Edited by: Rawlings DE, Johnson DB. 2007, Berlin Heidelberg: Springer-Verlag, 281-307.
Appia-Ayme C, Bengrine A, Cavazza C, Giudici-Orticoni MT, Bruschi M, Chippaux M, Bonnefoy V: Characterization and expression of the co-transcribed cyc1 and cyc2 genes encoding the cytochrome c4 (c552) and a high-molecular-mass cytochrome c from Thiobacillus ferrooxidans ATCC 33020. FEMS Microbiol Lett. 1998, 167: 171-177.
Bengrine A, Guiliani N, Appia-Ayme C, Jedlicki E, Holmes DS, Chippaux M, Bonnefoy V: Sequence and expression of the rusticyanin structural gene from Thiobacillus ferrooxidans ATCC33020 strain. Biochim Biophys Acta. 1998, 1443: 99-112.
Appia-Ayme C, Guiliani N, Ratouchniak J, Bonnefoy V: Characterization of an operon encoding two c-type cytochromes, an aa3-type cytochrome oxidase, and rusticyanin in Thiobacillus ferrooxidans ATCC 33020. Appl Environ Microbiol. 1999, 65: 4781-4787.
Yarzabal A, Brasseur G, Ratouchniak J, Lund K, Lemesle-Meunier D, DeMoss JA, Bonnefoy V: The high-molecular-weight cytochrome c Cyc2 of Acidithiobacillus ferrooxidans is an outer membrane protein. J Bacteriol. 2002, 184: 313-317. 10.1128/JB.184.1.313-317.2002.
Yarzabal A, Appia-Ayme C, Ratouchniak J, Bonnefoy V: Regulation of the expression of the Acidithiobacillus ferrooxidans rus operon encoding two cytochromes c, a cytochrome oxidase and rusticyanin. Microbiology. 2004, 150: 2113-2123. 10.1099/mic.0.26966-0.
Valdés J, Pedroso I, Quatrini R, Dodson RJ, Tettelin H, Blake R, Eisen JA, Holmes DS: Acidithiobacillus ferrooxidans metabolism: from genome sequence to industrial applications. BMC Genomics. 2008, 9: 597-10.1186/1471-2164-9-597.
Elbehti A, Brasseur G, Lemesle-Meunier D: First evidence for existence of an uphill electron transfer through the b c1 and NADH-Q oxidoreductase complexes of the acidophilic obligate chemolithotrophic ferrous ion-oxidizing bacterium Thiobacillus ferrooxidans. J Bacteriol. 2000, 182: 3602-3606. 10.1128/JB.182.12.3602-3606.2000.
Brasseur G, Bruscella P, Bonnefoy V, Lemesle-Meunier D: The bc1 complex of the iron-grown acidophilic chemolithotrophic bacterium Acidithiobacillus ferrooxidans functions in the reverse but not in the forward direction. Is there a second bc1 complex?. Biochim Biophys Acta. 2002, 1555: 37-43. 10.1016/S0005-2728(02)00251-7.
Brasseur G, Levican G, Bonnefoy V, Holmes D, Jedlicki E, Lemesle-Meunier D: Apparent redundancy of electron transfer pathways via bc1 complexes and terminal oxidases in the extremophilic chemolithoautotrophic Acidithiobacillus ferrooxidans. Biochim Biophys Acta. 2004, 1656: 114-126. 10.1016/j.bbabio.2004.02.008.
Levican G, Bruscella P, Guacunano M, Inostroza C, Bonnefoy V, Holmes DS, Jedlicki E: Characterization of the petI and res operons of Acidithiobacillus ferrooxidans. J Bacteriol. 2002, 184: 1498-1501. 10.1128/JB.184.5.1498-1501.2002.
Bruscella P, Appia-Ayme C, Levican G, Ratouchniak J, Jedlicki E, Holmes DS, Bonnefoy V: Differential expression of two bc1 complexes in the strict acidophilic chemolithoautotrophic bacterium Acidithiobacillus ferrooxidans suggests a model for their respective roles in iron or sulfur oxidation. Microbiology. 2007, 153: 102-110. 10.1099/mic.0.2006/000067-0.
Quatrini R, Appia-Ayme C, Denis Y, Ratouchniak J, Veloso F, Valdes J, Lefimil C, Silver S, Roberto F, Orellana O, Denizot F, Jedlicki E, Holmes DS, Bonnefoy V: Insights into the iron and sulfur energetic metabolism of Acidithiobacillus ferrooxidans by microarray transcriptome profiling. Hydrometallurgy. 2006, 83: 263-272. 10.1016/j.hydromet.2006.03.030.
Castelle C, Guiral M, Malarte G, Ledgham F, Leroy G, Brugna M, Giudici-Orticoni MT: A new Fe-oxidizing/O2-reducing supercomplex spanning both inner and outer membranes, isolated from the extreme acidophile Acidithiobacillus ferrooxidans. J Biol Chem. 2008, 283: 25803-25811. 10.1074/jbc.M802496200.
Taha TM, Kanao T, Takeuchi F, Sugio T: Reconstitution of iron oxidase from sulfur-grown Acidithiobacillus ferrooxidans. Appl Environ Microbiol. 2008, 74: 6808-6810. 10.1128/AEM.00787-08.
Friedrich CG, Bardischewsky F, Rother D, Quentmeier A, Fischer J: Prokaryotic sulfur oxidation. Curr Opin Microbiol. 2005, 8: 253-259. 10.1016/j.mib.2005.04.005.
Frigaard NU, Dahl C: Sulfur metabolism in phototrophic sulfur bacteria. Adv Microb Physiol. 2009, 54: 103-200. 10.1016/S0065-2911(08)00002-7.
Urich T, Bandeiras TM, Leal SS, Rachel R, Albrecht T, Zimmermann P, Scholz C, Teixeira M, Gomes CM, Kletzin A: The sulphur oxygenase reductase from Acidianus ambivalens is a multimeric protein containing a low-potential mononuclear non-haem iron centre. Biochem J. 2004, 381: 137-146. 10.1042/BJ20040003.
Harrison AP: Genomic and physiological diversity amongst strains of Thiobacillus ferrooxidans, and genomic comparison with Thiobacillus thiooxidans. Arch Microbiol. 1982, 131: 68-76. 10.1007/BF00451501.
Karavaiko GI, Turova TP, Kondrat'eva TF, Lysenko AM, Kolganova TV, Ageeva SN, Muntyan LN, Pivovarova TA: Phylogenetic heterogeneity of the species Acidithiobacillus ferrooxidans. Int J Syst Evol Microbiol. 2003, 53: 113-119. 10.1099/ijs.0.02319-0.
Peng H, Yang Y, Li X, Qiu G, Liu X, Huang J, Hu Y: Structure analysis of 16S rDNA sequences from strains of Acidithiobacillus ferrooxidans. J Biochem Mol Biol. 2006, 39: 178-182.
Ni YQ, Yang Y, Bao JT, He KY, Li HY: Inter- and intraspecific genomic variability of the 16S-23S intergenic spacer regions (ISR) in representatives of Acidithiobacillus thiooxidans and Acidithiobacillus ferrooxidans. FEMS Microbiol Lett. 2007, 270: 58-66. 10.1111/j.1574-6968.2007.00660.x.
Bouchal P, Zdrahal Z, Helanova S, Janiczek O, Hallberg KB, Mandl M: Proteomic and bioinformatic analysis of iron- and sulfur-oxidizing Acidithiobacillus ferrooxidans using immobilized pH gradients and mass spectrometry. Proteomics. 2006, 15: 4278-4285. 10.1002/pmic.200500719.
Ramirez P, Guiliani N, Valenzuela L, Beard S, Jerez CA: Differential Protein Expression during Growth of Acidithiobacillus ferrooxidans on Ferrous Iron, Sulfur Compounds, or Metal Sulfides. Appl Environ Microbiol. 2004, 70: 4491-4498. 10.1128/AEM.70.8.4491-4498.2004.
Wakai S, Tsujita M, Kikumoto M, Manchur MA, Kanao T, Kamimura K: Purification and characterization of sulfide:quinone oxidoreductase from an acidophilic iron-oxidizing bacterium, Acidithiobacillus ferrooxidans. Biosci Biotechnol Biochem. 2007, 71: 2735-2742. 10.1271/bbb.70332.
Kanao T, Kamimura K, Sugio T: Identification of a gene encoding a tetrathionate hydrolase in Acidithiobacillus ferrooxidans. J Biotechnol. 2007, 132: 16-22. 10.1016/j.jbiotec.2007.08.030.
Ramirez P, Toledo H, Guiliani N, Jerez CA: An exported rhodanese-like protein is induced during growth of Acidithiobacillus ferrooxidans in metal sulfides and different sulfur compounds. Appl Environ Microbiol. 2002, 68: 1837-1845. 10.1128/AEM.68.4.1837-1845.2002.
Valenzuela L, Beard S, Guiliani N, Jerez CA: Differencial expression proteomics of Acidithiobacillus ferrooxidans grown in different oxidizable substrates: study of the sulfate/thiosulfate/molybdate binding proteins. Proceedings of the 16th International Biohydrometallurgy Symposium: 25–29 September 2005; Cape Town, South Africa. Edited by: Harrison, STL, Rawlings DE, Petersen J. 2005, Compress Cape Town, South Africa, 773-78.
Castelle C, Guiral M, Giudici-Orticoni M-T: A new acid-stable, Fe-oxidizing/O2-reducing supercomplex spanning both inner and outer membranes, isolated from the extremophile Acidithiobacillus ferrooxidans. Biohydrometallurgy: from the single cell to the environment. Edited by: Schippers A, Sand W, Glombitza F, Willscher S. 2007, Stafa-Zurich: Trans Tech Publications Ltd, 572-572.
Arnesano F, Banci L, Bertini I, Martinelli M: Ortholog search of proteins involved in copper delivery to cytochrome c oxidase and functional analysis of paralogs and gene neighbors by genomic context. J Proteome Res. 2005, 4: 63-70. 10.1021/pr049862f.
Thony-Meyer L: Biogenesis of respiratory cytochromes in bacteria. Microbiol Mol Biol Rev. 1997, 61: 337-376.
Reniere ML, Torres VJ, Skaar EP: Intracellular metalloporphyrin metabolism in Staphylococcus aureus. Biometals. 2007, 20: 333-345. 10.1007/s10534-006-9032-0.
Kiley PJ, Beinert H: The role of Fe-S proteins in sensing and regulation in bacteria. Curr Opin Microbiol. 2003, 6: 181-185. 10.1016/S1369-5274(03)00039-0.
Elsen S, Swem LR, Swem DL, Bauer CE: RegB/RegA, a highly conserved redox-responding global two-component regulatory system. Microbiol Mol Biol Rev. 2004, 68: 263-279. 10.1128/MMBR.68.2.263-279.2004.
Wu J, Bauer CE: RegB/RegA, a global redox-responding two-component system. Adv Exp Med Biol. 2008, 631: 131-148. full_text.
Abdrakhmanova A, Zwicker K, Kerscher S, Zickermann V, Brandt U: Tight binding of NADPH to the 39-kDa subunit of complex I is not required for catalytic activity but stabilizes the multiprotein complex. Biochim Biophys Acta. 2006, 1757: 1676-1682. 10.1016/j.bbabio.2006.09.003.
Zickermann V, Zwicker K, Tocilescu MA, Kerscher S, Brandt U: Characterization of a subcomplex of mitochondrial NADH:ubiquinone oxidoreductase (complex I) lacking the flavoprotein part of the N-module. Biochim Biophys Acta. 2007, 1767: 393-400. 10.1016/j.bbabio.2007.03.005.
Persson B, Kallberg Y, Oppermann U, Jornvall H: Coenzyme-based functional assignments of short-chain dehydrogenases/reductases (SDRs). Chem Biol Interact. 2003, 143–144: 271-278. 10.1016/S0009-2797(02)00223-5.
Kervinen M, Patsi J, Finel M, Hassinen IE: A pair of membrane-embedded acidic residues in the NuoK subunit of Escherichia coli NDH-1, a counterpart of the ND4L subunit of the mitochondrial complex I, are required for high ubiquinone reductase activity. Biochemistry. 2004, 43: 773-781. 10.1021/bi0355903.
Kao MC, Nakamaru-Ogiso E, Matsuno-Yagi A, Yagi T: Characterization of the membrane domain subunit NuoK (ND4L) of the NADH-quinone oxidoreductase from Escherichia coli. Biochemistry. 2005, 44: 9545-9554. 10.1021/bi050708w.
Brandt U: Energy converting NADH:quinone oxidoreductase (complex I). Annu Rev Biochem. 2006, 75: 69-92. 10.1146/annurev.biochem.75.103004.142539.
Nakano T, Ikegami T, Suzuki T, Yoshida M, Akutsu H: A new solution structure of ATP synthase subunit c from thermophilic Bacillus PS3, suggesting a local conformational change for H+-translocation. J Mol Biol. 2006, 358: 132-144. 10.1016/j.jmb.2006.01.011.
Nicolle JLC, Simmons S, Bathe S, Norris PR: Ferrous iron oxidation and rusticyanin in halotolerant, acidophilic "Thiobacillus prosperus ". Microbiology. 2009, 155: 1302-1309. 10.1099/mic.0.023192-0.
Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF: Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004, 428: 37-43. 10.1038/nature02340.
Singer SW, Chan CS, Zemla A, VerBerkmoes NC, Hwang M, Hettich RL, Banfield JF, Thelen MP: Characterization of cytochrome 579, an unusual cytochrome isolated from an iron-oxidizing microbial community. Appl Environ Microbiol. 2008, 74: 4454-4462. 10.1128/AEM.02799-07.
Jeans C, Singer SW, Chan CS, Verberkmoes NC, Shah M, Hettich RL, Banfield JF, Thelen MP: Cytochrome 572 is a conspicuous membrane protein with iron oxidation activity purified directly from a natural acidophilic microbial community. ISME J. 2008, 2: 542-550. 10.1038/ismej.2008.17.
Dopson M, Baker-Austin C, Bond PL: Analysis of differential protein expression during growth states of Ferroplasma strains and insights into electron transport for iron oxidation. Microbiology. 2005, 151: 4127-4137. 10.1099/mic.0.28362-0.
Auernik KS, Maezato Y, Blum PH, Kelly RM: The genome sequence of the metal-mobilizing, extremely thermoacidophilic archaeon Metallosphaera sedula provides insights into bioleaching-associated metabolism. Appl Environ Microbiol. 2008, 74: 682-692. 10.1128/AEM.02019-07.
Auernik KS, Kelly RM: Identification of components of electron transport chains in the extremely thermoacidophilic crenarchaeon Metallosphaera sedula through iron and sulfur compound oxidation transcriptomes. Appl Environ Microbiol. 2008, 74: 7723-7732. 10.1128/AEM.01545-08.
Bathe S, Norris PR: Ferrous iron- and sulfur-induced genes in Sulfolobus metallicus. Appl Environ Microbiol. 2007, 73: 2491-2497. 10.1128/AEM.02589-06.
Wakai S, Kikumoto M, Kanao T, Kamimura K: Involvement of sulfide:quinone oxidoreductase in sulfur oxidation of an acidophilic iron-oxidizing bacterium, Acidithiobacillus ferrooxidans NASF-1. Biosci Biotechnol Biochem. 2004, 68: 2519-2528. 10.1271/bbb.68.2519.
Muller FH, Bandeiras TM, Urich T, Teixeira M, Gomes CM, Kletzin A: Coupling of the pathway of sulphur oxidation to dioxygen reduction: characterization of a novel membrane-bound thiosulphate:quinone oxidoreductase. Mol Microbiol. 2004, 53: 1147-1160. 10.1111/j.1365-2958.2004.04193.x.
Janiczek O, Zemanova J, Mandl M: Purification and some properties of thiosulfate dehydrogenase from Acidithiobacillus ferrooxidans. Prep Biochem Biotechnol. 2007, 37: 101-111. 10.1080/10826060701199015.
Pronk JT, Meijer WM, Hazeu W, van Dijken JP, Bos P, Kuenen JG: Growth of Thiobacillus ferrooxidans on Formic Acid. Appl Environ Microbiol. 1991, 57: 2057-2062.
Hedderich R, Hamann N, Bennati M: Heterodisulfide reductase from methanogenic archaea: a new catalytic role for an iron-sulfur cluster. Biol Chem. 2005, 386: 961-970. 10.1515/BC.2005.112.
Mander GJ, Pierik AJ, Huber H, Hedderich R: Two distinct heterodisulfide reductase-like enzymes in the sulfate-reducing archaeon Archaeoglobus profundus. Eur J Biochem. 2004, 271: 1106-1116. 10.1111/j.1432-1033.2004.04013.x.
Hamann N, Mander GJ, Shokes JE, Scott RA, Bennati M, Hedderich R: A cysteine-rich CCG domain contains a novel [4Fe-4S] cluster binding motif as deduced from studies with subunit B of heterodisulfide reductase from Methanothermobacter marburgensis. Biochemistry. 2007, 46: 12875-12885. 10.1021/bi700679u.
Duin EC, Madadi-Kahkesh S, Hedderich R, Clay MD, Johnson MK: Heterodisulfide reductase from Methanothermobacter marburgensis contains an active-site [4Fe-4S] cluster that is directly involved in mediating heterodisulfide reduction. FEBS Lett. 2002, 512: 263-268. 10.1016/S0014-5793(02)02281-0.
Acosta M, Beard S, Ponce J, Vera M, Mobarec JC, Jerez CA: Identification of putative sulfurtransferase genes in the extremophilic Acidithiobacillus ferrooxidans ATCC23270 genome: structural and functional characterization of the proteins. OMICS. 2005, 9: 13-29. 10.1089/omi.2005.9.13.
Klimmek O, Kreis V, Klein C, Simon J, Wittershagen A, Kroger A: The function of the periplasmic Sud protein in polysulfide respiration of Wolinella succinogenes. Eur J Biochem. 1998, 253: 263-269. 10.1046/j.1432-1327.1998.2530263.x.
Klimmek O, Stein T, Pisa R, Simon J, Kroger A: The single cysteine residue of the Sud protein is required for its function as a polysulfide-sulfur transferase in Wolinella succinogenes. Eur J Biochem. 1999, 263: 79-84. 10.1046/j.1432-1327.1999.00461.x.
Palenchar PM, Buck CJ, Cheng H, Larson TJ, Mueller EG: Evidence that ThiI, an enzyme shared between thiamin and 4-thiouridine biosynthesis, may be a sulfurtransferase that proceeds through a persulfide intermediate. J Biol Chem. 2000, 275: 8283-8286. 10.1074/jbc.275.12.8283.
Mueller EG, Palenchar PM, Buck CJ: The role of the cysteine residues of ThiI in the generation of 4-thiouridine in tRNA. J Biol Chem. 2001, 276: 33588-33595. 10.1074/jbc.M104067200.
Ikeuchi Y, Shigi N, Kato J, Nishimura A, Suzuki T: Mechanistic insights into sulfur relay by multiple sulfur mediators involved in thiouridine biosynthesis at tRNA wobble positions. Mol Cell. 2006, 21: 97-108. 10.1016/j.molcel.2005.11.001.
Dahl C, Schulte A, Stockdreher Y, Hong C, Grimm F, Sander J, Kim R, Kim SH, Shin DH: Structural and Molecular Genetic Insight into a Widespread Sulfur Oxidation Pathway. J Mol Biol. 2008, 384: 1287-1300. 10.1016/j.jmb.2008.10.016.
Silver M, Lundgren DG: Sulfur-oxidizing enzyme of Ferrobacillus ferrooxidans (Thiobacillus ferrooxidans). Can J Biochem. 1968, 46: 457-461.
Rohwerder T, Sand W: The sulfane sulfur of persulfides is the actual substrate of the sulfur-oxidizing enzymes from Acidithiobacillus and Acidiphilium spp. Microbiology. 2003, 149: 1699-1710. 10.1099/mic.0.26212-0.
Corbett CM, Ingledew WJ: Is Fe3+/2+ cycling an intermediate in sulphur oxidation by Fe2+-grown Thiobacillus ferrooxidans. FEMS Microbiol Lett. 1987, 41: 1-6. 10.1111/j.1574-6968.1987.tb02131.x.
Sugio T, Noguchi M, Tano T: Detoxification of sulfite produced during the oxidation of elemental sulfur by Thiobacillus ferrooxidans. Agric Biol Chem. 1987, 51: 1431-1433.
Harahuc L, Suzuki I: Sulfite oxidation by iron-grown cells of Thiobacillus ferrooxidans at pH 3 possibly involves free radicals, iron, and cytochrome oxidase. Can J Microbiol. 2001, 47: 424-430. 10.1139/cjm-47-5-424.
Suzuki I, Chan CW, Takeuchi TL: Oxidation of Elemental Sulfur to Sulfite by Thiobacillus thiooxidans Cells. Appl Environ Microbiol. 1992, 58: 3767-3769.
Vestal JR, Lundgren DG: The sulfite oxidase of Thiobacillus ferrooxidans (Ferrobacillus ferrooxidans). Can J Biochem. 1971, 49: 1125-1130.
Suzuki I: Sulfite:cytochrome c oxidoreductase of Thiobacilli. Methods Enzymol. 1994, 243: 447-454.
Sugio T, Katagiri T, Moriyama M, Zhen YL, Inagaki K, Tano T: Existence of a new type of sulfite oxidase which utilizes ferric ions as an electron acceptor in Thiobacillus ferrooxidans. Appl Environ Microbiol. 1988, 54: 153-157.
Sugio T, Hirose T, Ye LZ, Tano T: Purification and some properties of sulfite:ferric ion oxidoreductase from Thiobacillus ferrooxidans. J Bacteriol. 1992, 174: 4189-4192.
Kappler U, Dahl C: Enzymology and molecular biology of prokaryotic sulfite oxidation. FEMS Microbiol Lett. 2001, 203: 1-9.
Meyer B, Kuever J: Molecular analysis of the distribution and phylogeny of dissimilatory adenosine-5'-phosphosulfate reductase-encoding genes (aprBA) among sulfur-oxidizing prokaryotes. Microbiology. 2007, 153: 3478-3498. 10.1099/mic.0.2007/008250-0.
Meyer B, Kuever J: Homology modeling of dissimilatory APS reductases (AprBA) of sulfur-oxidizing and sulfate-reducing prokaryotes. PLoS ONE. 2008, 3: e1514-10.1371/journal.pone.0001514.
Hipp WM, Pott AS, Thum-Schmitz N, Faath I, Dahl C, Truper HG: Towards the phylogeny of APS reductases and sirohaem sulfite reductases in sulfate-reducing and sulfur-oxidizing prokaryotes. Microbiology. 1997, 143: 2891-2902. 10.1099/00221287-143-9-2891.
Yu Z, Lansdon EB, Segel IH, Fisher AJ: Crystal structure of the bifunctional ATP sulfurylase-APS kinase from the chemolithotrophic thermophile Aquifex aeolicus. J Mol Biol. 2007, 365: 732-743. 10.1016/j.jmb.2006.10.035.
Hanna E, MacRae IJ, Medina DC, Fisher AJ, Segel IH: ATP sulfurylase from the hyperthermophilic chemolithotroph Aquifex aeolicus. Arch Biochem Biophys. 2002, 406: 275-288. 10.1016/S0003-9861(02)00428-9.
Valdés J, Pedroso I, Quatrini R, Holmes DS: Comparative genome analysis of Acidithiobacillus ferrooxidans, A. thiooxidans and A. caldus: insights into their metabolism and ecophysiology. Hydrometallurgy. 2008, 94: 180-184. 10.1016/j.hydromet.2008.05.039.
Bisson R, Schiavo G: Slime mold cytochrome c oxidase. An example of environmental influence on subunit composition of a eukaryotic oxidase. Ann N Y Acad Sci. 1988, 550: 325-336. 10.1111/j.1749-6632.1988.tb35347.x.
Schiavo G, Bisson R: Oxygen influences the subunit structure of cytochrome c oxidase in the slime mold Dictyostelium discoideum. J Biol Chem. 1989, 264: 7129-7134.
Bisson R, Vettore S, Aratri E, Sandona D: Subunit change in cytochrome c oxidase: identification of the oxygen switch in Dictyostelium. Embo J. 1997, 16: 739-749. 10.1093/emboj/16.4.739.
Friedrich CG: Physiology and genetics of sulfur-oxidizing bacteria. Adv Microb Physiol. 1998, 39: 235-289. 10.1016/S0065-2911(08)60018-1.
Aiba H, Adhya S, de Crombrugghe B: Evidence for two functional gal promoters in intact Escherichia coli cells. J Biol Chem. 1981, 256: 11905-11910.
ArrayExpress transcriptomic database. [http://www.ebi.ac.uk/microarray-as/aer/]
Genesis. [http://genome.tugraz.at/genesisclient/genesisclient_description.shtml]
Peterson JD, Umayam LA, Dickinson T, Hickey EK, White O: The Comprehensive Microbial Resource. Nucleic Acids Res. 2001, 29: 123-125. 10.1093/nar/29.1.123.
Alm EJ, Huang KH, Price MN, Koche RP, Keller K, Dubchak IL, Arkin AP: The MicrobesOnline Web site for comparative genomics. Genome Res. 2005, 15: 1015-1022. 10.1101/gr.3844805.
Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, Bork P, von Mering C: STRING 8 – a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, D412-416. 10.1093/nar/gkn760. 37 Database
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology. The gene ontology consortium. Nature Genet. 2000, 25: 25-29. 10.1038/75556.
Acknowledgements
We thank Jeanine Ratouchniak for technical assistance. We acknowledge the Institut de Pharmacologie Moléculaire et Cellulaire (CNRS, Sophia-Antipolis, France) for the oligonucleotides spotting on the slides. Work supported by Fondecyt grants 1050063, 11060164 and 1090451, DI-UNAB, 34-06, DI-UNAB 15-06/I, a Microsoft Sponsored Research Award, Ecos/Conicyt, Geomex and Puces à ADN from the Centre National de la Recherche Scientifique and Biomine European project (sixth PCRD n° NM2.ct.2005.500329). We wish to thank A-L. Reysenbach and the European project partners for their contributions and/or advice to the work presented here.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
DH and EJ conceived the study; CA, RQ, DH and VB designed the experiments; CA and RQ carried out the experiments (each contributed equally); YD assisted in the microarray experiments; CA, RQ, DH and VB interpreted the results and wrote the paper. All authors read and approved the paper.
Raquel Quatrini, Corinne Appia-Ayme contributed equally to this work.
Electronic supplementary material
12864_2009_2278_MOESM1_ESM.xls
Additional file 1: Differentially expressed genes. Genes with a log2 ratio of median greater than 1.5 (corresponding to genes induced more than 2.8 fold) were considered differentially expressed in the two growth conditions. Functional categories are those defined by the TIGR database [94]. (Quatrini, R., C. Appia-Ayme, Y. Denis, E. Jedlicki, D. S. Holmes and V. Bonnefoy, BMC Genomics). (XLS 72 KB)
12864_2009_2278_MOESM2_ESM.pdf
Additional file 2: Alignment of SdrA with Ndu9 subunit of the NADH complex. The predicted classical NADH binding site in SdrA is highlighted in grey and catalytic residues involved in electron transfer in black. NduA9 and SdrA protein sequences ID in the alignment are as following: human (sp|Q16795|), horse (sp|Q5R5S0|), bovine (sp|P34943|), mouse (sp|Q9DC69|), Neurospora crassa (sp|P25284|), A. ferrooxidans SdrA1 (AFE_3108), A. ferrooxidans SdrA2 (AFE_2728). (Quatrini, R., C. Appia-Ayme, Y. Denis, E. Jedlicki, D. S. Holmes and V. Bonnefoy, BMC Genomics). (PDF 21 KB)
12864_2009_2278_MOESM3_ESM.pdf
Additional file 3: Q-PCR primers used in this study. (Quatrini, R., C. Appia-Ayme, Y. Denis, E. Jedlicki, D. S. Holmes and V. Bonnefoy, BMC Genomics). (PDF 67 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Quatrini, R., Appia-Ayme, C., Denis, Y. et al. Extending the models for iron and sulfur oxidation in the extreme Acidophile Acidithiobacillus ferrooxidans. BMC Genomics 10, 394 (2009). https://doi.org/10.1186/1471-2164-10-394
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-10-394