
Abstract
Background
The relative contributions of the cytotoxic phenotype of P. aeruginosa expressing type III secretory toxins and an immunocompromised condition lacking normal Toll-like receptor 4 (TLR4) signaling in the pathogenesis of acute lung injury and sepsis were evaluated in a mouse model for Pseudomonas aeruginosa pneumonia. By using lipopolysaccharide-resistant C3H/HeJ mice missing normal TLR4 signaling due to a mutation on the tlr4 gene, we evaluated how TLR4 signaling modulates the pneumonia caused by cytotoxic P. aeruginosa expressing type III secretory toxins.
Methods
We infected C3H/HeJ or C3H/FeJ mice with three different doses of either a cytotoxic P. aeruginosa strain (wild type PA103) or its non-cytotoxic isogenic mutant missing the type III secretory toxins (PA103ΔUT). Survival of the infected mice was evaluated, and the severity of acute lung injury quantified by measuring alveolar epithelial permeability as an index of acute epithelial injury and the water to dry weight ratios of lung homogenates as an index of lung edema. Bacteriological analysis and cytokine assays were performed in the infected mice.
Results
Development of acute lung injury and sepsis was observed in all mouse strains when the cytotoxic P. aeruginosa strain but not the non-cytotoxic strain was instilled in the airspaces of the mice. Only C3H/HeJ mice had severe bacteremia and high mortality when a low dose of the cytotoxic P. aeruginosa strain was instilled in their lungs.
Conclusion
The cytotoxic phenotype of P. aeruginosa is the critical factor causing acute lung injury and sepsis in infected hosts. When the P. aeruginosa is a cytotoxic strain, the TLR4 signaling system is essential to clear the batcteria to prevent lethal lung injury and bacteremia.
Similar content being viewed by others


Background
Pseudomonas aeruginosa (P. aeruginosa) frequently causes acute pneumonia in immunocompromised individuals. Patients who are mechanically ventilated are especially at high risk for developing P. aeruginosa pneumonia [1, 2]. In addition, patients with P. aeruginosa pneumonia are more likely to develop bacteremia, septic shock, and multiple organ failure, and have a higher mortality than patients with pneumonia due to other infectious agents [2, 3].
A poorer prognosis of patients with P. aeruginosa pneumonia is associated with specific virulence factors of P. aeruginosa. We have reported that the ability of these bacteria to cause acute epithelial injury involves the expression of virulent toxins that are translocated directly into eukaryotic cells via the type III secretion (TTS) system [4–9]. The TTS system is utilized by most virulent pathogenic gram-negative bacteria, including Yersinia, Salmonella, Shigella, and E. coli [10, 11]. The TTS system functions as a molecular syringe to deliver toxins directly into the cytosol of cells and inhibit host immune responses [12–14]. In animal models, cytotoxic P. aeruginosa strains expressing the TTS toxins were able to consistently induce septic shock by causing the dissemination of inflammatory mediators from injured lungs into the circulation [15]. The administration of anti-PcrV IgG blocked the TTS toxin translocation by P. aeruginosa and protected animals from lethal pneumonia [16–18]. Recently, several studies also demonstrated that patients infected with cytotoxic strains of P. aeruginosa expressing the TTS toxins have a high risk of mortality [19, 20].
In infections with gram-negative bacteria including P. aeruginosa, endotoxin (lipopolysaccharide, LPS), a component of bacterial outer membranes, has been reported to be responsible for the pathogenesis of acute organ injury and sepsis by provoking host inflammatory responses and inducing systemic inflammatory syndromes [21]. The acute immune response to LPS occurs mainly through Toll-like receptor 4 (TLR4) [22, 23]. In animal models of P. aeruginosa pneumonia, release and accumulation of inflammatory cytokines primarily occurs in the airspaces of the infected lung. The inflammatory cytokines produced in the lung are able to disseminate into the circulation across the lung epithelium damaged by the toxins secreted from cytotoxic P. aeruginosa, leading to septic shock [15]. The administration of interleukin-10, an anti-inflammatory cytokine, suppresses the systemic inflammatory mediators leaking into the circulation from the lungs and improves the survival of infected animals [15]. Therefore, two virulent components of P. aeruginosa, the TTS system and LPS, appeared to be important in causing sepsis associated with P. aeruginosa pneumonia.
In contrast, the inflammatory response to the airspace instillation of LPS alone does not cause acute lung epithelial injury [24]. In addition, neither lung injury nor septic shock can be induced by the airspace instillation of non-cytotoxic P. aeruginosa strains that are defective in the production of TTS toxins [15]. Therefore, whether the response to LPS through TLR4 becomes deleterious or beneficial for infected hosts appears to be influenced by the virulence of P. aeruginosa. Clinically, LPS-TLR4 signaling has been a therapeutic target for preventing acute lung injury and sepsis. However, the blockade of LPS-TLR4 signaling may be harmful because recognition of bacterial LPS through TLR4 and subsequent proinflammatory responses in infected hosts must be important for innate immunity against pathogens. In addition, the presence of endogenous ligands for TLRs has been reported [25, 26]. This implies that there are regulatory roles of TLR signaling in inflammatory and immune responses against not only infectious microbes but also endogenous harmful stimuli.
In this investigation, the contributions of the cytotoxic phenotype of P. aeruginosa and LPS-TLR4 signaling were evaluated in a mouse model of P. aeruginosa pneumonia. Using LPS-resistant C3H/HeJ mice missing normal TLR4 signaling due to a mutation on the tlr4 gene [27], we evaluated how TLR4 signaling modulated the development of P. aeruginosa pneumonia induced by the airspace instillation of either a cytotoxic P. aeruginosa strain or its non-cytotoxic isogenic mutant missing all TTS toxins. The outcomes suggested that the role of TLR4 signaling in infected hosts was important in clearance of the P. aeruginosa when the bacteria causing pneumonia is a cytotoxic strain expressing the type III secretory toxins.
Methods
Animals
Male C3H/FeJ and C3H/HeJ mice (8- to 12-week-old) were purchased from Jackson Laboratories (Bar Harbor, ME). Animals were housed in cages with filter tops in specific pathogen-free conditions. Sterile food and water were provided ad lib. All experiments were done in compliance with the Animal Care Committee Rules of the University of California at San Francisco, and all protocols were approved.
P. aeruginosa strains
P. aeruginosa parental PA103 strain and its isogenic mutant PA103ΔUT missing both ExoU and ExoT toxins were generously provided by Dara W. Frank (Medical College of Wisconsin, Milwaukee, WI) [15]. Bacteria from frozen stocks were streaked onto trypticase soy agar plates and grown in a deferrated dialysate of tryptic soy broth supplemented with 10 mM nitrilotriacetic acid (Sigma Chemical), 1% glycerol, and 100 mM monosodium glutamate and grown at 33°C for 13 h in a shaking incubator. Cultures were centrifuged at 8,500 × g for 5 minutes, and the bacterial pellet was washed twice and diluted into the appropriate number of CFU/ml in lactated Ringer's solution, determined by spectrophotometry. The bacterial concentration was confirmed by plating out the known dilutions of the samples on sheep blood agar plates.
Intratracheal instillation
Mice were briefly anesthetized with inhaled sevoflurane (Ultane, Abbot Laboratories, Abbot Park, Illinois, USA) and placed supine, at an angle of approximately 30°. Fifty microliters of bacterial solution were instilled slowly in the left lobe of the lungs through a gavage needle (Modified animal feeding needle, 24 G, Popper & Sons, Inc., New Hyde Park, NY) inserted into the trachea via the oropharynx. The proper insertion of the needle in the trachea was confirmed by palpation of the tip of the blunt needle through the skin and the observation of movement of the solution inside the syringe with respiratory efforts [28].
Quantification of acute lung injury
The severity of epithelial lung injury was quantified by measuring the efflux of the 131I-labeled albumin (the alveolar protein tracer) across the alveolar epithelium into the circulation, as previously described [28]. The alveolar instillate was made up of 0.5 μCi of 131I-labeled human serum albumin (Merck-Frost, Quebec, Canada) and 0.05 mg of anhydrous Evan's blue in 2 ml of lactated Ringer's solution with 5% BSA. An appropriate quantity of the specified P. aeruginosa was added to the 50 μl volume, if the experiment was to include bacteria. The total radioactivity (in counts/minute/g) in the instillate was measured in a γ-ray counter (Auto-Gamma, model 5550, Packard, Downers Grove, IL). Four hours after the instillation, mice were deeply anesthetized with intraperitoneal sodium pentobarbital. Blood was collected by a carotid arterial puncture and median sternotomies were done to harvest and measure the radioactivity of pleural fluids, lungs, tracheas, oropharynxes and stomachs. The percentage of radioactive albumin that left the lung and entered the circulation was calculated by multiplying the counts per gram measured in the terminal blood sample times the blood volume of the animal (body weight × 0.07). The lungs were homogenized in sterile containers with sterile 0.9% saline solution. Lung homogenates were placed in pre-weighed aluminium pans and dried to constant weight in an oven at 80°C for 3 days. The water to dry weight ratios of lung homogenates were used as an index of lung edema.
Bacteriological assay
Blood was plated onto sheep blood agar plates for quantitative assessment of CFU/ml. Lung homogenates were sequentially diluted then plated onto sheep blood agar plates. Colony forming units were calculated per gram of tissue.
Bronchoalveolar lavage (BAL)
Mouse lungs were lavaged by instilling and withdrawing 1 ml of phosphate buffered saline (PBS). Supernatants of centrifuged BAL fluids (BALF) were collected and processed for cytokine measurement.
Leukocyte counts in BALF
BALF samples were mixed with the solution including 0.01% crystal violet dye and 2.7% acetic acid for leukocyte staining and erythrocyte hemolysis. The number of leukocytes in BLAF was counted by a hemacytometer under a microscope.
Determination of cytokine levels
Biological activity of TNFα in plasma and BALF was measured by analyzing the cytotoxicity of TNFα on the fibrosarcoma cell line WEHI-13VAR, as reported previously [15, 29]. Cytokine ELISA was performed for IL-10, as previously described [28]. Sample concentrations were calculated by comparison with standard curves using recombinant cytokine.
Statistical analysis
Mantel-Cox log rank test was used for survival analysis. One-way analysis of variance (ANOVA) followed by the Bonferroni multiple comparison test or the Dunn's Multiple Comparisons test were used for comparisons among 3 groups. Student's t-test or Mann-Whitney test were used for comparisons between 2 groups. The Kruskal-Wallis non-parametric test with Dunn's test was used for the comparisons of leukocyte counts in BALF. Significance was accepted at p < 0.05.
Results
Survival study
To examine whether a lack of normal TLR4 signaling affects the mortality induced by P. aeruginosa pneumonia, we performed survival studies using LPS-resistant C3H/HeJ mice. LPS-sensitive C3H/FeJ mice (the background strain of C3H/HeJ) were used as control groups. Mice were intratracheally infected with wild type P. aeruginosa PA103 at doses of either 5 × 103 CFU (low dose), 6 × 104 CFU (medium dose), or 5 × 105 CFU (high dose), or with an isogenic mutant PA103ΔUT, which is missing the expression of TTS toxins (ExoU and ExoT), at a dose of 5 × 105 CFU (high dose).
After the instillation of a low dose (5 × 103 CFU) wild type PA103, 30% of LPS-resistant C3H/HeJ mice died in 3 days, while all C3H/FeJ survived for a week (Fig. 1a). However, statistically significant difference was not detected between the two strains of mice. After the instillation of a medium dose (6 × 104 CFU) of wild type PA103, 65% of LPS-resistant C3H/HeJ mice died in 4 days, while all C3H/FeJ survived for a week (Fig. 1b). There was statistically significant difference between C3H/HeJ and C3H/FeJ mice. After the instillation of high dose (5 × 105 CFU) wild type PA103, only 30% of C3H/FeJ and only 15% C3H/HeJ mice, respectively, survived for more than 3 days (Fig. 1c). The results suggest that, in the comparison between C3H/HeJ and C3H/FeJ mice, LPS-resistant C3H/HeJ mice were more susceptible to wild type PA103 at three different doses. The result implies that defective TLR4 signaling was not beneficial for the survival of infected mice with cytotoxic PA103.

Survival study. An inoculum of either PA103 (5 × 105 CFU or 6 × 104 CFU) or PA103ΔUT (5 × 105 CFU) was instilled into the lung of mice, then the survival was monitored for 7 days. n = 10 / group. a. PA103 (5 × 105 CFU). b. PA103 (6 × 104 CFU). c. PA103ΔUT (5 × 105 CFU). *, p < 0.05 versus C3H/FeJ PA103 6 × 104 CFU group.
Next, we examined whether the mortality of infected mice depended on the phenotypes of the TTS toxins of P. aeruginosa. An isogenic mutant PA103ΔUT missing ExoU and ExoT was intratracheally administered at a high dose (5 × 105 CFU) to the airspaces of C3H/HeJ and C3H/FeJ mice. Although instilled at a high dose, none of the mice died for a week (Fig. 1d). Therefore, the mortality we observed in the mice infected with a high dose of P. aeruginosa PA103 was totally dependent upon the TTS virulence of P. aeruginosa rather than the genotype of TLR4 in mice.
Acute lung injury
We quantified acute lung injury in the mice infected with P. aeruginosa. Acute lung epithelial damage was quantified by measuring the efflux of alveolar protein tracers from the lungs into the bloodstream (Fig. 2a). Lung edema was quantified by measuring the water to dry weight ratios of the lung (Fig. 2b). The instillation of a high dose (5 × 105 CFU) of PA103 led to a significant increase of lung epithelial injury (Fig. 2a) and severe increases of lung edema (Fig. 2b) in both groups of mice. When the mice were infected with a medium dose (6 × 104 CFU) of PA103, both severe lung epithelial injury and lung edema occurred in the both mouse strains (Fig. 2a). Although the injury levels in C3H/HeJ were slightly higher, these were not statistically significant. In contrast, the intratracheal instillation of PA103ΔUT even at a high dose (5 × 105 CFU) did not cause lung injury in either of the mice (Fig. 2a and 2b). This implies that, in our mouse model, the bacterial dose and virulence of the bacteria, but not the TLR4 phenotype of mice, determines the initial severity of acute lung injury. Because more C3H/HeJ mice infected with a medium dose (6 × 104 CFU) of PA103 died 3 days after infection in the survival studies (Fig. 1a), we analyzed lung edema 3 days after a medium dose instillation (6 × 104 CFU) of PA103 in the strains of mice. In contrast to C3H/FeJ mice, C3H/HeJ mice showed a significantly higher level of lung edema (Fig. 2c).

Acute lung injury. Two parameters of lung damage were considered 4 h after the instillation made of the protein tracer 131I-labeled albumin and either PA103 (5 × 105 CFU or 6 × 104 CFU), or PA103ΔUT (5 × 105 CFU), or no bacteria. a. Efflux of protein tracer into the bloodstream was calculated as a percentage of the total instilled amount to quantify lung epithelial injury. b. Water to dry weight ratios of lungs as index of lung edema. Data are means ± SE. n = 3 in control groups (no bacteria), and n = 5 in other groups. *, p < 0.05 versus C3H/FeJ, †, p < 0.05 versus no bacteria.
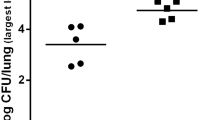
Lung bacterial clearance
We quantified the ability of the different strains of mice to clear the bacteria from their infected lungs and the severity of bacteremia during infection. Four hours after a high dose instillation (5 × 105 CFU) of wild type PA103, both strains of mice had similar quantities of bacteria in their lungs (Fig. 3a). However, 4 h after the instillation of a medium dose (6 × 104 CFU) of the wild type PA103, there was a significantly larger quantity of bacteria in the lungs of the C3H/HeJ mice. The quantity of bacteria in the lungs of the C3H/HeJ mice was significantly increased after 3 days of infection (6 × 104 CFU) compared to the quantity of bacteria in the C3H/FeJ mice (Fig. 3b). The lungs of C3H/HeJ infected with high dose (5 × 105 CFU) PA103ΔUT also showed a significantly higher level of bacterial burden in their lungs in comparison to the lungs of the C3H/FeJ mice (Fig. 3a) although all the mice survived for a week after instillation of the same dose of PA103ΔUT in the survival study (Fig. 1c). This result implies that C3H/HeJ mice could not clear the bacteria efficiently from the infected lungs even 3 days after the infection (Fig. 3b). Persistent lung infection observed in C3H/HeJ mice explained higher edema (Fig. 2c) and mortality (Fig. 1a) of the mice infected with a medium dose of PA103.

Bacteriology. Mice were infected with PA103 (5 × 105 CFU or 6 × 104 CFU) or PA103ΔUT (5 × 105 CFU). The number of viable bacteria remaining in the infected lungs and blood was counted 4 h and 2 days after the instillation. Lung homogenates were sequentially diluted with sterile water and placed on sheep blood agar plates for 24 h at 37°C. 100 μl of blood was plated on agar plates for 24 h at 37°C. a. Number of bacteria remaining in the lungs 4 h after infection. b. Number of bacteria remaining in the lungs 2 days after PA103 6 × 104 CFU infection. c. Bacteremia (CFU/ml of blood) 4 h after infection. d. Bacteremia (CFU/ml of blood) 2 days after PA103 6 × 104 CFU infection. n = 5 in groups. Data are means ± SE. *, p < 0.05 versus C3H/FeJ.
Bacteremia
We evaluated the severity of bacteremia in C3H/HeJ and C3H/FeJ mice. Only a high dose instillation of PA103 caused detectable bacteremia 4 h after the infection in either strain of mice (Fig. 3c). Among the two strains, C3H/HeJ mice showed a significantly higher level of bacteremia 4 h after infection (Fig. 3c), although more than 70% of mice in the both strains eventually died within a week after the instillation of bacteria (Fig. 1b). Neither the instillation of a high dose of PA103ΔUT nor the instillation of a medium dose of PA103 resulted in bacteremia 4 h after the infection (Fig. 3c). However, 3 days after the instillation of the medium dose PA103, C3H/HeJ mice showed a significantly greater level of bacteremia while C3H/FeJ mice had either minimal bacteremia or none, respectively (Fig. 3d). Therefore, the results imply that decreased bacterial clearance probably lead to a delay in the onset of bacteremia in C3H/HeJ mice.
Leukocytes in bronchoalveolar lavage fluids
We measured the number of leukocytes in BALFs which were collected 16 h after infection from the mice infected with either PA103 or PA103ΔUT at a medium dose (Fig. 4). More leukocytes were found in the BALF from the mice infected with PA103ΔUT than in the BALF from the mice infected with PA103. More leukocytes were found in the BALF of C3H/FeJ mice than in the C3H/FeJ mice. The number of leukocytes in the BALF of C3H/FeJ mice infected with PA103 was the lowest among the four groups, and significantly lower than that of C3H/HeJ mice infected with PA103ΔUT. The results implied that C3H/HeJ mice failed to recruit inflammatory cells efficiently into the site of infection. In addition, when infection was by virulent P. aeruginosa secreting TTS toxins, few leukocytes were detected in BALF, implying that virulent P. aeruginosa suppressed the proper inflammatory cell recruitment or eliminated the recruited leukocytes by its cytotoxicity.

The number of leukocytes in bronchoalveolar lavage fluids. The numbers of leukocytes were measured in the bronchoalveolar lavage fluids collected from the mice infected with either PA103 5 × 104 CFU or PA103ΔUT 5 × 104 CFU. Closed and open circles mean each data. n = 5 in groups. Boxes are the first quartile with medians as central bars. *, p < 0.05 versus C3H/HeJ infected with PA103ΔUT by the Kruskal-Wallis non-parametric test followed by the Dunn's test.
TNFα and IL-10 levels
We measured concentrations of TNFα in BALF and in the serum of infected animals. TNFα in BALF was almost undetectable in C3H/HeJ mice 4 h after the instillation of a high dose (5 × 105 CFU) of either wild type PA103 or of PA103ΔUT (Fig. 5a). Only in the C3H/FeJ mice there was a significant difference in the TNFα in BALFs between PA103 and PA103ΔUT infections (Fig. 5a). Only C3H/FeJ mice infected with a high dose of PA103 showed a significantly increased serum concentration of TNFα compared to those in the other strains of mice infected with either PA103 or PA103ΔUT (Fig. 5b).

Cytokines. TNFα and IL-10 levels were measured 10 h after P. aeruginosa infection (PA103 or PA103ΔUT 5 × 105 CFU). a. TNFα levels in bronchoalveolar lavage fluids. b. TNFα levels in serum. c. IL-10 levels in serum 10 h after PA103 infection. Data are means ± SE. n = 8 – 10/group. *, p < 0.05 versus C3H/HeJ.
We also measured the serum concentration of the anti-inflammatory cytokine IL-10 in the mice infected with a high dose of PA103. The concentration of IL-10 4 h after infection was higher in the C3H/HeJ mice, but the difference not statistically significant (Fig. 5c).
Discussion
This study was designed to determine the contributions of the TTS system and LPS of P. aeruginosa in the pathogenesis of acute lung injury and bacteria-induced death. To understand the role of TLR4 signaling in acute lung injury and sepsis, we compared LPS-resistant C3H/HeJ mice, which are refractory to the biological effects of LPS due to the mutation of tlr4, to LPS-sensitive C3H/FeJ mice [27]. The two strains of mice received tracheal instillation of either cytotoxic wild type P. aeruginosa PA103 or an isogenic strain PA103ΔUT which is missing two critical TTS toxins, ExoU and ExoT [15]. Three different doses of bacteria were tested in these mice. ExoU is a cytotoxin inducing necrotic cell death by a mechanism associated with its phospholipase activity [30]. ExoT is a 53-kDa exoenzyme S possessing ADP-ribosyltransferase and small GTPase-activating activities. ExoT blocks epithelial wound healing [31]. The administration of large doses of PA103ΔUT did not cause any lung injury nor affect the mortality of any strain of mice. Large doses of wild type PA103 caused acute injury, bacteremia and death in the both strains of mice. Although there appeared to be minor differences in survival, there were no statistically significant differences in the mortality rates between the strains of mice infected with large doses of PA103. These results suggest that the administration of large doses of bacteria overwhelms host defense, even in mice with an intact LPS-signaling pathway. Furthermore, although there was abnormal clearance of the non-cytotoxic PA103ΔUT in the C3H/HeJ mice in comparison to the C3H/FeJ mice, PA103ΔUT did not cause lung injury. As no lung injury occurred, PA103ΔUT did not disseminate, and all the mice survived. These results demonstrate that organ injury and mortality were not directly affected by P. aeruginosa LPS or the TLR4 signaling of the infected mice.
In contrast, an intact TLR4 signaling pathway was clearly essential for host protection from virulent PA103. More C3H/HeJ mice died from a medium dose of PA103, and some of C3H/HeJ mice still died even from the lowest dose of PA103. When lung injury was measured after a short interval, there was no statistical difference between the acute lung injury produced by similar doses in either the C3H/HeJ or C3H/FeJ mice. Therefore, a given dose of cytotoxic bacteria produced a reproducible quantity of lung injury, in a short interval, in both mice. However, when the infections were allowed to persist for a longer interval, the number of cytotoxic bacteria in the C3H/HeJ multiplied to a greater extent and caused lung edema later. The lack of bacterial clearance in the C3H/HeJ mice ultimately led to relatively larger quantities of lung injury, even when smaller doses of cytotoxic bacteria had been administered. The bacteria were able to multiply to injurious levels, implying that, in the absence of normal TLR4 signaling, cytotoxic PA103 can avoid the host immune clearance, disseminate into the bloodstream, and cause the death of the infected animals. The low leukocyte number and a lack of TNFα production in the infected airspace in the C3H/HeJ mice could indicate the lack of a normal inflammatory response that clears the bacteria from the infected site in these mice missing normal TLR4 signaling. Therefore, immune responses modulated by TLR4 signaling are important in the protecting hosts from cytotoxic P. aeruginosa expressing TTS toxins.
LPS is a static component of gram-negative bacteria, while the virulence associated with the TTS system relies on active energy production by live bacteria; the TTS apparatus can be upregulated and TTS toxins need to be translocated using bacterial energy [10, 11, 13]. Gram-negative bacteria such as E. coli and P. aeruginosa possess LPS as a component of their cell membranes, although there are many variations in its pattern [32]. However, only pathogenic strains of gram-negative bacteria possess the TTS system [33, 34]. In P. aeruginosa, only strains expressing the TTS system can cause significant acute necrotic cell death and tissue damage, while non-cytotoxic P. aeruginosa missing TTS but still possessing LPS do not lead to tissue damage [5, 7]. Recently, there have been reports of endogenous stimulators of TLRs [25, 26]. For TLR4, heparan sulfate, hyaluronan, HSP60 and HSP70, surfactant protein A, and β-defensin 2 have been all implicated as possible endogenous ligands or stimulators [25, 26, 35]. These facts suggest that TLRs play a role as surveillance receptors for tissue injury and tissue remodelling as well as for infection [35]. If TLR4 recognizes not only LPS but also degradation products of endogenous macromolecules from necrotic cells and microorganisms, and if this recognition provokes responses to limit tissue injury and induce remodelling, a lack of normal TLR4 signaling must be disadvantageous during infections involving severe tissue damage. The activation of this surveillance system must be more important when cytotoxic P. aeruginosa infects and causes tissue injury. Without having this surveillance system, hosts may not be able to survive infection involving severe tissue damage. Members of the TLR and interleukin-1 receptor (IL-1R) superfamily share an intracytoplasmic Toll-IL-1 receptor (TIR) domain, which mediates recruitment of the interleukin-1 receptor-associated kinase (IRAK) complex via TIR-containing adapter molecules. Recently, 3 unrelated children with an inherited IRAK-4 deficiency were reported. They were prone to infections caused by pyogenic bacteria [36]. Their blood and fibroblast cells did not activate NF-κB or mitogen-activated protein kinase (MAPK) and failed to induce downstream cytokines in response to any of the known ligands of TIR-bearing receptors. These findings suggest that in humans the normal TLR signaling pathway is crucial for protective immunity against specific virulent bacteria and is redundant against most other microorganisms. Our results suggest that, in P. aeruginosa infections, the identification of the phenotype or genotype of P. aeruginosa could be useful clinically.
Conclusions
In P. aeruginosa infections, the cytotoxic phenotype of P. aeruginosa is the most critical factor in causing acute lung injury and sepsis in infected hosts. TLR4 signaling is another important factor for the efficient clearance of bacteria in hosts infected with cytotoxic P. aeruginosa strains.
Abbreviations
- BAL:
-
Bronchoalveolar lavage
- BALF:
-
Bronchoalveolar lavage fluid
- Pseudomonas aeruginosa :
-
P. aeruginosa
- TTS:
-
type III secretion
- LPS:
-
ipopolysaccharide
- TLR:
-
Toll-like receptor
References
Chastre J, Fagon JY: Ventilator-associated pneumonia. Am J Respir Crit Care Med 2002, 165:867–903.
Heyland DK, Cook DJ, Griffith L, Keenan SP, Brun-Buisson C: The attributable morbidity and mortality of ventilator-associated pneumonia in the critically ill patient. The Canadian Critical Trials Group. Am J Respir Crit Care Med 1999, 159:1249–1256.
Brun-Buisson C, Doyon F, Carlet J, Dellamonica P, Gouin F, Lepoutre A, Mercier JC, Offenstadt G, Regnier B: Incidence, risk factors, and outcome of severe sepsis and septic shock in adults. A multicenter prospective study in intensive care units. French ICU Group for Severe Sepsis. JAMA 1995, 274:968–974.
Finck-Barbancon V, Goranson J, Zhu L, Sawa T, Wiener-Kronish JP, Fleiszig SM, Wu C, Mende-Mueller L, Frank DW: ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol Microbiol 1997, 25:547–557.
Fleiszig SM, Wiener-Kronish JP, Miyazaki H, Vallas V, Mostov KE, Kanada D, Sawa T, Yen TS, Frank DW: Pseudomonas aeruginosa -mediated cytotoxicity and invasion correlate with distinct genotypes at the loci encoding exoenzyme S. Infect Immun 1997, 65:579–586.
Kudoh I, Wiener-Kronish JP, Hashimoto S, Pittet JF, Frank D: Exoproduct secretions of Pseudomonas aeruginosa strains influence severity of alveolar epithelial injury. Am J Physiol 1994, 267:L551–556.
Sawa T, Ohara M, Kurahashi K, Twining SS, Frank DW, Doroques DB, Long T, Gropper MA, Wiener-Kronish JP: In vitro cellular toxicity predicts Pseudomonas aeruginosa virulence in lung infections. Infect Immun 1988, 66:3242–3249.
Wiener-Kronish JP, Frank DW, Sawa T: Mechanisms of lung epithelial cell injury by acute by Pseudomonas aeruginosa. In Molecular biology of acute lung injury (Edited by: Clark RSB, Carcillo JA). Boston: Kluwer Academic Publishers 2001, 149–161.
Wiener-Kronish JP, Sakuma T, Kudoh I, Pittet JF, Frank D, Dobbs L, Vasil ML, Matthay MA: Alveolar epithelial injury and pleural empyema in acute P. aeruginosa pneumonia in anesthetized rabbits. J Appl Physiol 1993, 75:1661–1669.
Galan JE, Collmer A: Type III secretion machines: bacterial devices for protein delivery into host cells. Science 1999, 284:1322–1328.
Hueck CJ: Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev 1998, 62:379–433.
Barinaga M: A shared strategy for virulence. Science 1996, 272:1261–1263.
Lee CA: Type III secretion systems: machines to deliver bacterial proteins into eukaryotic cells? Trends Microbiol 1997, 5:148–156.
Mecsas J, Strauss EJ: Molecular mechanisms of bacterial virulence: type III secretion and pathogenicity islands. Emerging Infect Dis 1996, 2:271–284.
Kurahashi K, Kajikawa O, Sawa T, Ohara M, Gropper MA, Frank DW, Martin TR, Wiener-Kronish JP: Pathogenesis of septic shock in Pseudomonas aeruginosa pneumonia. J Clin Invest 1999, 104:743–750.
Sawa T, Yahr TL, Ohara M, Kurahashi K, Gropper MA, Wiener-Kronish JP, Frank DW: Active and passive immunization with the Pseudomonas V antigen protects against type III intoxication and lung injury. Nat Med 1999, 5:392–398.
Shime N, Sawa T, Fujimoto J, Faure K, Allmond LR, Karaca T, Swanson BL, Spack EG, Wiener-Kronish JP: Therapeutic administration of anti-PcrV F(ab') 2 in sepsis associated with Pseudomonas aeruginosa . J Immunol 2001, 167:5880–5886.
Frank DW, Vallis A, Wiener-Kronish JP, Roy-Burman A, Spack EG, Mullaney BP, Megdoud M, Marks JD, Fritz R, Sawa T: Generation and characterization of a protective monoclonal antibody to Pseudomonas aeruginosa PcrV. J Infect Dis 2002, 186:64–73.
Hauser AR, Cobb E, Bodi M, Mariscal D, Valles J, Engel JN, Rello J: Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa . Crit Care Med 2002, 30:521–528.
Roy-Burman A, Savel RH, Racine S, Swanson BL, Revadigar NS, Fujimoto J, Sawa T, Frank DW, Wiener-Kronish JP: Type III Protein Secretion is Associated with Death in Lower Respiratory and Systemic Pseudomonas aeruginosa infection. J Infect Dis 2001, 183:1767–1774.
Parrillo JE, Parker MM, Natanson C, Suffredini AF, Danner RL, Cunnion RE, Ognibene FP: Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med 1990, 113:227–242.
Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S: Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 1999, 162:3749–752.
Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D: Endotoxin-tolerant mice have mutations in Toll-like receptor 4 ( tlr4 ). J Exp Med 1999, 189:615–625.
Wiener-Kronish JP, Albertine KH, Matthay MA: Differential responses of the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. J Clin Invest 1991, 88:864–875.
Beg AA: Endogenous ligands of Toll-like receptors: implications for regulating inflammatory and immune responses. Trends Immunol 2002, 23:509–512.
Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, Shirakawa AK, Farber JM, Segal DM, Oppenheim JJ, Kwak LW: Toll-like receptor 4-dependent activation of dendritic cells by β-defensin 2. Science 2002, 298:1025–1029.
Poltorak A, He X, Smirnova I, Liu MY, Huffel CV, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B: Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in tlr4 gene. Science 1998, 282:2085–2088.
Sawa T, Corry DB, Gropper MA, Ohara M, Kurahashi K, Wiener-Kronish JP: IL-10 improves lung injury and survival in Pseudomonas aeruginosa pneumonia. J Immunol 1997, 159:2858–2866.
Khabar KS, Siddiqui S, Armstrong JA: WEHI-13VAR: a stable and sensitive variant of WEHI 164 clone 13 fibrosarcoma for tumor necrosis factor bioassay. Immunol Lett 1995, 46:107–110.
Sato H, Frank DW, Hillard CJ, Feix JB, Pankhaniya RR, Moriyama K, Finck-Barbancon V, Buchaklian A, Lei M, Long RM, Wiener-Kronish J, Sawa T: The mechanism of action of the Pseudomonas aeruginosa -encoded type III cytotoxin, ExoU. EMBO J 2003, 22:2959–2969.
Geiser TK, Kazmierczak BI, Garrity-Ryan LK, Matthay MA, Engel JN: Pseudomonas aeruginosa ExoT inhibits in vitro lung epithelial wound repair. Cell Microbiol 2001, 3:223–236.
Rocchetta HL, Burrows LL, Lam JS: Genetics of O-antigen biosynthesis in Pseudomonas aeruginosa . Microbiol Mol Biol Rev 1999, 63:523–553.
Groisman EA, Ochman H: Pathogenicity islands: bacterial evolution in quantum leaps. Cell 1996, 87:791–794.
Mel SF, Mekalanos JJ: Modulation of horizontal gene transfer in pathogenic bacteria by in vivo signals. Cell 1996, 87:795–798.
Johnson GB, Brunn GJ, Tang AH, Platt JL: Evelutionalry clues to the functions of the Toll-like family as surveillance receptors. Trends Immunol 2003, 24:19–24.
Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C, Elbim C, Hitchcock R, Lammas D, Davies G, Al-Ghonaium A, Al-Rayes H, Al-Jumaah S, Al-Hajjar S, Al-Mohsen IZ, Frayha HH, Rucker R, Hawn TR, Aderem A, Tufenkeji H, Haraguchi S, Day NK, Good RA, Gougerot-Pocidalo MA, Ozinsky A, Casanova JL: Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 2003, 299:2076–2079.
Acknowledgements
This research was supported in part by the National Institutes of Health grants HL59239, AI44101 to Jeanine P. Wiener-Kronish; the National Institutes of Health grants HL067600 to Teiji Sawa; the American Lung Association RG-004-N to Teiji Sawa;, a postdoctoral research funds from Vaincre La Mucoviscidose, France, and from Bourse Lavoisier, French Ministry of Foreign Affairs to Karine Faure.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
Karine Faure and Teiji Sawa carried out animal studies. Temitayo Ajayi drafted and edited the manuscript. Junichi Fujimoto, Nobuaki Shime and Kiyoshi Moriyama participated in the animal studies. Teiji Sawa and Jeanine P. Wiener-Kronish conceived of the study, and participated in its design and coordination. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Faure, K., Sawa, T., Ajayi, T. et al. TLR4 signaling is essential for survival in acute lung injury induced by virulent Pseudomonas aeruginosa secreting type III secretory toxins. Respir Res 5, 1 (2004). https://doi.org/10.1186/1465-9921-5-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1465-9921-5-1