
Abstract
Background
Brazil's Atlantic Forest is a biodiversity hotspot endangered by severe habitat degradation and fragmentation. Habitat fragmentation is expected to reduce dispersal among habitat patches resulting in increased genetic differentiation among populations. Here we examined genetic diversity and differentiation among populations of two Heliconius butterfly species in the northern portion of Brazil's Atlantic Forest to estimate the potential impact of habitat fragmentation on population connectivity in butterflies with home-range behavior.
Results
We generated microsatellite, AFLP and mtDNA sequence data for 136 Heliconius erato specimens from eight collecting locations and 146 H. melpomene specimens from seven locations. Population genetic analyses of the data revealed high levels of genetic diversity in H. erato relative to H. melpomene, widespread genetic differentiation among populations of both species, and no evidence for isolation-by-distance.
Conclusions
These results are consistent with the hypothesis that the extensive habitat fragmentation along Brazil's Atlantic Forest has reduced dispersal of Heliconius butterflies among neighboring habitat patches. The results also lend support to the observation that fine-scale population genetic structure may be common in Heliconius. If such population structure also exists independent of human activity, and has been common over the evolutionary history of Heliconius butterflies, it may have contributed to the evolution of wing pattern diversity in the genus.
Similar content being viewed by others

Background
Landscape structure has a fundamental influence on the distribution of populations, affecting their demography and genetics [1]. Some populations may be found continuously distributed while others are patchily distributed across their range, both of which may ultimately lead to some degree of genetic differentiation. Such geographic patterns of genetic variation reflect both historical processes, such as natural selection, and contemporary gene flow [2].
Gene flow determines the potential for genetic differentiation among populations and for local adaptation and the spread of novel adaptations [3, 4]. In butterflies, as in other organisms, the nature and extent of gene flow is largely dependent on the mobility of individuals. Species with high vagility may disperse over large distances and therefore have extensive gene flow over large areas resulting in more homogeneous populations [5–8], whereas in species with low vagility, the effect of restricted dispersal will be evident at fine spatial scales [9–11]. Furthermore, gene flow may also be affected by a variety of ecological factors such as mating habits, gender-biased dispersal, diet specialization, habitat and population persistence, environmental factors and geographic distance [12].
Extensive studies using molecular markers on butterflies have shown how fragmentation leads to the reduction of gene flow among populations in different habitat patches and increases genetic differentiation among populations [9, 13–15]. In fact, intra- and inter-population genetic variability is generally more affected in small patches of habitat and in small populations [6, 16]. Fragmentation may even affect species with high vagility [8, 11], leading to a decrease in genetic diversity due to reduced gene flow between populations.
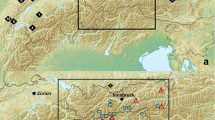
However, even though information on the relationship between fragmentation and genetic diversity in butterflies is available, many of these studies have been conducted with temperate species [6, 14, 17, 18]. To date, few such studies have been conducted with tropical species. One tropical region of particular interest for this type of study is Brazil's Atlantic Forest (Figure 1), a highly fragmented biodiversity hotspot that has a high level of species endemism [19], including butterflies [20, 21]. After more than 500 years of intensive human occupation, less than 7% of the original forest remains [22] and this region is now considered a priority area for conservation [23].

Location of Heliconius collecting sites in Rio Grande do Norte, Brazil, relative to patches of undisturbed habitat.
Heliconius butterflies are a well-studied group of tropical organisms [24–30], commonly found in New World tropical and subtropical forests [31]. Mark-recapture studies of Heliconius have shown that populations remain fairly stable over time, usually at low densities [26]. After a brief period of dispersal, Heliconius adults establish themselves in areas where they remain for the rest of their lives [32]. Thus, populations of Heliconius are organized in moderately sedentary units, with little movement of individuals, apparently as a result of home range behavior [32]. These behaviors suggest that gene flow among geographically separate populations may be low.
Yet, little is known about geographical structure of Heliconius populations and the effects of fragmentation on population connectivity. Most studies have employed mark-recapture methods to infer population structure [32–39], but some evidence from molecular markers is available. For instance, Kronforst & Fleming [7] analyzed population genetic structure of the highly vagile species Heliconius charithonia using allozymes and found low genetic diversity and no evidence of genetic subdivision in south Florida. Additional studies with allozymes have found little evidence for genetic differentiation in other Heliconius species [40–42]. In contrast, Kronforst & Gilbert [43] found evidence for extensive genetic differentiation and isolation by distance across multiple Heliconius species in Costa Rica using AFLP markers.
Population genetic structure in Heliconius also has important implications for the evolutionary dynamics of mimicry. Heliconius erato and H. melpomene are distantly-related species belonging to different clades within the genus [24, 44]. These two species are Müllerian mimics of one another throughout their shared range of Central and South America but they have both diversified, in parallel, into over 20 geographic wing pattern races. Historically, the co-mimetic radiations of H. erato and H. melpomene were thought to have occurred in parallel across time and space, possibly facilitated by Pleistocene rainforest refugia [31]. However, data from a variety of sources, including modern population genetic data [45–47], support an alternative 'mimetic advergence' hypothesis [48], with H. erato radiating first and establishing the diversity of wing patterns which H. melpomene later evolved to mimic. A remaining issue then is how H. erato originally diversified, given the strong stabilizing selection expected on warning coloration. One hypothesis is that stochastic events in local populations have occasionally allowed novel phenotypes to drift over the frequency threshold necessary to become a learned, and hence protected, warning pattern [49, 50] and this may be playing out as part of a larger "shifting-balance" type process [51–53]. This process requires small local population sizes as well as drift, and existing molecular data provide mixed evidence as to whether these conditions are generally met in Heliconius; allozymes have generally shown little structure while AFLPs have revealed more.
Our current analysis of population genetic structure in Heliconius was hence motivated by two factors. First, we were interested in determining the potential impact of the highly fragmented habitat along Brazil's Atlantic Forest on population connectivity in butterfly species with home range behavior. Second, we were interested in whether replicate analyses of population genetic structure in different geographic regions, and with a diversity of molecular markers, provide evidence for or against substantial population genetic structure in Heliconius. The generality of this phenomenon has important implications for the potential role of drift in color pattern diversification, as long as it has also occurred throughout the evolutionary history of Heliconius butterflies and independent of human activity. To address these questions, we analyzed the population genetic structure of co-mimics H. erato and H. melpomene throughout a portion of Brazil's Atlantic Forest using three types of molecular markers; microsatellites, AFLPs, and mitochondrial DNA sequences. Our results reveal substantial genetic differentiation among populations and intriguing differences between species and among molecular markers, reflecting the unique population biology of our study system and its geographic context.
Results and Discussion
Brazil's Atlantic Forest is a biodiversity hotspot endangered by extreme deforestation. To examine the potential impact of this habitat fragmentation on butterfly dispersal, we measured genetic diversity and differentiation among remnant forest patches for two co-mimetic Heliconius butterfly species, H. erato and H. melpomene. Our analysis included multiple populations of both species and three distinct classes of molecular markers, allowing us to compare and contrast patterns across species, populations, and types of molecular data.
Genetic Diversity
Across all marker types, H. erato displayed greater genetic diversity than H. melpomene (Table 1), which is consistent with previous genetic comparisons between the two species [43, 45, 47, 54]. Elevated genetic diversity in H. erato, relative to H. melpomene, is commonly attributed to the fact that H. erato is generally more abundant than H. melpomene[26, 48, 55, 56], which could result in a larger effective population size. Another potential contributing factor is that the geographic radiation of H. erato predates that of H. melpomene[45], allowing more time for the accumulation of standing genetic variation. It is unclear whether these explanations may apply to our sampling locations however. For instance, our collections revealed that H. melpomene is as abundant, or more abundant, than H. erato across most of our collecting sites. In addition, recent data suggest that H. melpomene may have originated in coastal Brazil while H. erato colonized this region after originating in western South America [47], which may mean that the two species have occupied this area for similar lengths of time. Regardless of the cause, our data show that H. erato is 14 times more variable than H. melpomene at mtDNA, seven times more variable at AFLP markers, and 1.3 times more variable at microsatellite loci (Table 1).
Differentiation and isolation-by-distance
Our analyses revealed widespread genetic differentiation in both species but substantial variation across marker types. Overall, H. erato displayed significant genetic differentiation at both AFLPs and mtDNA but not microsatellites while H. melpomene displayed significant genetic differentiation at microsatellites and AFLPs but not mtDNA (Table 1). Pairwise comparisons among populations revealed similar patterns. For instance, based on microsatellites, seven population comparisons were significant in H. melpomene while only two were significant in H. erato (Table 2). With AFLPs, virtually all pairwise comparisons were significant in both species (Table 3). Finally, mtDNA revealed 13 significant comparisons in H. erato and only four in H. melpomene (Table 4). Interestingly, mtDNA revealed much more genetic differentiation in H. erato than did the nuclear markers, suggesting females may be more sedentary than males in this species. While genetic differentiation appears to be widespread in both species, none of the tests for isolation-by-distance were significant (Table 1). This indicates that geographic distance alone is not a good indicator of genetic distance among populations.
Implications for habitat fragmentation and mimicry
Human activity in Northeastern Brazil over the last five hundred years has generated substantial habitat fragmentation in this region [57]. Today, much of Brazil's Atlantic Forest is highly fragmented with few stretches of continuous forest [19]. Because Heliconius butterflies are relatively sedentary, we expect this extensive fragmentation to limit migration among habitat patches and potentially generate genetic differentiation among populations over time. Consistent with this expectation, our analyses revealed widespread genetic differentiation among populations of both H. erato and H. melpomene. While it seems likely that the observed genetic differentiation is at least partially related to extensive habitat fragmentation, future comparative work in undisturbed habitats will be required to quantify the exact impact of fragmentation relative to baseline differentiation among populations in a natural setting.
The extent of population subdivision across the range of Heliconius species has potentially important implications beyond estimating the impact of fragmentation on population connectivity. For instance, it has been proposed that novel color patterns may occasionally arise and become locally abundant as a result of genetic drift [49, 51]. From there, these patterns may spread out to neighboring populations via a shifting balance type mechanism, thereby generating the geographic patchwork of wing pattern races which are particularly evident in H. erato and H. melpomene[51, 58]. The baseline conditions necessary for this process to operate are small local population sizes and genetic drift, but allozyme data from a variety of Heliconius species have shown that populations are not genetically differentiated, arguing against local population genetic structure and drift. In contrast, a recent study revealed widespread genetic differentiation among Heliconius populations from Costa Rica based on AFLP data [43]. Here we have shown that population genetic differentiation is common in Heliconius and evident in microsatellites and mtDNA sequence data, in addition to AFLPs. If Heliconius populations are generally subdivided, independent of human induced habitat fragmentation, this may have allowed drift to contribute to the establishment of novel warning pattern phenotypes over their evolutionary history.
Conclusions
Brazil's Atlantic Forest is a highly fragmented habitat and a priority for conservation efforts. Here we have shown that populations of two Heliconius butterfly species from the northern Atlantic Forest display widespread population genetic differentiation. These results are consistent with the expectation that fragmentation should reduce dispersal among neighboring habitat patches. The results also lend support to the observation that fine-scale population genetic structure may be common in Heliconius, which may have contributed to the evolution of mimetic diversity in the genus.
Methods
Sample collection
Between January 2007 and January 2008, we collected adult Heliconius erato and Heliconius melpomene specimens from various populations throughout the State of Rio Grande do Norte in northeastern Brazil (Figure 1). Our final sample set consisted of 136 H. erato from 8 locations and 146 H. melpomene from 7 locations (Table 5), with distances between sites ranging from 3 to 314 km. Seven sites were remnant patches of coastal Atlantic Forest and one was a cooler habitat island in the semi arid Caatinga scrub.
Microsatellite analysis
Using eight individuals from each species, we performed an initial screen of fourteen microsatellite loci: nine (Hel02, Hel04, Hel05, Hel08, Hel10-13, Hel15) described by Flanagan et al.[59], four (Hm06, Hm08, Hm16, Hm22) described by Mavárez & González [60], and one (He-Ca-001) described by Tobler et al.[61]. The microsatellites that amplified consistently were selected for analysis (Table 6). PCR amplification was performed using standard conditions and reagents following Flanagan et al.[59] and Mavárez & González [60]. Fluorescent-labeled PCR products were separated with an ABI3730 Genetic Analyzer (Applied Biosystems, Foster City, CA.) and sized and scored using ABI GeneMapper software v. 3.7.
For each locus we calculated standard population genetic statistics such as allele frequencies, expected heterozygosity (H E ), observed heterozygosity (H O ), and deviations from Hardy-Weinberg equilibrium using Arlequin 3.0 [62]. In addition, we used Arlequin 3.0 to estimate AMOVA-based [63] fixation indexes (FST) and to perform Mantel tests, comparing population pairwise FST values to straight line geographic distances, to test isolation-by-distance (IBD).
AFLP analysis
We genotyped each individual with AFLPs using a plant mapping kit (Applied Biosystems) and separated fragments with an ABI3730 Genetic Analyzer. Three selective primer combinations were used to generate fragments: EcoRI-ACT/MseI-CAT, EcoRI-ACT/MseI-CTG and EcoRI-ACA/MseI-CTG. We separated fragments with an ABI3730 Genetic Analyzer and scored fragments between 50 and 500 bp using ABI GeneMapper software v. 3.7. For each species, we calculated AFLP gene diversity using the Bayesian method of Zhivotovsky [64]. We calculated FST values and performed Mantel tests using Arlequin 3.0.
Mitochondrial DNA analysis
We PCR amplified and sequenced a 1600 bp region of mitochondrial DNA from each individual using the primers and methods described by Béltran et al.[65]. This region spans the 3' end of COI, tRNA-Leu, and COII. Contigs were assembled with Sequencher 3.1 (Gene Codes Corporation, Ann Arbor, MI) and aligned by eye. Arlequin 3.0 was used to calculate nucleotide diversity (π) and FST values as well as perform Mantel tests. DNA sequences were submitted to GenBank under accession numbers GU330064 - GU330070, GU330108 - GU330114, and HQ701917 - HQ702184.
Acknowledgements
We thank Katia Scortecci (Genome Lab-UFRN) for support during the early stages of this project, Luis Vicente Burle Maciel for assistance with the map, and Durrell Kapan and reviewers for comments on the manuscript. Butterflies were collected under MMA (Brazil) permit #10894-1 to MZC. This project was funded by grants from CAPES and the Graduate Program in Ecology at UFRN to PAM, FAPERN/CNPq (PPP/2007) to MZC, and NIH NIGMS grant GM068763 to MRK.
Abbreviations
- AFLP:
-
amplified fragment length polymorphism
- AMOVA:
-
analysis of molecular variance
- FST:
-
Wright's fixation index
- msat:
-
microsatellite
- mtDNA:
-
mitochondrial DNA
- PCR:
-
polymerase chain reaction
- SNP:
-
single nucleotide polymorphism.
References
Manel S, Schwartz MK, Luikart G, Taberlet P: Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol. 2003, 18: 189-197. 10.1016/S0169-5347(03)00008-9.
Fauvelot C, Planes S: Understanding origins of present-day genetic structure in marine fish: biologically or historically driven patterns?. Mar Biol. 2002, 141: 773-788. 10.1007/s00227-002-0869-7.
Slatkin M: Gene flow in natural populations. Annu Rev Ecol Syst. 1985, 16: 393-430. 10.1146/annurev.ecolsys.16.1.393.
Wright S: Evolution and the Genetics of Populations: Genetics and Biometric Foundations v. 4 (Variability within and Among Natural Populations); New Edition. 1984, Chicago: University of Chicago Press
Haag KL, de Araújo AM, Zaha A: Genetic structure of natural populations of Dryas iulia (Lepidoptera: Nymphalidae) revealed by enzyme polymorphism and mitochondrial DNA (mtDNA) restriction fragment length polymorphism (RFLP). Biochem Genet. 1993, 31: 449-460.
Krauss J, Schmitt T, Seitz A, Steffan-Dewenter I, Tscharntke T: Effects of habitat fragmentation on the genetic structure of the monophagous butterfly Polyommatus coridon along its northern range margin. Mol Ecol. 2004, 13: 311-320. 10.1046/j.1365-294X.2003.02072.x.
Kronforst MR, Fleming TH: Lack of genetic differentiation among widely spaced subpopulations of a butterfly with home range behaviour. Heredity. 2001, 86: 243-250. 10.1046/j.1365-2540.2001.00830.x.
Vandewoestijne S, Neve G, Baguette M: Spatial and temporal population genetic structure of the butterfly Aglais urticae L. (Lepidoptera, Nymphalidae). Mol Ecol. 1999, 8: 1539-1543. 10.1046/j.1365-294x.1999.00725.x.
Keyghobadi N, Roland J, Strobeck C: Genetic differentiation and gene flow among populations of the alpine butterfly, Parnassius smintheus, vary with landscape connectivity. Mol Ecol. 2005, 14: 1897-1909. 10.1111/j.1365-294X.2005.02563.x.
Peterson MA: Long-distance gene flow in the sedentary butterfly, Euphilotes enoptes (Lepidoptera: Lycaenidae). Evolution. 1996, 50: 1990-1999. 10.2307/2410757.
Williams BL, Brawn JD, Paige KN: Landscape scale genetic effects of habitat fragmentation on a high gene flow species: Speyeria idalia (Nymphalidae). Mol Ecol. 2003, 12: 11-20. 10.1046/j.1365-294X.2003.01700.x.
Scribner KT, Blanchong JA, Bruggeman DJ, Epperson BK, Lee CY, Pan YW, Shorey RI, Prince HH, Winterstein SR, Luukkonen DR: Geographical genetics: Conceptual foundations and empirical applications of spatial genetic data in wildlife management. J Wildlife Manage. 2005, 69: 1434-1453. 10.2193/0022-541X(2005)69[1434:GGCFAE]2.0.CO;2.
Clarke GM, O'Dwyer C: Genetic variability and population structure of the endangered golden sun moth, Synemon plana. Biol Conserv. 2000, 92: 371-381. 10.1016/S0006-3207(99)00110-X.
Keyghobadi N, Roland J, Strobeck C: Influence of landscape on the population genetic structure of the alpine butterfly Parnassius smintheus (Papilionidae). Mol Ecol. 1999, 8: 1481-1495. 10.1046/j.1365-294x.1999.00726.x.
Lushai G, Fjellsted W, Marcovitch O, Aagaard K, Sherratt TN, Allen JA, Maclean N: Application of molecular techniques to non-lethal tissue samples of endangered butterfly populations (Parnassiuss apollo L.) in Norway for conservation management. Biol Conserv. 2000, 94: 43-50. 10.1016/S0006-3207(99)00165-2.
Schmitt T, Seitz A: Influence of habitat fragmentation on the genetic structure of Polyommatus coridon (Lepidoptera: Lycaenidae): implications for conservation. Biol Conserv. 2002, 107: 291-297. 10.1016/S0006-3207(02)00066-6.
Debinski DM: Genetic diversity assessment in a metapopulation of the butterfly Euphydryas gillettii. Biol Conserv. 1994, 70: 25-31. 10.1016/0006-3207(94)90295-X.
Vandewoestijne S, Martin T, Liegeois S, Baguette M: Dispersal, landscape occupancy and population structure in the butterfly Melanargia galathea. Basic Appl Ecol. 2004, 5: 581-591. 10.1016/j.baae.2004.07.004.
Lele UJ, Viana VM, Verissimo A, Vosti S, Perkins K, Husain SA: Brazil. Forests in the balance: challenges of conservation with development (evaluation country case study series). 2000, Washington, D.C.: The World Bank
Brown KS, Freitas AVL: Lepidoptera. Biodiversidade do Estado de São Paulo, Brasil Invertebrados Terrestres. Edited by: Brandao CRF, Cancelo EM. 1999, Sao Paulo: FAPESP, 225-245.
Brown KS, Freitas AVL: Diversidade de Lepidoptera em Santa Teresa, Espírito Santo. Bol Mus Biol Mello Leitão. 2000, 11: 71-118.
Ribeiro MC, Metzger JP, Martensen AC, Ponzoni FJ, Hirota MM: The Brazilian Atlantic Forest: How much is left, and how is the remaining forest distributed? Implications for conservation. Biol Conserv. 2009, 142: 1141-1153. 10.1016/j.biocon.2009.02.021.
Myers N, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Kent J: Biodiversity hotspots for conservation priorities. Nature. 2000, 403: 853-858. 10.1038/35002501.
Brown KS: The biology of Heliconius and related genera. Annu Rev Entomol. 1981, 26: 427-456. 10.1146/annurev.en.26.010181.002235.
Gilbert LE: Ecological consequences of a coevolved mutualism between butterflies and plants. Coevolution of Animals and Plants. Edited by: Gilbert LE, Raven PH. 1975, Austin: University of Texas Press, 210-240.
Gilbert LE: Biodiversity of a Central American Heliconius community: pattern, process, and problems. Plant-Animal Interactions: Evolutionary Ecology in Tropical and Temperate Regions. Edited by: Price PW, Lewinsohn TM, Fernandes GW, Benson WW. 1991, New York: Wiley, 403-427.
Mallet J: Speciation, raciation, and colour pattern evolution in Heliconius butterflies: the evidence from hybrid zones. Hybrid Zones and the Evolutionary Process. Edited by: Harrison RG. 1993, New York: Oxford University Press, 226-260.
Mallet J, Jiggins CD, McMillan WO: Mimicry and warning colour at the boundary between races and species. Endless Forms: Species and Speciation. Edited by: Howard DJ, Berlocher SH. 1998, New York: Oxford University Press, 390-403.
Papa R, Martin A, Reed RD: Genomic hotspots of adaptation in butterfly wing pattern evolution. Curr Opin Genet Dev. 2008, 18: 559-564. 10.1016/j.gde.2008.11.007.
Sheppard PM, Turner JRG, Brown KS, Benson WW, Singer MC: Genetics and the evolution of Muellerian mimicry in Heliconius butterflies. Phil Trans R Soc Lond B. 1985, 308: 433-613. 10.1098/rstb.1985.0066.
Brown KS, Sheppard PM, Turner JRG: Quaternary refugia in tropical America: Evidence from race formation in Heliconius butterflies. Proc R Soc Lond B. 1974, 187: 369-378. 10.1098/rspb.1974.0082.
Mallet J: Dispersal and gene flow in a butterfly with home range behaviour: Heliconius erato (Lepidoptera: Nymphalidae). Oecologia. 1986, 68: 210-217. 10.1007/BF00384789.
Cook LM, Thomason EW, Young AM: Population structure, dynamics and dispersal of the tropical butterfly Heliconius charitonius. J Anim Ecol. 1976, 45: 851-863. 10.2307/3584.
Ehrlich PR, Gilbert LE: Population structure and dynamics of the tropical butterfly Heliconius ethilla. Biotropica. 1973, 5: 69-82. 10.2307/2989656.
Mallet J: Gregarious roosting and home range in Heliconius butterflies. Natl Geogr Res. 1986, 2: 198-215.
Quintero HE: Population dynamics of the butterfly Heliconius charitonius L. in Puerto Rico. Carib J Sci. 1988, 24: 155-160.
Ramos RR, Freitas AVL: Population biology and wing color variation in Heliconius erato phyllis (Nymphalidae). J Lep Soc. 1999, 53: 11-21.
Saalfeld K, Araújo AM: Studies on the genetics and ecology of Heliconius erato (Lepidoptera: Nymphalidae). I: demography of a natural population. Rev Brasil Biol. 1981, 41: 855-860.
Turner JRG: Experiments on the demography of tropical butterflies. II. Longevity and home-range behaviour in Heliconius erato. Biotropica. 1971, 3: 21-31. 10.2307/2989703.
Jiggins CD, McMillan WO, King P, Mallet J: The maintenance of species differences across a Heliconius hybrid zone. Heredity. 1997, 79: 495-505. 10.1038/hdy.1997.189.
Maurício-da-Silva L, Araújo AM: The genetic structure of Heliconius erato populations (Lepidoptera; Nymphalidae). Rev Brasil Genet. 1994, 17: 19-24.
Turner JRG, Johnson MS, Eanes WF: Contrasted modes of evolution in the same genome: allozymes and adaptive change in Heliconius. Proc Natl Acad Sci USA. 1979, 76: 1924-1928. 10.1073/pnas.76.4.1924.
Kronforst MR, Gilbert LE: The population genetics of mimetic diversity in Heliconius butterflies. Proc R Soc B. 2008, 275: 493-500. 10.1098/rspb.2007.1378.
Beltran M, Jiggins CD, Brower AVZ, Bermingham E, Mallet J: Do pollen feeding, pupal-mating and larval gregariousness have a single origin in Heliconius butterflies? Inferences from multilocus DNA sequence data. Biol J Linn Soc. 2007, 92: 221-239. 10.1111/j.1095-8312.2007.00830.x.
Flanagan NS, Tobler A, Davison A, Pybus OG, Kapan DD, Planas S, Linares M, Heckel D, McMillan WO: Historical demography of Mullerian mimicry in the neotropical Heliconius butterflies. Proc Natl Acad Sci USA. 2004, 101: 9704-9709. 10.1073/pnas.0306243101.
Mallet J, Jiggins CD, McMillan W: Mimicry meets the mitochondrion. Curr Biol. 1996, 6: 937-940. 10.1016/S0960-9822(02)00631-0.
Quek SP, Counterman BA, Albuquerque de Moura P, Cardoso MZ, Marshall CR, McMillan WO, Kronforst MR: Dissecting comimetic radiations in Heliconius reveals divergent histories of convergent butterflies. Proc Natl Acad Sci USA. 2010, 107: 7365-7370. 10.1073/pnas.0911572107.
Eltringham H: On specific and mimetic relationships in the genus Heliconius. Trans Entomol Soc Lond. 1916, 101-148.
Mallet J, Singer MC: Individual selection, kin selection, and the shifting balance in the evolution of warning colors: the evidence from butterflies. Biol J Linn Soc. 1987, 32: 337-350. 10.1111/j.1095-8312.1987.tb00435.x.
Turner JRG, Mallet JLB: Did forest islands drive the diversity of warningly coloured butterflies? Biotic drift and the shifting balance. Phil Trans R Soc Lond B. 1996, 351: 835-845. 10.1098/rstb.1996.0078.
Mallet J: Shift happens! Shifting balance and the evolution of diversity in warning colour and mimicry. Ecol Entomol. 2010, 35: 90-104. 10.1111/j.1365-2311.2009.01137.x.
Wright S: The roles of mutation, inbreeding, crossbreeding and selection in evolution. Proc Sixth Int Congr Genet. 1932, 1: 356-366.
Wright S: Evolution and the Genetics of Populations, Experimental Results and Evolutionary Deductions. 1977, Chicago: University of Chicago Press, 3:
Brower AVZ: Parallel race formation and the evolution of mimicry in Heliconius butterflies: A phylogenetic hypothesis from mitochondrial DNA sequences. Evolution. 1996, 50: 195-221. 10.2307/2410794.
Gilbert LE: The biology of butterfly communities. The Biology of Butterflies. Edited by: Vane-Wright RI, Ackery PR. 1984, London, UK: Academic Press, 41-54.
Mallet J: Causes and consequences of a lack of coevolution in Müllerian mimicry. Evol Ecol. 1999, 13: 777-806. 10.1023/A:1011060330515.
Silva JMC, Casteleti CHM: Status of the biodiversity of the Atlantic Forest of Brazil. The Atlantic Forest of South America: biodiversity status, threats, and outlook. Edited by: Galindo-Leal C, Camara IG. 2003, Washington, D.C.: Island Press, 43-59.
Blum MJ: Rapid movement of a Heliconius hybrid zone: Evidence for phase III of Wright's shifting balance theory?. Evolution. 2002, 56: 1992-1998.
Flanagan NS, Blum MJ, Davison A, Alamo M, Albarran R, Faulhaber K, Peterson E, McMillan WO: Characterization of microsatellite loci in neotropical Heliconius butterflies. Mol Ecol Notes. 2002, 2: 398-401. 10.1046/j.1471-8286.2002.00255.x.
Mavarez J, Gonzalez M: A set of microsatellite markers for Heliconius melpomene and closely related species. Mol Ecol Notes. 2006, 6: 20-23. 10.1111/j.1471-8286.2005.01115.x.
Tobler A, Kapan D, Flanagan NS, Gonzalez C, Peterson E, Jiggins CD, Johntson JS, Heckel DG, McMillan WO: First-generation linkage map of the warningly colored butterfly Heliconius erato. Heredity. 2005, 94: 408-417. 10.1038/sj.hdy.6800619.
Excoffier L, Laval G, Schneider S: Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform. 2005, 1: 47-50.
Excoffier L, Smouse PE, Quattro JM: Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics. 1992, 131: 479-491.
Zhivotovsky LA: Estimating population structure in diploids with multilocus dominant DNA markers. Mol Ecol. 1999, 8: 907-913. 10.1046/j.1365-294x.1999.00620.x.
Beltran M, Jiggins CD, Bull V, Linares M, Mallet J, McMillan WO, Bermingham E: Phylogenetic discordance at the species boundary: Comparative gene genealogies among rapidly radiating Heliconius butterflies. Mol Biol Evol. 2002, 19: 2176-2190.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
PAM, MZC and MRK planned the project; PAM and MZC collected specimens; PAM and SPQ generated molecular data; PAM and MRK analyzed the data; PAM, MZC and MRK wrote the paper; all authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Albuquerque de Moura, P., Quek, SP., Cardoso, M.Z. et al. Comparative population genetics of mimetic Heliconius butterflies in an endangered habitat; Brazil's Atlantic Forest. BMC Genet 12, 9 (2011). https://doi.org/10.1186/1471-2156-12-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2156-12-9