
Abstract
Neural tube defects (NTDs) are the second most common birth defect in humans. Despite many advances in the understanding of NTDs and the identification of many genes related to NTDs, the fundamental etiology for the majority of cases of NTDs remains unclear. Planar cell polarity (PCP) signaling pathway, which is important for polarized cell movement (such as cell migration) and organ morphogenesis through the activation of cytoskeletal pathways, has been shown to play multiple roles during neural tube closure. The disrupted function of PCP pathway is connected with some NTDs. Here, we summarize our current understanding of how PCP factors affect the pathogenesis of NTDs.
Similar content being viewed by others
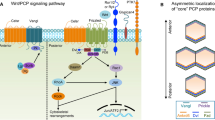

Background
Neural tube defects (NTDs), arise when the neural tube, the embryonic precursor of the brain and spinal cord, fails to close during neurulation. Defects in neural tube closure are the second most common human birth defects, after congenital heart defects [1]. Recent birth prevalence estimates show that NTDs account for 0.5 per 1000 in the United States during 2001-2004, 1 to 1.5 per 1000 in Western Australia during 2001-2006, and 2.8 per 1000 in Iran during 1998-2005, while prevalence in Shanxi, a province in North China, reach to 19.9 per 1000 during 2002-2004 [2].
The cranial region (anencephaly) or the low spine (open spina bifida and myelomeningocele) are most commonly affected [3]. NTDs affecting the brain are invariably lethal perinatally, whereas open spina bifida is compatible with postnatal survival but frequently results in serious handicap, because neurological impairment below the lesion leads to lack of sensation, inability to walk and incontinence [4].
Neural tube formation and NTDs classification
Neural tube closure is the result of neurulation, a process in which the neural plate bends upwards and eventually fuses to form the hollow tube that will become the brain and the spinal cord. The driving force of neural tube closure is provided and maintained by cells undergoing convergence and extension (CE) [5].
Both fish (such as zebrafish) and amphibian (such as Xenopus) embryos require this process [6, 7]. Neurulation is conserved between mammalian species [8] and can be conventionally divided into primary and secondary phases [9].
In primary neurulation, the fusion occurs along the spine and culminates in final closure at the posterior neuropore. Closure is initiated at the hindbrain/cervical boundary (Closure 1) and then spreads bi-directionally into the hindbrain and along the spinal region. Separate closure initiation sites occur at the midbrain-forebrain boundary (Closure 2) and at the rostral extremity of the forebrain (Closure 3). However, Closure 2 found in mice may be absent from human neurulation [10].
The secondary phase occurs at lower sacral and caudal levels, where the neural tube is formed in the tail bud without neural folding [4, 11].
Failure of Closure 1 leads to the most severe NTD, craniorachischisis, which combines an open neural tube encompassing the midbrain, hindbrain and entire spinal region. If Closure 1 is completed but closure of the cranial neural tube is incomplete, anencephaly develops, with cases exhibiting either defects confining in the midbrain (meroanencephaly) or lesions extending into the hindbrain (holoanencephaly) [12]. Failure of Closure 3 is uncommon but, when present, yields split face with anencephaly. In the spinal region, failure of final closure at the posterior neuropore yields open spina bifida (also called myelocele or myelomeningocele), in which the upper limit can be of varying axial level [9]. By contrast, defective secondary neurulation leads to 'closed' forms of spina bifida [9].
Human NTDs and possible causes
Epidemiological studies provide an opportunity to identify risk factors for NTDs, such as dietary or teratogenic agents, to which susceptibility may be modified by genetic predisposition [3, 13, 14]. Identification of causative factors is confounded by the fact that the majority of these malformations appears to result from a combination of genetic and non-genetic factors (environmental contributions) [3].
Many non-genetic factors may be associated with NTDs formation. They include: parental socioeconomic status [15, 16], parental age [17], parental race [18], hyperthermia during early pregnancy [19], maternal health (such as diabetes [20], obesity [21]), dietary agents or maternal nutrition (such as the uptake of folate [22–24], inositol [25, 26]), chemical teratogenic agents (such as valproic acid [27], retinoic acid [28], trichostatin A [29], exposure to pesticides [30] and selective serotonin-reuptake inhibitors [31] and so on).
As for genetic factors, the cumulative number of reported mouse genetic mutants with NTDs continues to rise steadily, from approximately 200 in 2007 [32] to approximately 245 in 2010 [33]. The different mouse gene mutations, naturally occurring or targeted mutations, are associated with various NTD phenotypes [3, 9, 32]. Many of the NTD-causing mouse mutations implicate specific signaling pathways such as PCP signaling, Sonic hedgehog (Shh) signaling, BMP signaling, Notch signaling, retinoid signaling and inositol metabolism [4]. Those signaling pathways are involved in the maintenance of the cell cycle, the regulation of the actin cytoskeleton, chromatin organization and epigenetic modifications including methylation and acetylation [3].
However, although there is evidence for a strong genetic component in the individual liability to NTDs in humans, little is known about the nature of these risk genes about their interactions with each other. In general, the risk genes are present in the affected individuals. However, it is unknown whether the same risk genes are shared by all population [33].
Meanwhile, gene-dosage can also affect neural tube closure. Chromosomal abnormalities, especially trisomy 13 and 18, are strongly associated with central nervous system malformations [34, 35], and a gene dosage imbalance of 16q12.1-q22.1 is also associated with spina bifida in the patient [36].
Recently, a major advance in understanding of the genetic basis of neurulation is the finding that the initiation of Closure 1 requires noncanonical Wnt signaling, the so-called planar cell polarity (PCP) signaling pathway [3].
PCP signaling pathway
PCP, which is within the plane of an epithelium, is not restricted to epithelial tissues, but is also found in mesenchymal cells during animal development [5].
There are two evolutionarily conserved sets of PCP factors that act together to coordinate PCP establishment: the Frizzled (Fz)/Flamingo (Fmi) core genes and the Fat/Dachsous (Ds) PCP system [5].
In Fat/Ds system, Dachsous (Ds) and Fat (Ft), together with a transmembrane Golgi complex protein, Four-jointed (Fj), set up a global polarity signal, which is then sensed and propagated by the asymmetric assembly of cell-surface complexes, transmitting signal between cells [37–40]. Members of the Fat/Ds group are expressed in gradients and their graded expression is under the control of canonical Wg-signaling [41, 42]. It has been suggested that Fat/Ds acts upstream of Fz/PCP signaling, largely based on data on the fly eye [39, 41]. However, recent genetic mosaic experiments in the Drosophila abdomen argue that these two systems may function in parallel rather than in series [43]. As there is no report about the relationship between Fat/Ds system and NTDs, in this review, we will not discuss the system in detail.
The Fz/Fmi system is the principal PCP signaling pathway and appears to be the "noncanonical" Wnt signaling pathway [44]. The components of the Fz/Fmi system include transmembrane proteins, such as Frizzled (Fz), Flamingo (Fmi, Celsr1 in in human and rodents), Strabismus/Van Gogh (Stbm/Vang) and intracellular proteins, such as Dishevelled (Dsh in Drosophila; Dvl in vertebrates), Prickle (Pk), and Diego (Dgo; Diversin in vertebrate and inversin in mouse). Scribble (Scrib) [45, 46] and Ptk [47, 48] are sometimes regarded as PCP proteins. All the components work together, through either coordination or antagonism. For example, Vang/Pk is thought to antagonize the Fz/Dvl signaling [49, 50].
The PCP system, to which Wnt5a and Wnt11 have been clearly linked in vertebrates, is related to the canonical Wnt signaling pathway, which interprets the directional signal to produce subcellular asymmetries [37, 44, 51–54] Downstream of the PCP system are so-called 'PCP effector', which are the novel proteins, Inturned, Fuzzy and Fritz [55, 56]. They mediate the PCP signaling in different tissues. This system can play an important role in polarized cell movement (cell migration) and organ morphogenesis through the activation of cytoskeletal pathways, such as the small GTPases RhoA and cdc42, Rho kinase, protein kinase C (PKC) and Jun N-terminal kinase (JNK) 1 [51, 57]. Activation of the PCP signaling in a given cell population is able to exert changes in neighboring cells that do not express PCP elements [58].
The role of PCP signaling pathway in NTDs
The genetic and molecular dissection of PCP began 29 years ago with the realization by Gubb and Garcia-Bellido that a small set of genes controls the polarity of cuticular hairs and bristles in Drosophila[44, 59].
At that time many vertebrate tissues and developmental processes have been shown to display typical PCP features [51, 60, 61]. Time-lapse studies in Xenopus revealed that PCP-dependent CE was required to narrow the distance between the elevating neural folds, allowing their apposition and fusion [62]. Other analyses in Xenopus [63, 64], zebrafish [65, 66] and mouse [67] also show that the PCP factors are key players in the process of CE movement during gastrulation and neurulation.
For a more detailed understanding of the PCP pathway in zebrafish gastrulation, Gong observed that PCP pathway plays a conserved role in vertebrate axis elongation, orienting both cell intercalation and mitotic division [68]. However, Ciruna et al have shown that PCP pathway is required for the reintegration of newly postmitotic cells into the neuroepithelium [69]. They also observed that loss of Vangl2 (trilobite) leads to an accumulation of apical daughter cells from recent mitoses in the center of the U-shaped, and incompletely closed, neural fold [69]. A striking demonstration that the failure to reintegrate these cells underlies the neural tube closure defect came from the observation that pharmacologically blocking cell division in the trilobite mutant late in gastrulation restores neural tube closure, presumably because without cell division there are no extruded cells [69]. By contrast, mitotic inhibitors did not rescue the CE phenotype caused by the trilobite mutation [44].
For the spatio-temporal expression, PCP is believed to initiate Closure 1 in mice [3]. In another perspective, the PCP pathway is believed to be responsible for caudal NTDs, though Dvl2-/- mice also display some rostral defects [5, 70], while the Shh pathway accounts for most of the rostral defects [5, 55]. However, in Patched1 null mice, both rostral and caudal defects are seen [71], suggesting that both pathways act at different stages during neurulation. When Shh pathway regulates neural plate bending and specification of the ventral neural cell fates, the PCP pathway drives neural tube closure [72].
PCP protein mutations and NTDs
When the correct expressivity of proteins in PCP signaling is disturbed, caused either by environmental factors or by genetic factors, some NTDs can occur.
Frizzled (Fz)
Fz, the first PCP gene to be defined molecularly, and also a member of the Wnt receptor family, codes seven transmembrane helices [73] and an amino-terminal cysteine-rich domain (CRD) that is sufficient and necessary for binding with the ligands of the Wnts [74–76]. It can also bind Dsh and recruit Dsh and Dgo to the membrane. In mammals, Fz genes have been implicated in a variety of developmental processes, including the nervous system formation. Fz3 is required for axonal outgrowth and guidance in the CNS [77, 78]. Fz3 can also play a role during sympathetic neuron development via the activation of β-catenin [79].
During the gastrulation in Xenopus, overexpression of Fz7 (Xfz7) in the dorsal equatorial region affects the CE movement and causes a delay of the mesodermal development [80]. In the mouse, Fz3 and Fz6 play a role in neural tube closure. Fz3-/-; Fz6-/- embryos exhibit craniorachischisis with nearly 100% penetrance, and these mice die within minutes after birth [81]. Fz1 and/or Fz2 mutations can cause defects in neural tube closure [82].
Flamingo (Fmi)/Starry night(Stan)/Celsr1
Three Fmi gene orthologs in human and rodents are named celser1 - celser3 respectively. Fmi genes encode proteins of the cadherin superfamily which are seven transmembrane proteins with nine cadherin repeats in the extracellular domain, and an uncharacterized intracellular C terminus. The Drosophila Fmi gene regulates epithelial planar cell polarity and dendritic field deployment [83, 84]. Recent studies show that the primary function of Fmi is to participate in the asymmetry of PCP [85, 86]. In mouse, the homozygous Celsr1 mutants (Crsh and Scy) exhibit severe neural tube defects, such as craniorachischisis, as a result of failure to initiate neural tube closure, providing evidence for the function of the Celsr family that are involved in a planar cell polarity pathway in vertebrate neurulation [87].
Strabismus (Stbm)/Van Gogh (Vang)/vangl
Vangl1 and Vangl2 are mammalian homologs of Drosophila gene Van Gogh (Vang), also known as Strabismus in which mutations disrupt the organization of various epithelial structures, causing characteristic swirled patterns of hairs on wing cells and misorientation of eye ommatidia [88]. Exon-intron structure of mammalian Vangl1 and Vangl2 orthologs was well conserved [89]. Vangl2 encodes a membrane protein comprising four transmembrane domains and a large intracellular domain with a PDZ-domain-binding motif at its carboxy terminus [90].
Vangl2 can modulate actin cytoskeleton through the small GTPases RhoA and Rac and the downstream Rho kinase. Thus it is partially responsible for a variety of changes in cell adhesion, polarity, and short-range tissue movements [91].
Studies of Stbm genes and the proteins that they encode in mice, flies, frogs and fish have shown that they have a crucial role in regulating planar cell polarity and convergent extension movements [88]. In fly mutated embryos, the polarity of the ommatidia of the compound eye and the hairs of the wing and thorax are disrupted, such that rather than pointing in the same direction, they point in multiple directions [92]. In zebrafish, trilobite mutant embryos (loss of Stbm) have defects in gastrulation movements and posterior migration of hindbrain neurons [65], resulting in ectopic neural progenitor accumulations and NTDs [69]. In Xenopus, the homolog of Stbm is called xstbm. The xstbm can regulate convergent extension in both dorsal mesoderm and neural tissue by either increasing or decreasing the Vangl2 function due to its optimal retard of convergent extension movements [93]. Reduction of xstbm function using a morpholino antisense oligo also causes the trunk shortening [94].
Loop-tail (LtapLp, also called as Lp, Ltap, Lpp1) gene is a semidominant mutation that affects neurulation in mice, which are characterized by a looped-tail appearance (pig tail) and wobbly head movements while homozygous embryos exhibit a neural tube closure defect that extends from the caudal midbrain to the tip of the tail [95]. A potential role of PCP in NTDs came to light following positional cloning of Vangl2 in the loop-tail mouse mutants that exhibit a severe NTD, craniorachischisis [90, 96]. Subsequently, several studies have shown that Vangl2 can also interact with different genes and cause several forms of NTDs. For example, Dvl3+/-; LtapLp/+ can cause craniorachischisis or exencephaly. Dvl3-/-; LtapLp/+mutants cause craniorachischisis [97]. Genetic interaction between Wnt5a and Ltap/Vangl2 could enhance the penetrance of neural tube closure and all Wnt5a-/-; LtapLp/+ mice exhibited craniorachischisis [98]. Sequence analysis has not been success thus far in identifying the mutations in human Vangl2 gene in patients with craniorachischisis [99], although the Vangl2 mutation was identified in stillborn or miscarried fetuses with neural-tube defects [100].
However, the mutation in Vangl1 was found in patients with familial and sporadic NTDs, who exhibited a caudal neural tube, including craniorachischisis. Furthermore, the result showed that the Vangl1 mutations disrupted the physical interaction with Dvl [101]. These data indicate that Vangl1 is a risk factor in human neural-tube defects. Later, mutations in Vangl1 were detected in spinal dysraphisms, providing further evidences to support the role of Vangl1 as a risk factor in the development of spinal NTDs [102].
Disheveled (Dsh/Dvl)
Disheveled proteins are important signaling components in both the canonical β-catenin/Wnt pathway [103], and the PCP pathway [97]. It is a cytoplasmic protein containing DIX, PDZ, DEP domains and is recruited to membrane by Fz, undergoing extensive phosphorylation. Homologues of Disheveled are Xdsh in Xenopus, and Dvl1, Dvl2 and Dvl3 in vertebrate. Disheveled is highly conserved and play an important role in CE movement. In PCP pathway, Disheveled acts in the downstream of Wnt11 and Wnt5a and the upstream of Ca2+/CamKII, JNK, and the Rho GTPase family members RhoA, Rac1, and Cdc42 [104].
In vertebrate, Dvl1, Dvl2 and Dvl3 participate in the CE movement. Dvl 1-/-[105], Dvl3-/-and Dvl1-/-; Dvl3-/- double mutants [7] do not display neural tube defects. Mice with targeted inactivation of the Dvl1 gene were found to exhibit alterations in sensorimotor gating and social interaction [105] and Dvl2 does not seem to play a similar role in the same way [70]. Dvl2-/- embryos displayed thoracic spina bifida, while virtually all Dvl1/2 double mutant embryos displayed a craniorachishisis, a completely open neural tube from the midbrain to the tail [7, 70]. For Dvl3, which is also required for signals in the PCP pathway to regulate the CE movement during the development of the neural tube, neurulation appeared normal both Dvl3-/- and LtapLp/+(Vangl2/Ltap) mutants, while defects were seen in both Dvl3+/-;LtapLp/+(7/22, 32%, 5 with craniorachischisis and 2 with exencephaly) and Dvl3-/-;LtapLp/+mutants (in a total of 16 mutants, 6 with craniorachischisis) [97]. These findings indicate that Dvl2 is the most important mammalian Dvl gene for neural tube closure and is sufficient by itself for normal neural tube closure. By contrast, Dvl1 and Dvl3 are not sufficient by themselves for a normal neural tube closure, but contribute significantly when Dvl2 is completely missing [7].
Diego (Dgo)/Diversin
Diego, comprises six ankyrin repeats and is co-localized with Flamingo at proximal/distal boundaries [106]. The homologue of Diego is Diversin in vertebrate and Inversin in mouse [44]. Diversin is also an essential component of the Wnt signaling pathway [107] and its centrosomal localization is crucial for its function in the Wnt signaling [108]. Diversin controls the balance between canonical and noncanonical Wnt signaling, with a higher diversin activity favoring PCP signaling and a lower diversin activity favoring canonical signaling [44].
In PCP pathway, Diversin act downstream of Wnt11 and Wnt5a and upstream of the small GTPases Rac and Rho [109]. In zebrafish [104] and Xenopus [62], knockdown of Diversin disrupts convegent extension. Div-ANK mRNA injection also disturbed CE in zebrafish embryos, which can be rescued by co-injection of mouse Inversin mRNA [104]. Moreover, combinations of low concentrations of Wnt11/5a Morpholino oligonucleotide (MO) and Div-ΔANK, which alone were virtually ineffective, acted synergistically in inducing strong CE phenotypes [104]. However, Diversin mRNA was unable to rescue the defects caused by Dishevelled lacking the DEP domain. and it's the same in reverse, although the two protein can interact [104].
Prickle (Pk)
Pk gene encodes a protein with a triple LIM domain and a novel domain that is present in human and murine. Caenorhabditis elegans has a homolog that is designated as PET. Three transcripts have been identified, Pk, PkM, and sple. In PCP signal pathway, Stbm/Vang and Pk antagonize Fz-Dsh activity [49, 50, 85]. Lack of both Pk and sple transcripts gives a phenotype that affects the whole body surface that is similar to those caused by deficiency of disheveled and Fz [110].
In zebrafish, both of homologs of Pk show a discrete and dynamic expression pattern during gastrulation. Both gain and loss of Pk1 function cause defects in convergent extension movement. In overexpression assays, Pk1 can inhibit the activation of Wnt/β-catenin signaling [111].
In Xenopus, orthologues of Pk is XPk, which expressed in tissues at the dorsal midline during gastrulation and early neurulation [112]. Both gain-of-function and loss-of-function of XPk severely perturbed gastrulation and caused a spina bifida in embryos, but no influence in mesodermal differentiation [113].
Global polarization
The appropriate function of the PCP pathway in neurulation can ensure a normal global polarization, which not only means that the cells in the plane coordinate with each other, but also demands that the tissues develop harmoniously within the whole body. One attractive model in PCP pathway is Fat/Ds system. However this system is not involved in the development of NTDs. Recent studies show that this system, the Fz/Fmi system, and also the principal PCP signaling pathway, function in parallel [43].
Asymmetric arrangement
The specific, highly controlled, asymmetric arrangement of these PCP core components, appearing to be highly sensitive to the orientation of the cell's sides with respect to the global axis of the epithelium, allows the polarity of the cell to be established within the plane of the epithelium and promotes the rearrangement of the cytoskeletal components of the cell [97]. Although the asymmetric localization of some of the PCP factors has been documented in some vertebrate tissues, for example, during zebrafish gastrulation and neurulation, a complete data set and thus an equivalent model to Drosophila do not yet exist [5]. The asymmetric distribution of core PCP components such as Pk1 in the neural plate has recently been shown to be essential for neural tube closure [114]. Another example is that the asymmetric localization of Pk and Dsh during zebrafish convergent extension processes [115]. The fluorescent fusion proteins during dorsal mesoderm CE movement have shown that Pk localizes at the anterior cell edge, whereas Dsh is enriched posteriorly. The asymmetrical localization of Pk and Dsh observed in zebrafish gastrula is similar to their localization in fly, suggesting that noncanonical Wnt signaling defines distinct anterior and posterior cell properties to bias cell intercalations [115].
Wnt signaling pathway
Wnt signaling plays a critical role in a vast array of biological process, including cell proliferation, migration, polarity establishment and stem cell self-renewal [103]. Wnt5a and Wnt11 are the core members in Wnt pathway and also are clearly linked to the PCP signaling pathway. It has been reported that Wnt5a/pipetail and Wnt11/silberblick control CE movement in zebrafish embryogenesis via the PCP pathway [116–119].
Wnt11 is thought to be involved in the CE movement taking place during gastrulation and perhaps more broadly during organogenesis [120]. Zebrafish Wnt11 mutants silberblick (Slb) have typical convergent extension phenotypes [117]. Wnt5a can genetically interact with Ltap/Vangl2 to regulate neural tube closure. All Wnt5a-/-;LtapLp/+ mutants exhibited craniorachischisis, indicating a drastic increase in penetrance as compared to the craniorachischisis phenotype displayed by Wnt5a-/- (1 in 34) or LtapLp/+ animals (0 in more than 100) [98].
Disheveled is a core component in both the PCP pathway and the Wnt pathway [103]. In zebrafish, slb phenotype, abnormal CE movement during gastrulation can be rescued by a truncated form of Disheveled [117]. In overexpression assays, Pk1 can inhibit activation of Wnt signaling during zebrafish CE movements of gastrulation [111].
Diversin, a homologue of Diego in vertebrate, is an essential component of the Wnt signaling pathway [107] and its centrosomal localization is crucial for its function in the Wnt signaling pathway [108]. Inversin, a homologue of Diego in mouse, can control the balance between canonical and noncanonical Wnt signaling [121]. A higher Inversin activity favors the noncanonical signaling (i.e. the PCP pathway) and a lower Inversin activity favors the canonical signaling [44].
Diversin, comprised six ankyrin repeats, can rescue CE phenotypes induced by Wnt11/5a MO. Also combinations of low concentrations of Wnt11/5a MO and Div-ΔANK, which alone were virtually ineffective, acted synergistically in inducing strong CE phenotypes, suggesting that Wnt5a and Wnt11 can control CE movement in zebrafish embryogenesis through Diversin [122].
Cilia
In vertebrates, many, if not all, epithelial cells have a single nonmotile cilium (the primary cilium), which is typically located in the center of the apical face of the cell [44]. Cilia are microtubule-based protrusions and are an important nexus for cellular signaling. They are apparently a critical junction between the signals that influence cell fate and the signals that influence cell movement [55].
Connections has recently been found between PCP and non-motile cilia based on the observation that several genes that affect vertebrate PCP also affect ciliary structure and/or function [123, 124].
Bardet-Biedl syndrome (BBS) is a pleiotropic disorder characterized by age-related retinal dystrophy, obesity, polydactyly, renal dysplasia, reproductive tract abnormalities and cognitive impairment. It is genetically heterogeneous, with mutations identified in several BBS genes. A connection has been found between BBS genes and PCP [38, 125]. 14% of Bbs4-/- mice display an open cephalic neural tube (exencephaly) [125]. MO knockdown of BBS4 in zebrafish leads to PCP phenotypes, including a failure of embryonic CE movement [44]. Other evidence suggesting a molecular connection between PCP and cilia comes from studies on the ciliary protein Inversin. This protein has been studied for some time in the context of cilia function, and it is also the core protein in PCP [55].
This connection between PCP signaling and a known ciliary protein became even more evident with the finding that the PCP proteins Vangl2 and DVL are localized at or near the base of cilia in vertebrate cells [125, 126].
The most recent link between PCP and cilia comes from experiments with Xenopus embryos in which disruption of Inturned or Fuzzy elicited prominent rostral neural tube closure defects in addition to more caudal neural tube defects. These defects were shown to arise from failure of both PCP and Shh signaling [126]. It is clear is that several signal transduction proteins must localize to cilia for Shh signal transduction to proceed normally [127]. This suggests that Inturned and Fuzzy play a role in ciliary structure or function.
The differences among species
Studies in Drosophila, zebrafish, Xenopus, mice, and human beings have revealed that similarities, as well as differences, exist in the PCP pathway and in the development of NTDs. The most important is that the principal PCP signaling pathway is highly conserved across species and tissues [5].
The numerous differences among species in anatomy, tissue types and morphogenetic processes, together with the existence of a number of distinct PCP components make it interesting to think about the difference in the development of TNDs among different species.
For example, Scrib and Ptk7, for which there is no evidence in Drosophila regarding a role in PCP, were associated with the PCP phenotypes in vertebrates when they were mutated, either alone and or in combination with other PCP gene mutations [128, 129]. Other examples are genes such as Inturned and Fuzzy. They are considered to be the PCP effector genes in Drosophila and have been found to be associated with a convergent extension phenotype in frog or fish embryos [126]. The full length transcript of mouse Scrib is about 5,547 bp and encodes a putative protein containing 1,665 amino acids, which exhibits 88% homologue with human SCRB1, 44% homologue with Drosophila Scribble and 36% homologue with C. elegan s protein LET-413 [129].
Most PCP genes have only one isoform in zebrafish, Xenopus, whereas in other species such as rodents, there are often numerous isoforms (for example, 3 Dvls, 2 Vangls, 2 Prickles, 3 Celsrs, etc). Furthermore, the expression of some isoforms is not overlapped. As such, the studies on PCP generation in mice have been hampered because of the redundancy of the PCP genes. These studies require a more detailed analysis using as many tissues as possible. Double and triple knockout mouse lines are often required and the necessary involvement of these models makes investigations lengthy and tedious [44].
From mouse to man
At the embryonic level, the events of neurulation appear extremely similar between mice and humans. As a result, mouse models are commonly used in the research of NTDs. There are over 200 different mouse genes that result in NTD phenotypes either through naturally occurring mutations or through the targeted mutations [9, 32]. Several mouse mutants involved in PCP signaling pathway for NTDs research, such as looptail[81, 130–132], circletai l [129, 133, 134], crash[87, 134], dishevelled knockout mouse [7, 70, 97], BBS- null mouse [125], frizzled 3 and frizzled 6 double mutants [81], Sfrp1, Sfrp2, and Sfrp5 compound mutant mice [135] and so on.
The human homologues of some of these mouse NTD genes have been examined in case-control association studies or directly sequenced in mutation screens, although with very few significant findings to date [3, 99–102].
So we have the reason to ask whether it is appropriate to use mouse models for the studies of human NTDs [3].
First, in the process of neural tube closure, Closure 2 in mice is thought to be absent in human neurulation [10], suggesting that neural tube closure may follow a somewhat different process in humans [3].
Secondly, many gene-specific homozygous null mouse embryos exhibit additional phenotypes besides NTDs, such as prenatally lethal heart defect. Such syndrome-like examples do not appear particularly close to the models of human NTDs [3]. Also, some mutations may be lethal to human fetus such as Vangl2 [100]. As a result, those embryonic lethal cases are unlikely to become the subject of successful studies.
Thirdly, detailed analysis of a few of the mouse mutants suggests that isolated NTDs can also result from the effect of hypomorphic alleles, combinations of heterozygous mutations, genetic background effects and/or gene-environmental interactions. This partial loss of function or multi-factorial etiologies may more closely resemble to human NTDs [3].
Outlook
Kibar and colleagues have identified three VANGL1 mutations (V239I, R274Q, and M328T) in patients with sporadic and familial neural-tube defects [101]. However, the phenotype associated with V239I varied among patients. Notably, the mother of the proband with the V239I de novo mutation did not have NTD. They think this finding is consistent with the proposed multi-factorial model for NTD formation. V239I has probably a partial or complete loss of function effect and it interacts with other genetic loci or unknown environmental factors to modulate the incidence and severity of the defect [101].
As discussed above, the development of NTDs is associated with multi-factors. To date, the concept is commonly accepted that the development of NTDs is related to the gene mutations and the gene interaction with other environment factors, which can explain some inexplicable phenomena related to deficiency [136], inositol [25, 137], diabetes [138], We think that the gene-environmental interaction is an important process in which the environmental factors can affect the gene expression and affect the process of transcription and translation.
Conclusions
In this paper, we have reviewed recent studies and highlighted an intimate relationship between PCP signaling pathway and the development of NTDs. The nature of this relationship remains to be further studied. What is certain is that the PCP, also called tissue polarity, is not only restricted to epithelial tissues, but is also found in mesenchymal cells throughout animal development. The PCP signaling pathway is highly conserved in various species, which mediates changes in cell polarity and cell motility in neurulation, through the activation of cytoskeletal pathways, such as RhoA and Rho kinase. Several components of the PCP pathway are expressed in the process of neural tube closure, and the disrupted function of the PCP pathway members in Xenopus, zebrafish and mouse are connected with various defects, and final lead to NTDs. In this process, the interaction of proteins within PCP pathway and PCP proteins with proteins in other pathways are also demonstrated. Although gene mutations in PCP that cause NTDs in humans are rarely reported, it is noted that environmental factors and other genetic factors may affect the expression of the PCP genes.
References
Detrait ER, George TM, Etchevers HC, Gilbert JR, Vekemans M, Speer MC: Human neural tube defects: Developmental biology, epidemiology, and genetics. Neurotoxicology and Teratology. 2005, 27: 515-524.
Gu X, Lin L, Zheng X: High prevalence of NTDs in Shanxi Province: A combined epidemiological approach. Birth Defects Research Part A: Clinical and Molecular Teratology. 2007, 79: 702-707. 10.1002/bdra.20397.
Greene NDE, Stanier P, Copp AJ: Genetics of human neural tube defects. Human Molecular Genetics. 2009, 18: R113-R129. 10.1093/hmg/ddp347.
Copp AJ, Greene NDE: Genetics and development of neural tube defects. The Journal of Pathology. 2010, 220: 217-230.
Simons M, Mlodzik M: Planar cell polarity signaling: from fly development to human disease. Annu Rev Genet. 2008, 42: 517-540. 10.1146/annurev.genet.42.110807.091432.
Kieserman EK, Wallingford JB: In vivo imaging reveals a role for Cdc42 in spindle positioning and planar orientation of cell divisions during vertebrate neural tube closure. J Cell Sci. 2009, 122: 2481-2490. 10.1242/jcs.042135.
Wang J, Hamblet NS, Mark S: Dishevelled genes mediate a conserved mammalian PCP pathway to regulate convergent extension during neurulation. Development. 2006, 133: 1767-1778. 10.1242/dev.02347.
Harrington MJ, Hong E, Brewster R: Comparative analysis of neurulation: First impressions do not count. Molecular Reproduction and Development. 2009, 76: 954-965. 10.1002/mrd.21085.
Copp AJ, Greene NDE, Murdoch JN: The genetic basis of mammalian neurulation. Nat Rev Genet. 2003, 4: 784-793. 10.1038/nrg1181.
Greene NDE, Copp AJ: Development of the vertebrate central nervous system: formation of the neural tube. Prenatal Diagnosis. 2009, 29: 303-311. 10.1002/pd.2206.
Cai W, Zhao H, Guo J, Li Y, Yuan Z, Wang W: Retinoic acid-induced lumbosacral neural tube defects: myeloschisis and hamartoma. Child's Nervous System. 2007, 23: 549-554. 10.1007/s00381-006-0289-y.
Wyszynski DF: Neural tube defects: from origin to treatment. 2006, Oxford University Press
Mitchell LE: Epidemiology of neural tube defects. American Journal of Medical Genetics Part C: Seminars in Medical Genetics. 2005, 135C: 88-94. 10.1002/ajmg.c.30057.
Au KS, Ashley-Koch A, Northrup H: Epidemiologic and genetic aspects of spina bifida and other neural tube defects. Developmental Disabilities Research Reviews. 2010, 16: 6-15. 10.1002/ddrr.93.
Grewal J, Carmichael SL, Song J, Shaw GM: Neural tube defects: an analysis of neighbourhood- and individual-level socio-economic characteristics. Paediatric and Perinatal Epidemiology. 2009, 23: 116-124. 10.1111/j.1365-3016.2008.00992.x.
Canfield MA, Ramadhani TA, Shaw GM: Anencephaly and spina bifida among Hispanics: Maternal, sociodemographic, and acculturation factors in the National Birth Defects Prevention Study. Birth Defects Research Part A: Clinical and Molecular Teratology. 2009, 85: 637-646. 10.1002/bdra.20582.
Vieira AR, Castillo Taucher S: Maternal age and neural tube defects: evidence for a greater effect in spina bifida than in anencephaly. Rev Med Chil. 2005, 133: 62-70.
Njamnshi AK, Djientcheu VdP, Lekoubou A: Neural tube defects are rare among black Americans but not in sub-Saharan black Africans: The case of Yaounde -- Cameroon. Journal of the Neurological Sciences. 2008, 270: 13-17. 10.1016/j.jns.2008.01.010.
Moretti ME, Bar-Oz B, Fried S, Koren G: Maternal Hyperthermia and the Risk for Neural Tube Defects in Offspring: Systematic Review and Meta-Analysis. Epidemiology. 2005, 16: 216-219. 10.1097/01.ede.0000152903.55579.15. 210.1097/1001.ede.0000152903.0000155579.0000152915
Loeken MR: Current perspectives on the causes of neural tube defects resulting from diabetic pregnancy. American Journal of Medical Genetics Part C: Seminars in Medical Genetics. 2005, 135C: 77-87. 10.1002/ajmg.c.30056.
Ray JG, Wyatt PR, Vermeulen MJ, Meier C, Cole DEC: Greater maternal weight and the ongoing risk of neural tube defects after folic acid flour fortification. Obstet Gynecol. 2005, 105: 261-265. 10.1097/01.AOG.0000151988.84346.3e.
Fleming A, Copp AJ: Embryonic Folate Metabolism and Mouse Neural Tube Defects. Science. 1998, 280: 2107-2109. 10.1126/science.280.5372.2107.
Kiberstis P: An Absorbing Tale. Science. 2006, 314: 1659-
De Wals P, Tairou F, Van Allen MI: Reduction in Neural-Tube Defects after Folic Acid Fortification in Canada. New England Journal of Medicine. 2007, 357: 135-142. 10.1056/NEJMoa067103.
Greene ND, Copp AJ: Inositol prevents folate-resistant neural tube defects in the mouse. Nat Med. 1997, 3: 60-66. 10.1038/nm0197-60.
Cogram P, Hynes A, Dunlevy LPE, Greene NDE, Copp AJ: Specific isoforms of protein kinase C are essential for prevention of folate-resistant neural tube defects by inositol. Human Molecular Genetics. 2004, 13: 7-14.
Gurvich N, Berman MG, Wittner BS, Gentleman RC, Klein PS, Green JBA: Association of valproate-induced teratogenesis with histone deacetylase inhibition in vivo. FASEB J. 2005, 19: 1166-1168.
Liu J, Qi J, Zhu J: Effects of Retinoic Acid on the Expressions of Vangl1 and Vangl2 in Mouse Fetuses. Journal of Neurogenetics. 2008, 22: 167-179. 10.1080/01677060802049605.
Menegola E, Di Renzo F, Broccia ML: Inhibition of histone deacetylase activity on specific embryonic tissues as a new mechanism for teratogenicity. Birth Defects Research Part B: Developmental and Reproductive Toxicology. 2005, 74: 392-398. 10.1002/bdrb.20053.
Brender JD, Felkner M, Suarez L, Canfield MA, Henry JP: Maternal Pesticide Exposure and Neural Tube Defects in Mexican Americans. Annals of Epidemiology. 2010, 20: 16-22. 10.1016/j.annepidem.2009.09.011.
Alwan S, Reefhuis J, Rasmussen SA, Olney RS, Friedman JM: Use of Selective Serotonin-Reuptake Inhibitors in Pregnancy and the Risk of Birth Defects. New England Journal of Medicine. 2007, 356: 2684-2692. 10.1056/NEJMoa066584.
Harris MJ, Juriloff DM: Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Research Part A: Clinical and Molecular Teratology. 2007, 79: 187-210. 10.1002/bdra.20333.
Harris MJ, Juriloff DM: An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Research Part A: Clinical and Molecular Teratology. 2010, 88: 653-669. 10.1002/bdra.20676.
Sepulveda W, Corral E, Ayala C, Be C, Gutierrez J, Vasquez P: Chromosomal abnormalities in fetuses with open neural tube defects: prenatal identification with ultrasound. Ultrasound in Obstetrics and Gynecology. 2004, 23: 352-356. 10.1002/uog.964.
Goetzinger KR, Stamilio DM, Dicke JM, Macones GA, Odibo AO: Evaluating the incidence and likelihood ratios for chromosomal abnormalities in fetuses with common central nervous system malformations. American Journal of Obstetrics and Gynecology. 2008, 199: 285.e281-285.e286.
Gustavsson P, Schoumans J, Staaf J, Borg Å, Nordenskjöld M, Annerén G: Duplication 16q12.1-q22.1 characterized by array CGH in a girl with spina bifida. European Journal of Medical Genetics. 50: 237-241.
Klein TJ, Mlodzik M: Planar cell polarization: an emerging model points in the right direction. Annu Rev Cell Dev Biol. 2005, 21: 155-176. 10.1146/annurev.cellbio.21.012704.132806.
Yen H-J, Tayeh MK, Mullins RF, Stone EM, Sheffield VC, Slusarski DC: Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer's vesicle cilia function. Human Molecular Genetics. 2006, 15: 667-677. 10.1093/hmg/ddi468.
Ma D, Yang C-h, McNeill H, Simon MA, Axelrod JD: Fidelity in planar cell polarity signalling. Nature. 2003, 421: 543-547. 10.1038/nature01366.
Lawrence PA, Casal J, Struhl G: Cell interactions and planar polarity in the abdominal epidermis of Drosophila. Development. 2004, 131: 4651-4664. 10.1242/dev.01351.
Yang C-h, Axelrod JD, Simon MA: Regulation of Frizzled by Fat-like Cadherins during Planar Polarity Signaling in the Drosophila Compound Eye. Cell. 2002, 108: 675-688. 10.1016/S0092-8674(02)00658-X.
Matakatsu H, Blair SS: Interactions between Fat and Dachsous and the regulation of planar cell polarity in the Drosophila wing. Development. 2004, 131: 3785-3794. 10.1242/dev.01254.
Casal J, Lawrence PA, Struhl G: Two separate molecular systems, Dachsous/Fat and Starry night/Frizzled, act independently to confer planar cell polarity. Development. 2006, 133: 4561-4572. 10.1242/dev.02641.
Wang Y, Nathans J: Tissue/planar cell polarity in vertebrates: new insights and new questions. Development. 2007, 134: 647-658. 10.1242/dev.02772.
Montcouquiol M, Rachel RA, Lanford PJ, Copeland NG, Jenkins NA, Kelley MW: Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature. 2003, 423: 173-177. 10.1038/nature01618.
Davis EE, Katsanis N: Cell Polarization Defects in Early Heart Development. Circulation Research. 2007, 101: 122-124. 10.1161/CIRCRESAHA.107.157446.
Golubkov VS, Chekanov AV, Cieplak P: The Wnt/Planar Cell Polarity Protein-tyrosine Kinase-7 (PTK7) Is a Highly Efficient Proteolytic Target of Membrane Type-1 Matrix Metalloproteinase. Journal of Biological Chemistry. 2010, 285: 35740-35749. 10.1074/jbc.M110.165159.
Savory JGA, Mansfield M, Rijli FM, Lohnes D: Cdx mediates neural tube closure through transcriptional regulation of the planar cell polarity gene Ptk7. Development. 2011
Henderson DJ, Phillips HM, Chaudhry B: Vang-like 2 and Noncanonical Wnt Signaling In Outflow Tract Development. Trends in Cardiovascular Medicine. 2006, 16: 38-45. 10.1016/j.tcm.2005.11.005.
Jenny A, Reynolds-Kenneally J, Das G, Burnett M, Mlodzik M: Diego and Prickle regulate Frizzled planar cell polarity signalling by competing for Dishevelled binding. Nat Cell Biol. 2005, 7: 691-697. 10.1038/ncb1271.
Veeman MT, Axelrod JD, Moon RT: A Second Canon: Functions and Mechanisms of [beta]-Catenin-Independent Wnt Signaling. Developmental Cell. 2003, 5: 367-377. 10.1016/S1534-5807(03)00266-1.
Strutt D: Frizzled signalling and cell polarisation in Drosophila and vertebrates. Development. 2003, 130: 4501-4513. 10.1242/dev.00695.
Adler PN: Planar Signaling and Morphogenesis in Drosophila. Developmental Cell. 2002, 2: 525-535. 10.1016/S1534-5807(02)00176-4.
Tree DRP, Ma D, Axelrod JD: A three-tiered mechanism for regulation of planar cell polarity. Seminars in Cell and Developmental Biology. 2002, 13: 217-224. 10.1016/S1084-9521(02)00042-3.
Wallingford JB: Planar cell polarity, ciliogenesis and neural tube defects. Human Molecular Genetics. 2006, 15: R227-R234. 10.1093/hmg/ddl216.
Gray RS, Abitua PB, Wlodarczyk BJ: The planar cell polarity effector Fuz is essential for targeted membrane trafficking, ciliogenesis and mouse embryonic development. Nat Cell Biol. 2009, 11: 1225-1232. 10.1038/ncb1966.
Zhou W, Lin L, Majumdar A: Modulation of morphogenesis by noncanonical Wnt signaling requires ATF/CREB family-mediated transcriptional activation of TGF[beta]2. Nat Genet. 2007, 39: 1225-1234. 10.1038/ng2112.
Perez-Pomares JM: Myocardial-Coronary Interactions: Against the Canon. Circ Res. 2008, 102: 513-515. 10.1161/CIRCRESAHA.108.173039.
Gubb D, García-Bellido A: A genetic analysis of the determination of cuticular polarity during development in Drosophila melanogaster. Journal of Embryology and Experimental Morphology. 1982, 68: 37-57.
Keller R: Shaping the Vertebrate Body Plan by Polarized Embryonic Cell Movements. Science. 2002, 298: 1950-1954. 10.1126/science.1079478.
Wallingford JB: Closing in on vertebrate planar polarity. Nat Cell Biol. 2004, 6: 687-689. 10.1038/ncb0804-687.
Wallingford JB, Harland RM: Neural tube closure requires Dishevelled-dependent convergent extension of the midline. Development. 2002, 129: 5815-5825. 10.1242/dev.00123.
Tada M, Smith JC: Xwnt11 is a target of Xenopus Brachyury: regulation of gastrulation movements via Dishevelled, but not through the canonical Wnt pathway. Development. 2000, 127: 2227-2238.
Wallingford JB, Rowning BA, Vogeli KM, Rothbacher U, Fraser SE, Harland RM: Dishevelled controls cell polarity during Xenopus gastrulation. Nature. 2000, 405: 81-85. 10.1038/35011077.
Jessen JR, Topczewski J, Bingham S: Zebrafish trilobite identifies new roles for Strabismus in gastrulation and neuronal movements. Nat Cell Biol. 2002, 4: 610-615.
Marlow F, Zwartkruis F, Malicki J: Functional Interactions of Genes Mediating Convergent Extension, knypekandtrilobite, during the Partitioning of the Eye Primordium in Zebrafish. Developmental Biology. 1998, 203: 382-399. 10.1006/dbio.1998.9032.
Ybot-Gonzalez P, Savery D, Gerrelli D: Convergent extension, planar-cell-polarity signalling and initiation of mouse neural tube closure. Development. 2007, 134: 789-799. 10.1242/dev.000380.
Gong Y, Mo C, Fraser SE: Planar cell polarity signalling controls cell division orientation during zebrafish gastrulation. Nature. 2004, 430: 689-693. 10.1038/nature02796.
Ciruna B, Jenny A, Lee D, Mlodzik M, Schier AF: Planar cell polarity signalling couples cell division and morphogenesis during neurulation. Nature. 2006, 439: 220-224. 10.1038/nature04375.
Hamblet NS, Lijam N, Ruiz-Lozano P: Dishevelled 2 is essential for cardiac outflow tract development, somite segmentation and neural tube closure. Development. 2002, 129: 5827-5838. 10.1242/dev.00164.
Goodrich LV, Milenković L, Higgins KM, Scott MP: Altered Neural Cell Fates and Medulloblastoma in Mouse patched Mutants. Science. 1997, 277: 1109-1113. 10.1126/science.277.5329.1109.
De Marco P, Merello E, Mascelli S, Capra V: Current perspectives on the genetic causes of neural tube defects. neurogenetics. 2006, 7: 201-221. 10.1007/s10048-006-0052-2.
Vinson CR, Adler PN: Directional non-cell autonomy and the transmission of polarity information by the frizzled gene of Drosophila. Nature. 1987, 329: 549-551. 10.1038/329549a0.
Povelones M, Nusse R: The role of the cysteine-rich domain of Frizzled in Wingless-Armadillo signaling. EMBO J. 2005, 24: 3493-3503. 10.1038/sj.emboj.7600817.
Carron C, Pascal A, Djiane A, Boucaut J-C, Shi D-L, Umbhauer M: Frizzled receptor dimerization is sufficient to activate the Wnt/{beta}-catenin pathway. J Cell Sci. 2003, 116: 2541-2550. 10.1242/jcs.00451.
Povelones M, Howes R, Fish M, Nusse R: Genetic Evidence That Drosophila frizzled Controls Planar Cell Polarity and Armadillo Signaling by a Common Mechanism. Genetics. 2005, 171: 1643-1654. 10.1534/genetics.105.045245.
Wang Y, Zhang J, Mori S, Nathans J: Axonal Growth and Guidance Defects in Frizzled3 Knock-Out Mice: A Comparison of Diffusion Tensor Magnetic Resonance Imaging, Neurofilament Staining, and Genetically Directed Cell Labeling. J Neurosci. 2006, 26: 355-364. 10.1523/JNEUROSCI.3221-05.2006.
Lyuksyutova AI, Lu C-C, Milanesio N: Anterior-Posterior Guidance of Commissural Axons by Wnt-Frizzled Signaling. Science. 2003, 302: 1984-1988. 10.1126/science.1089610.
Armstrong A, Ryu YK, Chieco D, Kuruvilla R: Frizzled3 Is Required for Neurogenesis and Target Innervation during Sympathetic Nervous System Development. The Journal of Neuroscience. 2011, 31: 2371-2381. 10.1523/JNEUROSCI.4243-10.2011.
Djiane A, Riou J, Umbhauer M, Boucaut J, Shi D: Role of frizzled 7 in the regulation of convergent extension movements during gastrulation in Xenopus laevis. Development. 2000, 127: 3091-3100.
Wang Y, Guo N, Nathans J: The Role of Frizzled3 and Frizzled6 in Neural Tube Closure and in the Planar Polarity of Inner-Ear Sensory Hair Cells. J Neurosci. 2006, 26: 2147-2156. 10.1523/JNEUROSCI.4698-05.2005.
Yu H, Smallwood PM, Wang Y, Vidaltamayo R, Reed R, Nathans J: Frizzled 1 and frizzled 2 genes function in palate, ventricular septum and neural tube closure: general implications for tissue fusion processes. Development. 2010, 137: 3707-3717. 10.1242/dev.052001.
Das G, Reynolds-Kenneally J, Mlodzik M: The Atypical Cadherin Flamingo Links Frizzled and Notch Signaling in Planar Polarity Establishment in the Drosophila Eye. Developmental Cell. 2002, 2: 655-666. 10.1016/S1534-5807(02)00147-8.
Usui T, Shima Y, Shimada Y: Flamingo, a Seven-Pass Transmembrane Cadherin, Regulates Planar Cell Polarity under the Control of Frizzled. Cell. 1999, 98: 585-595. 10.1016/S0092-8674(00)80046-X.
Strutt D, Strutt H: Differential activities of the core planar polarity proteins during Drosophila wing patterning. Developmental Biology. 2007, 302: 181-194. 10.1016/j.ydbio.2006.09.026.
Strutt H, Strutt D: Differential Stability of Flamingo Protein Complexes Underlies the Establishment of Planar Polarity. Current Biology. 2008, 18: 1555-1564. 10.1016/j.cub.2008.08.063.
Curtin JA, Quint E, Tsipouri V: Mutation of Celsr1 Disrupts Planar Polarity of Inner Ear Hair Cells and Causes Severe Neural Tube Defects in the Mouse. Current Biology. 2003, 13: 1129-1133. 10.1016/S0960-9822(03)00374-9.
Torban E, Kor C, Gros P: Van Gogh-like2 (Strabismus) and its role in planar cell polarity and convergent extension in vertebrates. Trends in Genetics. 2004, 20: 570-577. 10.1016/j.tig.2004.09.003.
Katoh Y, Katoh M: Comparative genomics on Vangl1 and Vangl2 genes. Int J Oncol. 2005, 26: 1435-1440.
Kibar Z, Vogan KJ, Groulx N, Justice MJ, Underhill DA, Gros P: Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nat Genet. 2001, 28: 251-255. 10.1038/90081.
Pérez-Pomares JM: Myocardial-Coronary Interactions. Circulation Research. 2008, 102: 513-515. 10.1161/CIRCRESAHA.108.173039.
Klein TJ, Mlodzik M: PLANAR CELL POLARIZATION: An Emerging Model Points in the Right Direction. Annual Review of Cell and Developmental Biology. 2005, 21: 155-176. 10.1146/annurev.cellbio.21.012704.132806.
Goto T, Keller R: The Planar Cell Polarity Gene Strabismus Regulates Convergence and Extension and Neural Fold Closure in Xenopus. Developmental Biology. 2002, 247: 165-181. 10.1006/dbio.2002.0673.
Darken RS, Scola AM, Rakeman AS, Das G, Mlodzik M, Wilson PA: The planar polarity gene strabismus regulates convergent extension movements in Xenopus. EMBO J. 2002, 21: 976-985. 10.1093/emboj/21.5.976.
Kibar Z, Underhill DA, Canonne-Hergaux F, Gauthier S, Justice MJ, Gros P: Identification of a New Chemically Induced Allele (Lpm1Jus) at the Loop-Tail Locus: Morphology, Histology, and Genetic Mapping. Genomics. 2001, 72: 331-337. 10.1006/geno.2000.6493.
Murdoch JN, Doudney K, Paternotte C, Copp AJ, Stanier P: Severe neural tube defects in the loop-tail mouse result from mutation of Lpp1, a novel gene involved in floor plate specification. Human Molecular Genetics. 2001, 10: 2593-2601. 10.1093/hmg/10.22.2593.
Etheridge SL, Ray S, Li S: Murine Dishevelled 3 Functions in Redundant Pathways with Dishevelled 1 and 2 in Normal Cardiac Outflow Tract, Cochlea, and Neural Tube Development. PLoS Genet. 2008, 4: e1000259-10.1371/journal.pgen.1000259.
Qian D, Jones C, Rzadzinska A: Wnt5a functions in planar cell polarity regulation in mice. Developmental Biology. 2007, 306: 121-133. 10.1016/j.ydbio.2007.03.011.
Doudney K, Moore GE, Stanier P: Analysis of the planar cell polarity gene Vangl2 and its co-expressed paralogue Vangl1 in neural tube defect patients. American Journal of Medical Genetics Part A. 2005, 136A: 90-92. 10.1002/ajmg.a.30766.
Lei Y-P, Zhang T, Li H, Wu B-L, Jin L, Wang H-Y: VANGL2 Mutations in Human Cranial Neural-Tube Defects. New England Journal of Medicine. 2010, 362: 2232-2235. 10.1056/NEJMc0910820.
Kibar Z, Torban E, McDearmid JR: Mutations in VANGL1 Associated with Neural-Tube Defects. New England Journal of Medicine. 2007, 356: 1432-1437. 10.1056/NEJMoa060651.
Kibar Z, Bosoi CM, Kooistra M: Novel mutations in VANGL1 in neural tube defects. Human Mutation. 2009, 30: E706-E715. 10.1002/humu.21026.
Gao C, Chen Y-G: Dishevelled: The hub of Wnt signaling. Cellular Signalling. 2010, 22: 717-727. 10.1016/j.cellsig.2009.11.021.
Matsui T, Raya Á, Kawakami Y: Noncanonical Wnt signaling regulates midline convergence of organ primordia during zebrafish development. Genes & Development. 2005, 19: 164-175. 10.1101/gad.1253605.
Lijam N, Paylor R, McDonald MP: Social Interaction and Sensorimotor Gating Abnormalities in Mice Lacking Dvl1. Cell. 1997, 90: 895-905. 10.1016/S0092-8674(00)80354-2.
Feiguin F, Hannus M, Mlodzik M, Eaton S: The Ankyrin Repeat Protein Diego Mediates Frizzled-Dependent Planar Polarization. Developmental Cell. 2001, 1: 93-101. 10.1016/S1534-5807(01)00010-7.
Schwarz-Romond T, Asbrand C, Bakkers J: The ankyrin repeat protein Diversin recruits Casein kinase Iepsilon to the beta-catenin degradation complex and acts in both canonical Wnt and Wnt/JNK signaling. Genes Dev. 2002, 16: 2073-2084. 10.1101/gad.230402.
Itoh K, Jenny A, Mlodzik M, Sokol SY: Centrosomal localization of Diversin and its relevance to Wnt signaling. J Cell Sci. 2009, 122: 3791-3798. 10.1242/jcs.057067.
Habas R, Dawid IB, He X: Coactivation of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation. Genes & Development. 2003, 17: 295-309. 10.1101/gad.1022203.
Gubb D, Green C, Huen D: The balance between isoforms of the prickle LIM domain protein is critical for planar polarity in Drosophila imaginal discs. Genes Dev. 1999, 13: 2315-2327. 10.1101/gad.13.17.2315.
Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT: Zebrafish Prickle, a Modulator of Noncanonical Wnt/Fz Signaling, Regulates Gastrulation Movements. Current Biology. 2003, 13: 680-685. 10.1016/S0960-9822(03)00240-9.
Wallingford JB, Goto T, Keller R, Harland RM: Cloning and expression of Xenopus Prickle, an orthologue of a Drosophila planar cell polarity gene. Mechanisms of Development. 2002, 116: 183-186. 10.1016/S0925-4773(02)00133-8.
Takeuchi M, Nakabayashi J, Sakaguchi T: The prickle-Related Gene in Vertebrates Is Essential for Gastrulation Cell Movements. Current Biology. 2003, 13: 674-679. 10.1016/S0960-9822(03)00245-8.
Narimatsu M, Bose R, Pye M: Regulation of Planar Cell Polarity by Smurf Ubiquitin Ligases. Cell. 2009, 137: 295-307. 10.1016/j.cell.2009.02.025.
Yin C, Kiskowski M, Pouille P-A, Farge E, Solnica-Krezel L: Cooperation of polarized cell intercalations drives convergence and extension of presomitic mesoderm during zebrafish gastrulation. J Cell Biol. 2008, 180: 221-232. 10.1083/jcb.200704150.
Hammerschmidt M, Pelegri F, Mullins MC: Mutations affecting morphogenesis during gastrulation and tail formation in the zebrafish, Danio rerio. Development. 1996, 123: 143-151.
Heisenberg C-P, Tada M, Rauch G-J: Silberblick/Wnt11 mediates convergent extension movements during zebrafish gastrulation. Nature. 2000, 405: 76-81. 10.1038/35011068.
Kilian B, Mansukoski H, Barbosa FC, Ulrich F, Tada M, Heisenberg CP: The role of Ppt/Wnt5 in regulating cell shape and movement during zebrafish gastrulation. Mech Dev. 2003, 120: 467-476. 10.1016/S0925-4773(03)00004-2.
Lele Z, Bakkers J, Hammerschmidt M: Morpholino phenocopies of the swirl, snailhouse, somitabun, minifin, silberblick, and pipetail mutations. genesis. 2001, 30: 190-194. 10.1002/gene.1063.
Nagy II, Railo A, Rapila R: Wnt-11 signalling controls ventricular myocardium development by patterning N-cadherin and beta-catenin expression. Cardiovascular Research. 2010, 85: 100-109. 10.1093/cvr/cvp254.
Germino GG: Linking cilia to Wnts. Nat Genet. 2005, 37: 455-457. 10.1038/ng0505-455.
Moeller H, Jenny A, Schaeffer H-J: Diversin regulates heart formation and gastrulation movements in development. Proceedings of the National Academy of Sciences. 2006, 103: 15900-15905. 10.1073/pnas.0603808103.
Bisgrove BW, Yost HJ: The roles of cilia in developmental disorders and disease. Development. 2006, 133: 4131-4143. 10.1242/dev.02595.
Singla V, Reiter JF: The Primary Cilium as the Cell's Antenna: Signaling at a Sensory Organelle. Science. 2006, 313: 629-633. 10.1126/science.1124534.
Ross AJ, May-Simera H, Eichers ER: Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet. 2005, 37: 1135-1140. 10.1038/ng1644.
Park TJ, Haigo SL, Wallingford JB: Ciliogenesis defects in embryos lacking inturned or fuzzy function are associated with failure of planar cell polarity and Hedgehog signaling. Nat Genet. 2006, 38: 303-311. 10.1038/ng1753.
Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DYR, Reiter JF: Vertebrate Smoothened functions at the primary cilium. Nature. 2005, 437: 1018-1021. 10.1038/nature04117.
Lu X, Borchers AGM, Jolicoeur C, Rayburn H, Baker JC, Tessier-Lavigne M: PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature. 2004, 430: 93-98. 10.1038/nature02677.
Murdoch JN, Henderson DJ, Doudney K: Disruption of scribble (Scrb1) causes severe neural tube defects in the circletail mouse. Human Molecular Genetics. 2003, 12: 87-98. 10.1093/hmg/ddg014.
Kibar Z, Salem S, Bosoi CM: Contribution of VANGL2 mutations to isolated neural tube defects. Clinical Genetics. 2010, no-no
Torban E, Patenaude A-M, Leclerc S: Genetic interaction between members of the Vangl family causes neural tube defects in mice. Proceedings of the National Academy of Sciences. 2008, 105: 3449-3454. 10.1073/pnas.0712126105.
Torban E, Wang H-J, Groulx N, Gros P: Independent Mutations in Mouse Vangl2 That Cause Neural Tube Defects in Looptail Mice Impair Interaction with Members of the Dishevelled Family. Journal of Biological Chemistry. 2004, 279: 52703-52713. 10.1074/jbc.M408675200.
Murdoch JN, Rachel RA, Shah S: Circletail, a New Mouse Mutant with Severe Neural Tube Defects: Chromosomal Localization and Interaction with the Loop-Tail Mutation. Genomics. 2001, 78: 55-63. 10.1006/geno.2001.6638.
Doudney K, Stanier P: Epithelial cell polarity genes are required for neural tube closure. American Journal of Medical Genetics Part C: Seminars in Medical Genetics. 2005, 135C: 42-47. 10.1002/ajmg.c.30052.
Satoh W, Matsuyama M, Takemura H, Aizawa S, Shimono A: Sfrp1, Sfrp2, and Sfrp5 regulate the Wnt/β-catenin and the planar cell polarity pathways during early trunk formation in mouse. genesis. 2008, 46: spcone-spcone.
Burren KA, Savery D, Massa V: Gene-environment interactions in the causation of neural tube defects: folate deficiency increases susceptibility conferred by loss of Pax3 function. Human Molecular Genetics. 2008, 17: 3675-3685. 10.1093/hmg/ddn262.
Wilson MP, Hugge C, Bielinska M, Nicholas P, Majerus PW, Wilson DB: Neural tube defects in mice with reduced levels of inositol 1,3,4-trisphosphate 5/6-kinase. Proceedings of the National Academy of Sciences. 2009, 106: 9831-9835. 10.1073/pnas.0904172106.
Salbaum JM, Kappen C: Neural tube defect genes and maternal diabetes during pregnancy. Birth Defects Research Part A: Clinical and Molecular Teratology. 2010, 88: 601-611. 10.1002/bdra.20680.
Acknowledgements
This work was supported by National Science Foundation of China (No.30825039 and No.30973236 to Dezhi Mu), and Program of Changjiang Scholars and Innovative Research Team in University (IRT0935).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests disclosure
The authors declare that they have no competing interests.
Authors' contributions
GW was participated in data and information collection and part of the writing.
XH performed part of text writing and the editing of the whole manuscript.
YH wrote the part of the manuscript and information collection. DM was in charge of the whole project and participated in the manuscript writing. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wu, G., Huang, X., Hua, Y. et al. Roles of planar cell polarity pathways in the development of neutral tube defects. J Biomed Sci 18, 66 (2011). https://doi.org/10.1186/1423-0127-18-66
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1423-0127-18-66