
Abstract
One of the most obvious characteristics of the aging process is the progressive decline in the regenerative potential of tissues. Adult somatic stem cells are critical for rejuvenating tissues and persist throughout the lifespan of organisms. However, stem cell function declines during the aging process in tissues such as the brain, blood, skin, intestinal epithelium, bone, and skeletal muscle. This demise may contribute to tissue degeneration, organismal aging, and age-related diseases. A series of organismal models have emerged as valuable systems to study stem cell aging in vivo. Here, we review the age-associated changes of stem cells and the different organismal models used to define stem cell aging.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Organismal aging is associated to a progressive imbalance in tissue and organ homeostasis and a timely decline in their regeneration capacity, which is accompanied by the loss of physiological integrity that, in turn, results in susceptibility to death. Stem cell exhaustion has been defined as one of the hallmarks of aging and determines the tissue repair ability of organisms [1••]. Stem cells are divided into two groups: embryonic stem cells (ESCs) and adult stem cells. ESCs are pluripotent and are found in early stage of embryos. Since ESCs do not undergo replicative senescence, they are considered to be immortal in culture [2, 3]. Adult organisms have two types of stem cells: (1) adult somatic stem cells with regenerative potential, which are found in several tissues and (2) germline stem cells (GSCs), which can generate gametes for reproduction [4]. GSCs are designed to maintain an unlimited proliferative capacity to fulfill their biological purpose: to be passed from one generation to the next. Adult somatic stem cells are critical for rejuvenating tissues and persist in the adult body of an organism throughout its lifespan. Adult somatic stem cells reside in various adult tissues such as liver, brain, and bone marrow. In contrast to ESCs, they can only differentiate into a limited type of cells. Adult somatic stem cell function decreases with age, and this failure may contribute to age-related diseases [1••, 5, 6]. For instance, neural stem cells (NSCs) from older animals are less able to self-renew and more skewed toward astrocytes [7]. A decline in the proliferation of NSCs and neurogenesis during aging has been correlated with impairment of olfactory discrimination [8]. Notably, postnatally generated neurons are absent in advanced stages of Huntington’s disease [9]. Another example is the decline of hematopoietic stem cell (HSCs) function with age. This failure could have a significant impact on organismal aging since decreased hematopoiesis with age results in a diminished generation of adaptive cells and increased myeloid malignancies and anemia [6]. In this review, we will focus on age-associated changes of somatic and germline stem cells and the emerging models for the study of their aging in vivo.
In Vivo Models for Stem Cell Aging
A series of model organisms have started to reveal the general principles of stem cell aging (Fig. 1). These findings generally come from studies in the budding yeast (Saccharomyces cerevisiae), invertebrates (the nematode Caenorhabditis elegans and the fruit fly Drosophila melanogaster), and rodents (Table 1). S. cerevisiae has been used to identify genes that modulate replicative lifespan [10], a process observed in many dividing cells such as adult stem cells. The yeast cell divides asymmetrically by forming a bud which gives rise to the daughter cell. As the cell wall of the daughter cell is formed de novo, the mother cell retains the same wall, evidenced by scars from each budding event on the cell wall surface. Accumulation of budding scars is a marker of replicative senescence. Ultimately, the mother cell is not able to reproduce anymore, and sterility is the final phenotype of yeast aging [10]. In addition, the asymmetric cell divisions invoked by the budding yeast resemble stem cell self-renewal. In yeast asymmetric cell division, the daughter cell inherits a limited amount of damaged macromolecules from the mother cell [11] enabling the generation of a rejuvenated, germ-like, daughter cell lineage. Thus, this phenomenon can be considered a model for GSCs. One interesting example for successful use of yeast and its extrapolation to stem cell aging is the role of sirtuins, which are histone-deacetylating enzymes involved in gene silencing. The sirtuin family of proteins impinges upon replicative lifespan of yeast [12]. For example, Sir2 was shown to be involved in the stabilization of repetitive DNA in yeast. During aging and DNA damage, Sir2 specifically localizes to the sites of low genomic integrity (genomic instability), and SIRT1, the mammalian orthologue of Sir2, may play a major role in age-dependent gene expression [13]. Thus, data from yeast have suggested a major role for epigenetic changes in aging. Finally, yeast can also be used as a model for chronological aging [14]. In these studies, yeast cells are induced into a non-dividing state by withdrawal of nutrients.

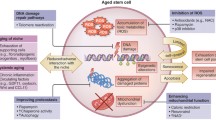
The impact of aging on adult stem cells. Aging causes a decrease in stem cell functionality that leads to a decline in tissue rejuvenation and repair. Age-associated changes of stem cells include (1) genomic changes, which arise from increased DNA damage as well as a shortening of the telomeres, (2) loss of proteostasis through altered chaperone levels and deregulation of autophagy rate which might lead to accumulation of damaged proteins, (3) epigenetic changes in methylation and chromatin-remodeling factors, (4) mitochondrial dysfunction as a consequence of ROS accumulation and mtDNA mutations, (5) cellular senescence, and (6) impaired nutrient sensing regulation
Metazoan models provide the means to study stem cell decline during organismal aging. C. elegans and D. melanogaster are established models for the study of GSCs. Fly and mouse models are the main organisms used for somatic stem cell aging research. D. melanogaster has been mostly used to study the intestinal stem cell (ISCs) population in the midgut, although other somatic stem cell compartments are starting to be identified. Mouse models are suitable for the study of a wide number of stem cell types. These models have helped to establish not only general principles of stem cell aging but also differences between the different somatic stem cells [8, 15]. Although these general age-related changes may induce a decline in stem cell function at some extent, they do not always result in a change in the stem cell number. For instance, the number of NSCs and melanocyte stem cells decreases with age in mammals. However, the number of HSCs increases with age in some mouse strains [16]; whereas, their functionality declines (e.g., HSCs from old mice are skewed to differentiate toward the myeloid lineage [17]). Metazoans also allow the study of the modulation of specific stem cells by either their niche or other tissues in a non-autonomous manner (i.e., through systemic signals in response to environmental and nutrient changes). Of special relevance are heterochronic parabiotic experiments in mice that demonstrated that aging of muscle stem cells (also known as satellite cells) is regulated by systemic factors. In this experimental paradigm, in which aged mice are surgically connected to young mice, muscle stem cells are rejuvenated by the exposure to a youthful environment [18].
Age-Associated Changes of Stem Cells
Somatic stem cell exhaustion is one of the hallmarks of aging [1••]. Besides stem cell exhaustion, López-Otín and colleagues have pointed out other hallmarks of aging (i.e., genomic instability, telomere shortening, epigenetic alterations, loss of proteostasis, mitochondrial dysfunction, cellular senescence, altered intercellular communication, impaired nutrient sensing regulation) [1••]. Somatic stem cells exhibit a series of these age-related changes that could trigger stem cell dysfunction/death and, in turn, a progressive decline in regeneration capacity [15] (Fig. 1). In this section, we discuss age-related cellular changes of somatic stem cells.
Genomic Instability and Telomere Attrition
Similar to other cell types, somatic stem cells also undergo both DNA damage and telomere shortening with age [6]. Markers of DNA damage have been observed in stem cell populations during aging [19, 20]. Increased DNA damage contributes to the decline in the self-renewal ability of both epidermal and melanocyte stem cells during the aging process [21, 22]. In addition to elevated DNA damage, a decrease in DNA damage response (DDR), which is crucial for repair mechanism, has also been reported in stem cells [23, 24]. Increased DNA damage in stem cells may result in their senescence and production of defective daughter cells [25, 26]. It has been shown that deletion of Atm, a DNA damage sensor, results in increased ROS levels and loss of quiescence in HSCs. Eventually, this defect causes depletion of the HSC pool [27].
Stem cells may be prone to telomere shortening due to their high self-renewal ability. Both human and mice studies have shown telomere shortening in stem cells during the aging process [28, 29]. The importance of telomere length in stem cell homeostasis has emerged especially in NSCs. In these cells, telomere shortening results in interrupted neuronal differentiation. Moreover, reduction of NSC function has been also observed in telomerase-deficient (Terc−/−) mice, a deficiency that results in short telomeres [29].
Epigenetic Alterations
Epigenetic alterations (e.g., histone modifications such as acetylation, ubiquitination, and methylation) contribute to the aging process [1••]. Several studies have suggested an important role of epigenetic alterations in stem cell aging [30, 31]. However, the analysis of epigenomic changes requires a large amount of cells, which is not always feasible for somatic stem cells. Therefore, epigenetic research on stem cells usually depends on measurement of tissue compartments in which these cells are found.
One of the examples of epigenetic changes in stem cells indicates that DNA methylation levels increase in a site-specific manner in HSCs with age [30]. Chambers and colleagues [31] showed that epigenomic changes in aged stem cells are related with alterations in chromatin-modifying enzymes and their cofactors. Microarray data from HSCs from young and old mice have revealed a decrease in the expression levels of switch/sucrose nonfermentable (SWI/SNF) chromatin-remodeling complex subunits, specific histone deacetylases (HDAC1, 5, and 6), and a DNA methyltransferase (DNMT3b) [31]. In addition, age-dependent changes in histone modification could switch expression of self-renewal genes in HSCs [32•].
Loss of Proteostasis
The integrity of the proteome (proteostasis) declines in a progressive manner with age [1••]. During the aging process, cells accumulate damaged and misfolded proteins. These damaged proteins impair cell functionality and tissue homeostasis and, therefore, must be scavenged. The accumulation of damaged proteins with age is associated to a decline in the intracellular ability of maintaining proteostasis. Several nodes of the proteostasis network such as autophagy-lysosomal potential, proteasomal activity, and chaperone function decline during organismal aging [33–37]. This demise in the proteostasis network could also determine stem cell exhaustion (for extensive review, see [8]).
A series of cellular responses such as the heat shock (HSR) or the endoplasmic reticulum (UPRER) and mitochondrial (UPRmt) unfolded protein responses are activated to maintain the quality of the proteome when aberrant proteins accumulate. These responses are essential mechanisms to ensure proper cytosolic protein folding and reduce proteotoxic stress. Activation of these mechanisms and increased levels of chaperones are associated with enhanced protection against proteotoxic stress [38]. However, their activity also decreases during the aging process [39, 40]. Supporting evidence suggests that chaperones such as heat shock proteins (HSPs) may play a role in stemness and differentiation. HSPA8 (a non-inducible HSP) negatively influences the stability of proapoptotic Bim mRNA, increasing HSCs survival and preventing their differentiation [41]. Bim is required for apoptosis during hematopoiesis. HSP70 indirectly triggers erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1 [42], an essential transcriptional factor for differentiation and maturation within the erythroid lineage. Several components of the UPRER have a key role during differentiation. For instance, XBP1 induces osteogenic and plasma differentiation [43], IRE1 increases lymphopoiesis of B cells [44], and CHOP promotes differentiation of B cells, erythrocytes, osteocytes, and chondrocytes [45–48]. Moreover, the UPRER, as a stress-coordinated pathway, has an important role in the modulation of differentiation of mouse intestinal epithelial stem cells [49•]. The transition from stem cell to transit-amplifying cells of the intestine is accompanied by induced activity of the UPRER. Perk-eIF2α induces endoplasmic reticulum stress that, in turn, promotes loss of stemness. In organoid cultures of primary intestinal epithelium, inhibition of Perk-eIF2α leads to accumulation of stem cells [49•]. Taken together, these observations make it difficult to correlate high levels of HSPs or proteotoxicity protection with somatic stem cell differentiation, and further insights into the impact of these pathways on stem cell identity are necessary. In addition, it will be fascinating to define whether the levels of chaperones and/or the ability to activate proteotoxic cellular stress responses decrease in somatic stem cells with age. Notably, a recent study has shown that a decline in the UPRmt contributes to HSC aging [50]. Inactivation of SIR7, a regulator of the UPRmt, reduces quiescence, increases mitochondrial protein folding stress, and compromises the regenerative potential of HSCs. Moreover, SIR7 expression is decreased in HSCs, whereas SIRT7 up-regulation enhances the regenerative potential of aged HSCs [50].
When misfolded proteins cannot be “rescued” by chaperones and stress responses, they are degraded through the proteasome and/or autophagy. Increased levels of proteasome components result in enhanced replicative lifespan and resistance to proteotoxic stress in yeast [51]. Although the impact of increased proteasome activity as a determinant of ESC function has been shown, whether somatic stem cells also have an enhanced proteasome activity remains to be elucidated. However, maintenance of proteasomal activity in somatic stem cells may critically impact organismal aging. On the other hand, more insights into the impact of autophagy in somatic stem cells have been shown. In vitro studies with somatic stem cells such as human mesenchymal stem cells (hMSCs) [52], HSCs, dermal stem cells (DSCs), and epidermal stem cells [53] suggest that autophagy activity is increased in these cells compared to their differentiated counterparts. In fetal and postnatal mouse HSCs, a deficiency in essential autophagy genes such as FIP200 or Atg7 increases ROS levels and deregulates proliferation, suggesting that autophagy is required for stemness in fetal and postnatal mouse HSCs [54–57]. Moreover, deletion of ATG7 triggers the depletion of HSC pools [56]. FoxO3, a forkhead transcription factor linked to both longevity and stem cell homeostasis [58], maintains the expression of pro-autophagy genes in adult mouse HSCs to allow a quick autophagic response upon stress [59••]. Notably, old mouse HSCs have higher basal levels of autophagy activity, a characteristic required for their cloning efficiency, and are able to induce autophagy like young HSCs. However, autophagy activity in young HSCs is not required for their cloning efficiency [59••]. This observation is controversial as previous data showed the opposite effect [54–57]. The difference might be that one study [59••] used a drug to block autophagy in normally developed adult mouse HSCs, whereas the latter study used a genetic model to block autophagy that causes severe defects early in life leading to death, thus looking at fetal/early stages. The higher levels of autophagy activity in old adult mouse HSCs were due to an attenuated nutrient (2-NBD glucose) uptake [59••]. That old adult mouse HSCs maintain an autophagy potential similar to young mouse HSCs and exhibit higher levels of autophagy for their survival confronts the traditional view where compromised autophagy is established as a determinant of aging [34].
In NSCs or cardiac stem cells (CSCs), autophagy activity increases upon their differentiation [60–62]. This enhanced autophagy might be due to a specific increase in the requirements of their differentiated counterparts, such as neurons, to recycle their cellular components. During the initial period of neuronal differentiation (E15.5 mouse embryos), the expression of the autophagy genes Atg7, Becn1, Ambra1, and LC3 are increased in vivo in the mouse embryonic olfactory bulb (OB) [60]. Blocking autophagy either genetically or chemically impairs NSCs and CSCs differentiation [60–62]. Inhibition of autophagy by wortmannin or 3-MA decreases neurogenesis of OB-derived stem cells [60]. In addition, Ambra1 loss-of-function mice show decreased neural markers in the E13.5 OB [60]. However, FIP200, an essential component of autophagy, is required for NSC proliferation [63]. Ex-vivo treatment of E8.5 mouse embryos with the autophagy activator rapamycin increases the expression of cardiomyocyte markers in the second heart field [61, 62]. Taken together, these observations suggest a higher degree of protection, at least, in somatic stem cells to cytotoxic stresses. Consistent with this idea, impairment of autophagy in epidermal stem cells, DSCs, and HSCs leads to increased susceptibility to cytotoxic stress such as etoposide, doxorubicid, or UV [53].
Accumulation of ROS and Mitochondrial Dysfunction
Several studies indicate that increased accumulation of ROS determines stem cell dysfunction and fate decision during the aging process [64–66]. In mice, the amount of HSCs with low ROS levels decrease in an age-dependent manner [67]. Furthermore, increased ROS levels result in disruption of proliferation and self-renewal of HSCs [68]. The treatment with N-acetyl-l-cysteine (NAC), an oxidative stress inhibitor, rescues the defects in HSC and NSC function in a progeroid mice model [69, 70].
Another well-characterized hallmark of aging is mitochondrial dysfunction [1••]. This demise can be caused by mitochondrial DNA mutation or changes in mitochondrial function such as alterations in nutrient sensing and energy metabolism [71]. mtDNA mutagenesis in mice affects HSCs during the fetal development stage. mtDNA mutations also affect the number of quiescent NSCs in vivo and their self-renewal ability in vitro [69]. Furthermore, HSCs exhibit a decrease in their capacity of nutrient uptake in an age-dependent manner [59••].
Senescence
Cellular senescence is defined as irreversible cell cycle arrest. Indirect evidence indicates that signaling pathways related with cellular senescence increase with age in somatic stem cells [15]. However, senescence of these cells has not been clearly shown. Notably, a mutant mouse strain deficient in Klotho, a protein involved in mineral homeostasis, shows senescence markers in stem cells of the gut and the skin [72]. These cells show foci of γ-H2AX staining, a hallmark of senescence induced by DNA damage. Similarly, aged HSCs and muscle stem cells have increased levels of γ-H2AX foci [19, 20]. However, DNA damage could also lead to apoptosis. Therefore, it cannot be ruled out that these cells are undergoing programmed cell death.
Impaired Nutrient Sensing Regulation
The use of invertebrate models for aging research led to the discovery of several longevity pathways [37]. Among these signaling networks, we find evolutionary conserved pathways that have emerged as regulators of somatic stem cell function [8]. One of these pathways is the reduced insulin/insulin-like growth factor 1 (IGF-1) signaling (IIS). IGF-1 is the secondary mediator of the somatotrophic axis in mammals, which is regulated by the growth hormone. The intracellular signaling pathway of IGF-1 is the same as the one triggered by insulin. The major downstream effector of IIS is the FOXO family of transcription factors. Notably, FOXOs regulate the functionality of somatic stem cells such as HSCs and NSCs. A combined deficiency of FoxO1/FoxO3/FoxO4 depletes the HSC and NSC pools in mice [73, 74]. Deficiency of FoxOs increases ROS levels in both NSCs and HSCs [73, 74], which may increase protein misfolding. FoxOs protect from oxidative stress, promoting the expression of antioxidant enzymes [75]. FoxO3 is essential for regulating this process in mice, since FoxO3 deficiency alone increases ROS levels and depletes the pool of HSCs and NSCs [76, 77]. Notably, the loss of FoxO3 causes increased neurogenesis during development followed by NSC depletion in adulthood of mice [76]. Interestingly, FoxO3-bound genes thoroughly overlap with those bound by the proneuronal bHLH transcription factor ASCL/MASH1 in cultured neural progenitor cells [78]. FoxO3 represses the expression of specific ASCL1 neurogenic targets restraining neurogenesis. Therefore, FoxO3 may contribute to maintain the homeostasis of the NSC pool by negatively regulating neurogenesis. In flies, IIS is also required for ISC proliferation and may regulate division of these cells during aging and tissue repair [79, 80]. Insulin-like peptides (dILP)-expressing cells in the brain modulate ISC proliferation and age-associated dysplasia [80]. Furthermore, dFOXO modulates the aging of muscle in flies [81, 82]. Circular muscle releases factors that induce ISC proliferation and, therefore, is considered a component of the ISC niche [83]. Thus, IIS may also modulate ISC function through their niche.
Dietary restriction (DR), defined as reduced food-intake without malnutrition, also extends lifespan in multiple species [36, 84]. The target of rapamycin (mTOR) is required for mediating the effects of DR on longevity. mTOR associates with other proteins to form two different complexes: mTORC1 and mTORC2. mTORC1 activity is inhibited by DR resulting in longevity [85]. Recent findings indicate a role of DR on stem cell proliferation. DR increases functionality of mouse ISCs via a non-cell-autonomous mechanism acting through adjacent Paneth cells of the ISC niche. DR down-regulates mTORC1 activity in Paneth cells, but not in ISCs, eventually creating an environment where ISC function is enhanced [86•]. Decreased activity of mTORC1 in Paneth cells up-regulates the levels of bone stromal antigen 1 (Bst-1), a protein that promotes cell proliferation in bone marrow. Regulation of Bst-1 levels by mTORC1 is required for the enhanced ISC function upon DR. Similarly, the regenerative potential of muscle stem (satellite) cells increases upon DR in both young and old mice [87]. The number of satellite cells per fibers is enhanced after DR treatment during 3 months. DR also increases neurogenesis [88], but the effects are not compatible for in vivo application since a high intake deprivation is necessary. Taken together, these data could help understand the pro-longevity effects of DR suggesting an impact on stem cell function.
Germline Stem Cells
Both C. elegans and D. melanogaster are well-established models for the study of GSCs. In the female germline stem cell niche of flies, the primary niche-associated factor decapentaplegic (DPP) promotes GSC self-renewal. Interestingly, GSCs themselves regulate the restriction of DPP function through activation of the epidermal growth factor receptor (EGFR) mitogen-activated protein kinase (MAPK) in the nearby somatic cells [89]. In aged flies, dILPs regulate the homeostasis and proliferation of GSCs through autonomous and non-autonomous mechanisms in both females and males [90–92]. Furthermore, IIS declines in the ovary from aged females [90]. Thus, age-associated changes in the levels of local or circulating dILPs may contribute to the decline in the number and functionality of GSCs. In addition, DR also enhances GSC maintenance [93].
Notably, signals from the GSCs regulate organismal lifespan in both worms and flies [94]. Genetic ablation of the germline, but not the entire gonad, leads to lifespan extension in worms [95]. This phenotype may be caused by increased proteome stability within the post-mitotic soma [96, 97]. Inhibiting germline proliferation induces an increase in the somatic levels of the proteasome subunit RPN-6 that, in turn, promotes proteasome activity and extends lifespan [96, 97]. Increased levels of RPN-6 are modulated by DAF-16, the worm FOXO orthologue [98, 99•]. Extended lifespan induced by the lack of germline could be explained by the disposable soma theory of aging in reverse. Hypothetically, the need for repairing and preventing damage to the germline dominates resource allocation strategies, while the somatic tissues age and deteriorate [100]. Abrogation of the germline would stimulate a re-allocation of metabolic resources to the somatic tissues.
Conclusions
In this review, we have discussed the different general principles that might define adult stem cell aging. Besides the insights gained into the aging of these cells, the molecular mechanisms underlying stem cell exhaustion are still unknown. It will be fascinating to continue exploring the interconnectedness between stem cell exhaustion and the other candidate hallmarks of aging. Although cumulative evidence suggests that delaying age-associated changes in stem cells could have beneficial effects in the health and lifespan of an organism, more research in this field is needed to facilitate the development of specific adult stem-cell-based therapies. However, considering the links between enhanced stem cell proliferation and malignant cells, some detrimental effects of this approach should not be discarded. Additionally, it is important to remark the contribution of the decline of post-mitotic and progenitor cells to age-associated diseases and loss of tissue homeostasis. Thus, it is important to understand the interplay between stem, progenitor, and post-mitotic cells to develop efficient healthspan-promoting therapies.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Lopez-Otin C et al. The hallmarks of aging. Cell. 2013;153:1194–217. This review classifies, for the first time, nine tentative hallmarks of aging, which are common in different organisms.
Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–6.
Thomson JA et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–7.
Li L, Xie T. Stem cell niche: structure and function. Annu Rev Cell Dev Biol. 2005;21:605–31.
Oh J et al. Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nat Med. 2014;20:870–80.
Signer RA, Morrison SJ. Mechanisms that regulate stem cell aging and life span. Cell Stem Cell. 2013;12:152–65.
Bondolfi L et al. Impact of age and caloric restriction on neurogenesis in the dentate gyrus of C57BL/6 mice. Neurobiol Aging. 2004;25:333–40.
Vilchez D et al. Proteostasis and aging of stem cells. Trends Cell Biol. 2014;24:161–70.
Ernst A et al. Neurogenesis in the striatum of the adult human brain. Cell. 2014;156:1072–83.
Mortimer RK, Johnston JR. Life span of individual yeast cells. Nature. 1959;183:1751–2.
Aguilaniu H et al. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science. 2003;299:1751–3.
Kennedy BK et al. Mutation in the silencing gene SIR4 can delay aging in S. cerevisiae. Cell. 1995;80:485–96.
Wood JG et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–9.
Fabrizio P, Longo VD. The chronological life span of Saccharomyces cerevisiae. Aging cell. 2003;2:73–81.
Jones DL, Rando TA. Emerging models and paradigms for stem cell ageing. Nat Cell Biol. 2011;13:506–12.
de Haan G et al. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: correlation between lifespan and cycling activity. Blood. 1997;89:1543–50.
Rossi DJ et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005;102:9194–9.
Conboy IM et al. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–4.
Charville GW, Rando TA. Stem cell ageing and non-random chromosome segregation. Philos Trans R Soc Lond Ser B Biol Sci. 2011;366:85–93.
Rossi DJ et al. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–9.
Inomata K et al. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell. 2009;137:1088–99.
Sotiropoulou PA et al. Bcl-2 and accelerated DNA repair mediates resistance of hair follicle bulge stem cells to DNA-damage-induced cell death. Nat Cell Biol. 2010;12:572–82.
Behrens A et al. Impact of genomic damage and ageing on stem cell function. Nat Cell Biol. 2014;16:201–7.
Fortini P et al. The response to DNA damage during differentiation: pathways and consequences. Mutat Res. 2013;743–744:160–8.
Erol A. Deciphering the intricate regulatory mechanisms for the cellular choice between cell repair, apoptosis or senescence in response to damaging signals. Cell Signal. 2011;23:1076–81.
Mandal PK et al. DNA damage response in adult stem cells: pathways and consequences. Nat Rev Mol Cell Biol. 2011;12:198–202.
Ito K et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–51.
Allen ND, Baird DM. Telomere length maintenance in stem cell populations. Biochim Biophys Acta. 2009;1792:324–8.
Flores I et al. The longest telomeres: a general signature of adult stem cell compartments. Genes Dev. 2008;22:654–67.
Beerman I et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell. 2013;12:413–25.
Chambers SM et al. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5:e201.
Sun D et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14:673–88. In this study, genomic fragments containing histone modifications, transcriptomes and DNA methylation are compared between young and old HSCs giving a data set which can be used for future stem cell epigenomic profiling.
Koga H et al. Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res Rev. 2011;10:205–15.
Rubinsztein DC et al. Autophagy and aging. Cell. 2011;146:682–95.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76.
Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol. 2011; 3.
Vilchez D et al. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun. 2014;5:5659.
Morimoto RI. The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb Symp Quant Biol. 2011;76:91–9.
Gidalevitz T, et al. The stress of protein misfolding: from single cells to multicellular organisms. Cold Spring Harb Perspect Biol. 2011; 3.
Vabulas RM et al. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb Perspect Biol. 2010;2:a004390.
Matsui H et al. Cytokines direct the regulation of Bim mRNA stability by heat-shock cognate protein 70. Mol Cell. 2007;25:99–112.
Ribeil JA et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature. 2007;445:102–5.
Iwakoshi NN et al. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol Rev. 2003;194:29–38.
Zhang K et al. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. 2005;115:268–81.
Cui K et al. Novel interaction between the transcription factor CHOP (GADD153) and the ribosomal protein FTE/S3a modulates erythropoiesis. J Biol Chem. 2000;275:7591–6.
Pereira RC et al. CCAAT/enhancer binding protein homologous protein (DDIT3) induces osteoblastic cell differentiation. Endocrinology. 2004;145:1952–60.
Skalet AH et al. Rapid B cell receptor-induced unfolded protein response in nonsecretory B cells correlates with pro- versus antiapoptotic cell fate. J Biol Chem. 2005;280:39762–71.
Yang L et al. Multiple signals induce endoplasmic reticulum stress in both primary and immortalized chondrocytes resulting in loss of differentiation, impaired cell growth, and apoptosis. J Biol Chem. 2005;280:31156–65.
Heijmans J et al. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Reports. 2013;3:1128–39. This study indicates that the unfolded protein response in the ER plays a key role in the differentiation of intestinal epithelial stem cells.
Mohrin M et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347:1374–7.
Kruegel U et al. Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet. 2011;7:e1002253.
Oliver L et al. Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev. 2012;21:2779–88.
Salemi S et al. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell Res. 2012;22:432–5.
Liu F et al. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood. 2010;116:4806–14.
Mortensen M et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci U S A. 2010;107:832–7.
Mortensen M et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208:455–67.
Mortensen M et al. Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy. 2011;7:1069–70.
Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14:83–97.
Warr MR et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494:323–7. This study shows that FOXO3A-dependent autophagy induction protects blood haematopoietic stem cells (HSC) during metabolic stress and that, contrary to what has been previously reported, autophagy is dispensable for the cloning efficiency of HSC.
Vazquez P et al. Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy. 2012;8:187–99.
Zhang J et al. FRS2alpha-mediated FGF signals suppress premature differentiation of cardiac stem cells through regulating autophagy activity. Circ Res. 2012;110:e29–39.
Zhang J et al. The fibroblast growth factor signaling axis controls cardiac stem cell differentiation through regulating autophagy. Autophagy. 2012;8:690–1.
Wang C et al. FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat Neurosci. 2013;16:532–42.
Harris JM et al. Glucose metabolism impacts the spatiotemporal onset and magnitude of HSC induction in vivo. Blood. 2013;121:2483–93.
Takubo K et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12:49–61.
Yu WM et al. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell. 2013;12:62–74.
Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–63.
Ito K et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002.
Ahlqvist KJ et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012;15:100–9.
Norddahl GL et al. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell. 2011;8:499–510.
Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123:951–7.
Liu H et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–6.
Paik JH et al. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell. 2009;5:540–53.
Tothova Z et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–39.
Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008;20:126–36.
Renault VM et al. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell. 2009;5:527–39.
Yalcin S et al. Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress-mediated homeostasis of hematopoietic stem cells. J Biol Chem. 2008;283:25692–705.
Webb AE et al. FOXO3 shares common targets with ASCL1 genome-wide and inhibits ASCL1-dependent neurogenesis. Cell Reports. 2013;4:477–91.
Amcheslavsky A et al. Tissue damage-induced intestinal stem cell division in Drosophila. Cell Stem Cell. 2009;4:49–61.
Biteau B et al. Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet. 2010;6:e1001159.
Demontis F, Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010;143:813–25.
Wessells RJ et al. Insulin regulation of heart function in aging fruit flies. Nat Genet. 2004;36:1275–81.
Lin G et al. Paracrine wingless signalling controls self-renewal of Drosophila intestinal stem cells. Nature. 2008;455:1119–23.
Fontana L et al. Extending healthy life span—from yeast to humans. Science. 2010;328:321–6.
Stanfel MN et al. The TOR pathway comes of age. Biochim Biophys Acta. 2009;1790:1067–74.
Yilmaz OH et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature. 2012;486:490–5. This study reports a link between stem cell identity and the nutritional status of an organism.
Cerletti M et al. Short-term calorie restriction enhances skeletal muscle stem cell function. Cell Stem Cell. 2012;10:515–9.
Park HR, Lee J. Neurogenic contributions made by dietary regulation to hippocampal neurogenesis. Ann N Y Acad Sci. 2011;1229:23–8.
Liu M et al. The Drosophila female germline stem cell lineage acts to spatially restrict DPP function within the niche. Sci Signal. 2010;3:ra57.
Hsu HJ, Drummond-Barbosa D. Insulin levels control female germline stem cell maintenance via the niche in Drosophila. Proc Natl Acad Sci U S A. 2009;106:1117–21.
LaFever L, Drummond-Barbosa D. Direct control of germline stem cell division and cyst growth by neural insulin in Drosophila. Science. 2005;309:1071–3.
Ueishi S et al. Male germline stem cell division and spermatocyte growth require insulin signaling in Drosophila. Cell Struct Funct. 2009;34:61–9.
Mair W et al. Dietary restriction enhances germline stem cell maintenance. Aging Cell. 2010;9:916–8.
Kenyon C. A pathway that links reproductive status to lifespan in Caenorhabditis elegans. Ann N Y Acad Sci. 2010;1204:156–62.
Khodakarami A, et al. Mediation of organismal aging and somatic proteostasis by the germline. Front Mol Biosci. 2015; 2.
Shemesh N, et al. Germline stem cell arrest inhibits the collapse of somatic proteostasis early in Caenorhabditis elegans adulthood. Aging cell. 2013.
Vilchez D et al. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature. 2012;489:263–8.
Vilchez D, et al. FOXO4 is necessary for neural differentiation of human embryonic stem cells. Aging cell. 2013.
Vilchez D et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature. 2012;489:304–8. This study shows that human embryonic stem cells have high proteasome activity, mediated by high levels of the proteasome subunit PSMD11, and that this high proteasome activity protects their proteome by preventing the accumulation of misfolded proteins.
Kirkwood TB. Evolution of ageing. Nature. 1977;270:301–4.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) (DFG Research Grant VI742/1-1), the University of Cologne Advanced Postdoc Grant, the Cologne Graduate School of Ageing Research, the Alexander von Humboldt Foundation, and the European Commission (FP7-PEOPLE-2013-CIG).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
The authors declare that they have no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Age-related Stem Cell Modifiers
Rights and permissions
About this article

Cite this article
Koyuncu, S., Irmak, D., Saez, I. et al. Defining the General Principles of Stem Cell Aging: Lessons from Organismal Models. Curr Stem Cell Rep 1, 162–169 (2015). https://doi.org/10.1007/s40778-015-0017-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-015-0017-1