
Abstract
Purpose of Review
Mitochondria play various roles that are important for cell function and survival; therefore, significant mitochondrial dysfunction may have chronic consequences that extend beyond the cell. Mitochondria are already susceptible to damage, which may be exacerbated by environmental exposures. Therefore, the aim of this review is to summarize the recent literature (2012–2022) looking at the effects of six ubiquitous classes of compounds on mitochondrial dysfunction in human populations.
Recent Findings
The literature suggests that there are a number of biomarkers that are commonly used to identify mitochondrial dysfunction, each with certain advantages and limitations. Classes of environmental toxicants such as polycyclic aromatic hydrocarbons, air pollutants, heavy metals, endocrine-disrupting compounds, pesticides, and nanomaterials can damage the mitochondria in varied ways, with changes in mtDNA copy number and measures of oxidative damage the most commonly measured in human populations. Other significant biomarkers include changes in mitochondrial membrane potential, calcium levels, and ATP levels.
Summary
This review identifies the biomarkers that are commonly used to characterize mitochondrial dysfunction but suggests that emerging mitochondrial biomarkers, such as cell-free mitochondria and blood cardiolipin levels, may provide greater insight into the impacts of exposures on mitochondrial function. This review identifies that the mtDNA copy number and measures of oxidative damage are commonly used to characterize mitochondrial dysfunction, but suggests using novel approaches in addition to well-characterized ones to create standardized protocols. We identified a dearth of studies on mitochondrial dysfunction in human populations exposed to metals, endocrine-disrupting chemicals, pesticides, and nanoparticles as a gap in knowledge that needs attention.
Similar content being viewed by others

Introduction
The mitochondrion is a fundamental component of the cell that plays a vital part in energy metabolism. In addition to generating energy, mitochondria are also important in multiple cell signaling cascades, metabolite generation, the homeostasis of various minerals and lipids, calcium storage, the immune response, the synthesis of steroids and heme groups, and apoptosis [1–5]. Given these diverse functions, mitochondria are a critical component of cellular homeostasis and survival.
Despite the various roles they perform within the cell, mitochondria are particularly vulnerable to damage. This is due in part to their proximity to reactive oxygen species (ROS). Oxidative phosphorylation, the main source of ATP generation, occurs in the inner mitochondrial membrane [6]. During this process, electrons leak from complexes I, II, and III and react with oxygen to form superoxide. The superoxide radical is then converted to hydrogen peroxide by superoxide dismutase, and together, hydrogen peroxide and superoxide are considered mitochondrial ROS [7, 8, 9]. Due to the proximity of its production, excess ROS can result in damage to mitochondrial biomolecules, induce mitochondrial DNA mutations, alter membrane permeability and structure, and change calcium ion (Ca2+) homeostasis [8, 10, 11]. Damage to mitochondrial DNA (mtDNA) is particularly concerning, as the mitochondria have reduced DNA repair capacity in comparison to the nucleus [12]. This is likely due to the reliance on polymerase γ for both replication and repair of mtDNA and a limited repair mechanism, primarily base excision repair, when dealing with mtDNA damage [13, 14]. This is significant because persistent mtDNA damage can have further downstream effects on the mitochondrion.
Due to their susceptibility to damage, mitochondria are highly sensitive to environmental toxicants. The charged difference between the mitochondrial matrix and the cytosol allows for positively charged and lipophilic chemicals to accumulate within the mitochondrial matrix [15, 16]. The damage caused by these chemicals within the mitochondria can manifest in multiple ways. Often, the damage leads to the disruption of the mitochondrial electron transport chain (ETC), which results in excess generation of ROS, and decreased ATP levels [7, 17]. Other types of damage can include dysregulation of Ca2+, changes in membrane permeability, and structural damage to the mitochondria [18, 19]. The different types of damage interact to exacerbate detrimental effects and can result in cell death. Hence, the goal of this review is to characterize the effect of various environmental toxicants on mitochondrial dysfunction, focusing on human population research published within the past 5 years when available. Tables 1 and 2 summarize the literature cited in this review in human populations and experimental studies, respectively.
Mitochondrial Biomarkers for Environmental Health
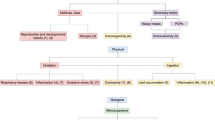
Given the importance of the mitochondria and its susceptibility to damage, there is a growing need for sensitive biomarkers to detect mitochondrial dysfunction from environmental toxicants (Fig. 1). One of the most common biomarkers used in human population studies is changes in the mtDNA copy number (mtDNAcn). mtDNAcn is the number of mitochondrial genomes in a cell, and is positively correlated with the size and the number of mitochondria [20]. Each cell contains hundreds to thousands of mitochondria, each of which contains many copies of the mitochondrial genome. mtDNAcn can change depending on the energetic demands of the cells. For instance, muscle cells contain around 7000 copies of mtDNA per cell, which is higher compared to that of cells with a lower metabolic capacity [21]. Under environmental stressors, significant changes in mtDNAcn may indicate a biological response to excess ROS production and mtDNA damage and dysfunction [22, 23]. In fact, changes in mtDNAcn are associated with neurodegenerative, cardiovascular, and chronic kidney diseases, making them a relevant biomarker of mitochondrial dysfunction [24, 25, 26]. Moreover, measurement of mtDNAcn uses relatively simple techniques, making it an accessible biomarker for large human population studies [24, 27]. However, the mtDNAcn biomarker has some limitations. Conflicting associations have been observed in human population studies between chemical exposures and mtDNAcn which may be attributed to population characteristics, as well as the exposure concentration and duration. Furthermore, both an excess and a dearth of mtDNA can represent mitochondrial dysfunction, so consistency in the direction of effect across studies may not be informative. Additionally, significant variations between individuals and within an individual’s cell-specific mtDNAcn have been detected, which may be due to the various biological states that can lead to either an increase or a decrease in mtDNAcn [30•]. In particular, the magnitude and duration of oxidative stress and damage within the mitochondria may lead to varying responses in mtDNAcn. For instance, mitochondrial insult may initially result in mtDNA replication to compensate for the damage, leading to an increased copy number. However, it is also possible that past a certain threshold, the mitochondria are no longer able to compensate for the damage, leading to mitochondrial membrane permeability and apoptosis, which results in a decrease in the copy number [28, 29]. These different reasons give rise to the concern than the mtDNAcn values may be over interpreted [30•].

Common biomarkers used to identify and measure mitochondrial dysfunction. mtDNA mitochondrial DNA, Ca2+ calcium ions, 8-OHdG 8-oxo-2′-deoxyguanosine
Heteroplasmy is another mitochondrial biomarker that describes the proportion of mutated mtDNA within a cell and may be used to indicate the severity of damage to the mitochondria [31, 32]. While a small amount of heteroplasmy (< 1%) in the mtDNA is normal, when the mtDNA undergoes damage, it may alter mitochondrial gene expression, leading to a higher proportion of mutations [32]. Hence, toxicant-induced mitochondrial damage may lead to a higher mtDNA mutation load, i.e., increased heteroplasmy, making it a relevant biomarker. In fact, recently published literature has demonstrated that heteroplasmy can be measured in human populations and is associated with changes in birth outcomes, respiratory functions, blood pressure, and depressive symptoms [33••, 34–36]. Heteroplasmy can also provide insight into mtDNA function through examination of heteroplasmic sites in coding regions [37]. However, for a biochemical defect to be detected, the proportion of mutated DNA must exceed a threshold level, and each cell, tissue, organ, and person has its own individual threshold, making it hard to compare across different populations [32, 38]. As a consequence, not many studies use heteroplasmy as a biomarker to measure the response to environmental toxicant exposure.
The mitochondrial respiratory chain is made up of five transmembrane enzyme complexes that work together with electron transfer carriers, ubiquinone, and cytochrome c, to produce ATP during oxidative phosphorylation. These complexes may be a target of environmental toxicants that alter their expression, concentration, or maximum activity [39]. During the process of oxidative phosphorylation, the complexes aid in the maintenance of an electrochemical gradient through a series of redox reactions. This electrochemical gradient generates the mitochondrial membrane potential and is an essential component of energy production. Either through the disruption of the complexes, perturbation of the electron transfer carriers or proteins, and/or damage to the membranes, external chemicals can alter the membrane potential, which may affect ATP and induce cell death [40, 41]. Changes in both the activity of the respiratory chain complexes and membrane potential are useful biomarkers because they help elucidate the mechanisms of toxicant-induced mitochondrial dysfunction. However, these measurements often require large quantities of fresh samples, which are beyond the capabilities of most cohort studies. Furthermore, a significant limitation is that the probes often used to measure these changes can be affected by the cellular membrane potential, mitochondrial pH, and changes in ATP production [41–44]. Nonetheless, more techniques are being developed to measure these mitochondrial bioenergetics functions in humans [45••].
Changes in oxidative phosphorylation, among other mitochondrial defects, often have downstream effects that are also commonly measured as biomarkers. The oxidation of guanine in mtDNA and the subsequent formation of 8-hydroxy-2′-deoxyguanosine (8-OHdG) is one of the main forms of free radical–induced DNA lesions [46]. High concentrations of mitochondrial 8-OHdG are indicative of oxidative DNA damage, and therefore are a common biomarker used to measure mitochondrial dysfunction [47]. Exposure to environmental toxicants can often lead to higher concentrations of ROS within the mitochondria and is associated with higher concentrations of 8-OHdG. The assays used to measure 8-OHdG are well established and are widely used to represent mitochondrial dysfunction in human populations. However, 8-OHdG is also detected in nuclear DNA, so mtDNA often needs to be separated prior to quantification. Additionally, there have been discrepancies between chromatographic and immunoassay approaches used to measure 8-OHdG within human samples [49].
Ca2+ levels play an important role in membrane potential regulation, ROS homeostasis, and oxidative phosphorylation within the mitochondria [50]. As a consequence, impaired mitochondrial Ca2+ transfer alters the production of ATP and downregulates mitochondrial metabolism, while high concentrations of mitochondrial Ca2+ suggest a disruption of the electrochemical gradient [50, 51]. Toxicant-induced overload of Ca2+ concentrations is associated with oxidative stress, a collapse in membrane potential, and eventually cell death [52]. While Ca2+ levels in in vitro models are commonly used to measure mitochondrial dysfunction, an important consideration is that this assay is unable to differentiate if toxicant-induced effects were a cause or consequence of the phenotype [39]. Additionally, there have been discrepancies in the Ca2+ levels measured using fluorescent dyes and genetically encoded calcium indicators, which may be attributed to the fact that mitochondria from different cell types uptake Ca2+ in different concentrations, making it hard to cover the full range using one type of sensor [48].
In addition to these measures of mitochondrial dysfunction, the alteration of cardiolipin is an emerging mitochondrial biomarker. Cardiolipin is a mitochondrion-exclusive phospholipid and plays an important role in mitochondrial protein transport, membrane morphology, cellular signaling, and bioenergetics [53, 54•]. While there has yet to be research examining associations between chemical exposure and cardiolipin levels, studies have found associations between cardiolipin alterations and diseases in human populations [54•, 55]. This suggests that it might be a relevant biomarker to account for when examining mitochondrial dysfunction.
Additionally, the presence of circulating cell-free mitochondria in blood may serve as an alternative matrix for the biomarkers discussed above. Cell-free mitochondria are the presence of whole and functioning mitochondria out of the cell, which has been detected within human blood [56•, 57]. In addition to whole mitochondria, cell-free mtDNA fragments are also detected in human blood, either encapsulated within extracellular vesicles or free-circulating. While the mechanisms and functions of cell-free mitochondria are relatively unknown, elevated levels of plasma cell-free mtDNA are associated with stress, inflammatory diseases, cancers, and sepsis in human populations [58•, 59, 60]. The emergence of standardized ways of measuring this biomarker may allow for wider use when looking at associations with toxicant-induced mitochondrial damage. The use of mitochondrial biomarkers in human population and experimental studies has provided great insight into the impact of environmental agents on mitochondrial function and health.
Known Mitochondrial Disruptors
Much of our present knowledge on the critical role of mitochondria in health comes from the few chemicals whose mechanisms of toxicity on the mitochondria are well characterized. Acute poisoning from these highly specific mitochondrial toxicants leads to nausea, headaches, seizures, cardiac failure, and, in extreme cases, death. Cyanide is a potent mitochondrial inhibitor that binds to complex IV, specifically the a3 portion of cytochrome oxidase, within the ETC [61]. From there, cyanide competes with oxygen and binds to the Fe-Cu center which inhibits activity and energy production [62]. Rotenone, a pesticide and insecticide, is another mitochondrial inhibitor that affects the electron transfer from the Fe-S centers in complex I. This leads to the inhibition of oxidative phosphorylation and consequently a limited production of ATP, which further induces apoptosis in cells. Moreover, rotenone-induced apoptosis is closely related to mitochondrial ROS formation which may cause mitochondrial damage [63, 64]. Azidothymidine is an anti-HIV drug that accumulates within the mitochondrial intermembrane space where it disrupts the ATP/ADP translocator and enhances the production of ROS [65, 66]. Doxorubicin is an anticancer drug that also generates ROS; however, it does so by interacting with complex I and the proteins involved in oxidative phosphorylation [67, 68]. The resulting oxidative stress then goes on to cause mitochondrial injury and apoptosis. Lastly, exposure to benzene, a common industrial chemical and environmental toxicant, consistently increases mtDNAcn and alters mitochondrial pathways, possibly in response to the oxidative stress caused by benzene within the mitochondria [69–72]. Among all these classic mitochondrial disruptors, a common theme is disruption of energy production and oxidative stress. Understanding the well-established mechanisms of mitochondrial disruption caused by these chemicals has allowed researchers to investigate the role of other ubiquitous and well-known toxicants on mitochondrial dysfunction.
Polycyclic Aromatic Hydrocarbons
Polycyclic aromatic hydrocarbons (PAHs) are a class of compounds that are common byproducts of incomplete combustion. They are frequently detected following incineration of industrial, domestic, and agricultural products and emissions from vehicles [73]. Once emitted, PAHs may bind to or form small particles in the air which subsequently lead to human exposure. PAHs are highly lipophilic toxicants and therefore readily accumulate in the mitochondria due to their high lipid content [74]. In fact, PAHs are also shown to preferentially bind to the mtDNA at 40–90 times greater than nuclear DNA [74, 75]. Moreover, the mitochondrial cytochrome P450 system may bioactivate PAHs to make them more toxic in the organelle [76]. PAHs may also be activated through mitochondrial aldo–keto reductase and/or manganese superoxide dismutase which causes the production of ROS [77]. In vitro studies have shown that exposure to PAHs triggers mitochondrial oxidative damage in blood lymphocytes and affects the mitochondrial redox machinery which leads to higher concentrations of ROS [78]. This excess generation of ROS and associated oxidative stress within the mitochondria may act as a regulator of the mtDNAcn [29, 79], leading to mtDNAcn changes in populations exposed to PAHs.
The literature examining the associations between PAH exposure and mtDNAcn within human populations is inconclusive. Higher urinary PAH metabolites were associated with higher mtDNAcn in peripheral blood samples of asphalt workers [80] and in leukocytes of coke oven workers [77]. Urinary PAH metabolites were also positively associated with increased peripheral blood mtDNAcn in an urban population in China [81]. Prenatal exposure to PAHs measured through maternal urinary metabolites was associated with increased mtDNAcn in cord blood in China [82]. Conversely, other studies have also shown negative associations between PAH exposure and mtDNAcn. Increased urinary PAH metabolites were associated with decreased mtDNAcn in college student sperm samples [83] and leukocytes of non-smoking women [84]. Occupational exposures to PAHs in different coke oven workers showed significantly lower mtDNAcn in peripheral blood compared to the control groups [85•, 86, 87]. This relationship was also detected in the blood of individuals that lived in homes with a higher PAH concentration in their house dust [88]. The differences in mtDNAcn may be attributed to varied exposure levels between the different studies; however, because exposures to PAHs were measured in different matrices, we cannot directly compare across studies.
Particulate Air Pollutants and Black Carbon
Air pollution is a complex mixture that consists of a variety of physical and chemical components depending on the sources [89]. While airborne PAHs are due to combustion of fuel sources, the presence of other chemical substances, gases, or particulate matter within the air is attributed primarily to vehicle exhaust and industry emissions. In this section, we will focus on the compounds, other than PAHs, that have clearly displayed toxic effects on the mitochondria. Mitochondria are susceptible to air pollutants particularly due to their lack of repair capacity and their enhanced vulnerability to ROS. Experimental studies have shown that exposure to air pollutants leads to oxidative stress, changes in mitochondrial membrane potential, and decreases in mtDNAcn in cells [90–92] and lower mtDNAcn, lower mitochondrial consumption rate, and mitochondrial structural abnormalities in mice [92, 93].
Air pollutants are some of the most well-studied exposures in relation to mitochondria in humans. Studies have shown that increased prenatal exposure to particulate matter (PM) was associated with increased levels of mitochondrial urinary 8-OHdG in maternal and umbilical cord blood, suggesting oxidative stress within the mitochondria [94]. Moreover, during the air quality intervention for the Beijing Olympic Games, a reduction in ambient air pollutant levels led to a significant decreased in urinary 8-OHdG levels in schoolchildren [95].
Similar to PAHs, particulate air pollutants have a varied effect on mtDNAcn, possibly as a response to the excess ROS within the mitochondria. Increased PM2.5 (PM with a diameter of 2.5 µm or less), PM10 (PM with a diameter of 10 µm or less), and black carbon (BC) exposure was associated with a decrease in mtDNAcn in the blood of an elderly Flemish truck driver population and leukocytes of an elderly Belgian population [96–98]. Moreover, studies have also shown that prenatal exposure to NO2, PM10, and PM2.5 are associated with decreased placental mtDNAcn [84, 98, 99, 100] and cord blood mtDNAcn [101, 102]. Other studies, however, have shown that occupational PM exposure was associated with increased whole-blood mtDNAcn in steel workers [103, 104] and BC exposure was positively associated with whole-blood mtDNAcn in older adults [105]. Exposure levels, duration of exposure, and life stages of the participants in these studies are highly varied, which may contribute to differences in study findings. Lastly, in addition to changes in mtDNAcn, PM2.5 and NO2 have shown to be positively associated with mtDNA methylation in blood and placenta [104, 106•, 107] and DNA methylation in mitochondrion-related genes in umbilical cord blood [108]. Moreover, PM2.5 was associated with an increase in heteroplasmy on genes coding for NADH dehydrogenase and subunits for ATP synthase in mtDNA [109]. PM10 exposure was also associated with transcriptomic pathways related to mitochondrial genome maintenance, ETC, and tricarboxylic acid (TCA) cycle in whole blood, suggesting that the pathways were upregulated to compensate for the PM-induced damage [110]. Prenatal exposure to PM2.5 has also been shown to be positively associated with a decrease in mitochondrial function in blood and placenta [106•, 107].
Heavy Metals
Heavy metals, specifically cationic metals, are shown to preferentially accumulate within the mitochondria through the calcium transporter due to their similarity to the Ca2+ ion [111]. Moreover, the mitochondrial membrane contains unsaturated lipids which enhance its susceptibility to metals, such as arsenic (As), compared to other organelles [112]. Human population studies have shown that exposure to manganese (Mn), aluminum (Al), and lead (Pb) in the prenatal period has resulted in an increase in mtDNAcn in cord blood, and exposure to Pb was associated with an increase in maternal mtDNAcn [113•, 114, 115•, 116]. Conversely, exposure to thallium and As was associated with a decrease in mtDNAcn in cord blood leukocytes, and magnesium (Mg) exposure was associated with decreased maternal and cord blood mtDNAcn [116–118]. Smith et al. (2021) also reported a non-linear relationship between prenatal Mg exposure and cord blood mtDNAcn, as well as between barium, Pb, and mercury (Hg) exposure and maternal mtDNAcn. Interestingly, they did not find any significant associations between As, cadmium (Cd), cesium, Mn, selenium, and zinc exposure and mtDNAcn [116].
Much of the literature examining the effect of metals on mitochondrial dysfunction details experiments conducted in in vitro and animal models, and therefore, this section of the review, as well as for the following chemical classes, will focus on elucidating mechanisms behind this toxicity that might be relevant to humans. The most common dysfunction induced by heavy metals is the production of elevated mitochondrial ROS. The Fenton reaction, where transition metals such as iron and copper (Cu) catalyze the generation of hydroxyl radicals from hydrogen peroxide, has been commonly implicated in the production of ROS [119, 120]. Cu, Cd, Pb, Mn, Hg, As, and Al have all shown to increase ROS which in turn triggers mitochondrial dysfunction and subsequent apoptotic and autophagic death in both in vitro systems and rodent models [62, 111, 121–129]. In human populations, high Cd exposure was associated with higher 8-OHdG and citrate (a urinary metabolite associated with mitochondrial metabolism) levels [130].
In addition to producing excess ROS, Cu, Cd, and As decreased the transmembrane potential and ATP levels in human cell lines and rats [111, 122, 124, 128, 131, 132]. This is possibly through the inhibition of ADP, which induces ion permeability of the inner mitochondrial membrane [133]. Once the membrane potential is lost, cytochrome c is released and caspases may be activated, leading to apoptosis of the mitochondria [128, 134]. In addition, Cd treatment also inhibits mitochondrial respiratory chain enzymes within human osteoblasts [122] and leads to organelle swelling causing the inhibition of respiration in rats [135].
Another mechanism of toxicity for other heavy metals such as Pb, Mn, As, and Hg is via Ca2+-dependent signaling pathways. Mitochondria have been implicated as major sites for Pb2+ and Mn2+ accumulation [127, 136], following which both Pb2+ and Mn2+ can substitute for Ca2+ in the Ca2+ uniporter and TCA cycle dehydrogenases, respectively, and cause Ca2+ dysregulation in the mitochondria [62]. This in turn induces Ca2+ efflux, which leads to decreased NADH levels in the mitochondria and eventually apoptosis.
Endocrine-Disrupting Chemicals
Endocrine-disrupting chemicals (EDCs) are a class of compounds that modulate hormone action primarily by mimicking naturally occurring hormones, binding to their respective receptors and changing downstream pathways [137]. There are a wide variety of chemicals that are classified as EDCs, including phthalates, parabens, and bisphenols. These are commonly used as plasticizers in consumer products but are also used in pharmaceuticals, cosmetics, and personal care products [138]. As EDCs affect different cellular processes, including those related to energy production and utilization, it is thought that EDC disruption of energy homeostasis may be associated with mitochondrial dysfunction [139•].
Exposures to phthalates and bisphenols have been shown to be associated with changes in mtDNA methylation [140]. Specifically, EDCs such as alkylphenol 4-nonylphenol (NP), di(2-ethylhexyl) phthalate (DEHP), monoethylhexyl phthalate (MEHP), and bisphenol A (BPA) are associated with elevated oxidative stress through increased ROS production, changes in redox homeostasis, and production of extracellular superoxide [139•, 140–146]. This in turn affects the mtDNAcn as described for toxicants above. Human studies have shown that exposure to phthalates is positively associated with mtDNAcn in sperm and bisphenol S (BPS) is positively associated with mtDNAcn in children [147•, 148].
In addition to oxidative stress, studies have shown that BPA exposure was associated with a decrease in mitochondrial respiratory complex activity and consequently a decrease in mitochondrial membrane potential and ATP production in human lymphoblasts and rat models [146, 149, 150]. BPA and BPS may also alter the expression of regulatory genes related to mitochondrial energy metabolism, mitochondrial fusion and division, and mitochondrial fatty acid metabolism in rats [145, 149, 151]. Additionally, DEHP exposure is associated with mitochondrial ultrastructural abnormalities in quail [141].
Pesticides
Pesticides are a large class of chemical compounds with a wide range of properties that lend themselves to different modes of action when inducing mitochondrial toxicity. Organophosphate (OP) and organochlorine (OC) pesticides are classes of chemicals that are highly lipophilic and can therefore easily enter and accumulate within the mitochondria similar to PAHs. In fact, OP pesticides with hydrophobic properties have an increased mitochondrial translocator protein–binding affinity [152]. Once in the mitochondria, both OP and OC pesticides have been shown to reduce the mitochondrial membrane potential, produce mtDNA damage, promote oxidative damage, and reduce mitochondrial ATP in cell lines and zebra fish [152, 153, 156]. In addition to these other mechanisms, Budnik et al. (2013] also showed that exposure to OC pesticides was significantly associated with elevated serum levels of circulating mtDNA, suggesting decreased integrity of mtDNA in exposed individuals. Additionally, prenatal exposure to benzothiazoles, a class of compounds that are used as fumigants, is associated with changes in mtDNAcn in cord blood [158]. In this study, investigators observed a positive association with exposure measured in the first trimester, which was then reversed in the third trimester.
Paraquat and atrazine, two widely used pesticides, induce mitochondrial toxicity through very similar mechanisms. Both paraquat and atrazine produce ROS which induces mitochondrial toxicity [159, 160]. Both compounds adversely affect the electron transfer within the ETC to form a superoxide anion which forms an excess of ROS in various animal systems [159–163]. Exposure to paraquat and atrazine has also been shown to decrease mitochondrial membrane potential in pigs and mice [160, 164]. In addition to these mechanisms, atrazine has been shown to activate the mitochondrial unfolded protein response, as well as increase mitochondrial damage and vacuolar degeneration, and decrease mitochondrial cristae and volume density in Caenorhabditis elegans [163].
Nanomaterials
Nanomaterials are particles that range from 1 to 100 nm that may be formed naturally or engineered. Nanomaterials are found in numerous consumer products including cosmetics, tires, and electronics. Once in the body, due to their small size, nanomaterials are easily transported across cell membranes where they can accumulate within the mitochondria [165, 166•] and lead to the disruption of the mitochondrial membrane potential and structure [166•, 167]. Nanomaterials are distinct from the previous classes of chemicals in that they are primarily physical rather than chemical stressors. Studies have shown that exposure to silver nanoparticles, hydroxyapatite nanoparticles, cadmium telluride quantum dots, graphene, fullerene, and carbon nanotubules leads to a significant decrease in mitochondrial membrane potential and ADP-induced depolarization through increased permeability of the mitochondrial inner membrane and induction of mitochondrial permeability transition [168–172] in both human and rat in vitro systems. Exposure to nanomaterials also leads to increased intracellular Ca2+ levels and overproduction of ROS in human cells [171, 172, 173]. They are also associated with a change in levels and activities of enzymes of the ETC [171, 174]. In addition to the changes within the ETC, the presence of iron-rich nanoparticles and graphene oxide in mitochondria is associated with deformed cristae and ruptured membranes in human heart samples and zebra fish models [175, 176]. This in vitro evidence suggests that nanoparticles are associated with mitochondrial toxicity, and therefore could be important for human health effects. Hence, more research in human populations is key towards understanding the mitochondrial health impacts of nanoparticles.
Conclusion
A large body of human population and experimental research suggests that multiple classes of environmental toxicants can induce mitochondrial stress and disrupt mitochondrial function (Fig. 2, Tables 1 and 2). Several chronic diseases are characterized by system- or organ-specific mitochondrial dysfunction. As discussed throughout, disparate toxicants can induce common types of mitochondrial damage and responses. For instance, excess production of ROS, a ubiquitous response across different chemical classes, is commonly tied to other mitochondrial biomarkers and dysfunction such as alterations of mitochondrial membrane permeability, calcium homeostasis, and ATP production [177–179]. Moreover, the presence of excess ROS within the mitochondria can induce a positive feedback loop in the mitochondrial environment, leading to more ROS release [180, 181]. Superfluous ROS may affect the normal functioning of mitochondria, cells, and organisms and is tied to cardiovascular diseases [182], autism spectrum disorder [183], neurodegenerative diseases [181, 184], obesity [185], and diabetes [178]. Another common response to the different forms of mitochondrial damage is a decrease in mitochondrial energetics, as demonstrated through reduction in ATP levels and oxygen consumption. This decrease has also been associated with the onset of chronic kidney diseases [186], heart diseases [187, 188], neurodegenerative diseases [189–191], liver diseases [192], and diabetes [193]. Lastly, persistent mtDNA damage caused by chemical exposure may inhibit replication, RNA transcription, and mitochondrial function. Therefore, it is associated with neurodegenerative diseases [194, 195], cardiovascular diseases [196, 197], liver diseases [198], inflammatory diseases [199], kidney diseases [200, 201], and obesity [202].

The environmental toxicant–induced mitochondrial dysfunction pathways discussed within this review. Blue boxes outlining the environmental toxicants represent associations shown in both human populations and experimental models, whereas red boxes represent associations found only in experimental models
A wealth of experimental evidence indicates the ability of environmental toxicant exposures, such as PAHs and air pollutants, to induce mitochondrial dysfunction. However, there is a greater need for more studies examining the role of additional chemicals such as heavy metals, EDCs, pesticides, and nanomaterials in mitochondrial dysfunction within human populations. Understanding the associations between toxicant exposure and mitochondrial dysfunction in humans may help elucidate potential mechanisms through which these chemicals induce toxicity. Moreover, recognizing these mechanisms may aid in the development of therapeutics that target the mitochondrial dysfunction and prevent disease advancement [203, 204].
As described within this review, most of the human population studies linking exposure to mitochondrial dysfunction used blood or placental mtDNAcn as a biomarker. While changes in mtDNAcn can suggest mitochondrial dysfunction and may be associated with health outcomes [25, 96], they are not a perfect representation of mitochondrial content or biogenesis and there is inherent variability in copy number associated with the cell type composition within a tissue or biospecimen [30•]. Furthermore, the inconsistent directionality of changes in mtDNAcn may make it difficult to interpret the nature of the adverse effects. Additional research is needed to untangle the complex impacts of toxicants on mtDNAcn and their significance within human populations. Therefore, with the advent of new techniques and biomarkers such as cell-free mitochondria [56•, 205] and cardiolipin levels in blood [206], there is a need to apply these novel approaches and generate a standardized protocol to continue to characterize the mechanisms behind and consequences of toxicant-induced mitochondrial dysfunction.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018;69:62–72.
Coffman JA, Coluccio A, Planchart A, Robertson AJ. Oral–aboral axis specification in the sea urchin embryo: III. Role of mitochondrial redox signaling via H2O2. Dev Biol. 2009;330:123–30.
Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163:560–9.
Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–17.
Meyer JN, Chan SSL. Sources, mechanisms, and consequences of chemical-induced mitochondrial toxicity. Toxicology. 2017;391:2–4.
Russell OM, Gorman GS, Lightowlers RN, Turnbull DM. Mitochondrial diseases: hope for the future. Cell. 2020;181:168–88.
Zhao R-Z, Jiang S, Zhang L, Yu Z-B. Mitochondrial electron transport chain, ROS generation and uncoupling (review). Int J Mol Med. Spandidos Publications 2019;44:3–15.
Zia A, Farkhondeh T, Pourbagher-Shahri AM, Samarghandian S. The roles of mitochondrial dysfunction and reactive oxygen species in aging and senescence. Curr Mol Med. 2021;22:37–49.
Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. 2015;6:472–85.
Blajszczak C, Bonini MG. Mitochondria targeting by environmental stressors: implications for redox cellular signaling. Toxicology. 2017;391:84–9.
Guo C, Sun L, Chen X, Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen Res. 2013;8:2003–14.
Phillips NR, Sprouse ML, Roby RK. Simultaneous quantification of mitochondrial DNA copy number and deletion ratio: a multiplex real-time PCR assay. Sci Rep. 2014;4:3887.
Copeland WC. The mitochondrial DNA polymerase in health and disease. Subcell Biochem. 2010;50:211–22.
Bohr VA, Stevnsner T, de Souza-Pinto NC. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene. 2002;286:127–34.
Frantz M-C, Wipf P. Mitochondria as a target in treatment. Environ Mol Mutagen. 2010;51:462–75.
Meyer JN, Leung MCK, Rooney JP, Sendoel A, Hengartner MO, Kisby GE, et al. Mitochondria as a target of environmental toxicants. Toxicol Sci. 2013;134:1–17.
Trotta AP, Gelles JD, Serasinghe MN, Loi P, Arbiser JL, Chipuk JE. Disruption of mitochondrial electron transport chain function potentiates the pro-apoptotic effects of MAPK inhibition. J Biol Chem. 2017;292:11727–39.
Bauer TM, Murphy E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circulation Research. Am Heart Assoc. 2020;126:280–93.
Lemasters JJ, Theruvath TP, Zhong Z, Nieminen A-L. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787:1395–401.
Lee H-C, Wei Y-H. Mitochondrial role in life and death of the cell. J Biomed Sci. 2000;7:2–15.
Castellani CA, Longchamps RJ, Sun J, Guallar E, Arking DE. Thinking outside the nucleus: mitochondrial DNA copy number in health and disease. Mitochondrion. 2020;53:214–23.
Wallace DC. Mitochondrial DNA in evolution and disease. Nature. 2016;535:498–500.
Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402.
Filograna R, Mennuni M, Alsina D, Larsson N-G. Mitochondrial DNA copy number in human disease: the more the better? FEBS Lett. 2021;595:976–1002.
Ashar FN, Zhang Y, Longchamps RJ, Lane J, Moes A, Grove ML, et al. Association of mitochondrial DNA copy number with cardiovascular disease. JAMA Cardiology. 2017;2:1247–55.
Tin A, Grams ME, Ashar FN, Lane JA, Rosenberg AZ, Grove ML, et al. Association between mitochondrial DNA copy number in peripheral blood and incident CKD in the atherosclerosis risk in communities study. J Am Soc Nephrol. 2016;27:2467–73.
Longchamps RJ, Castellani CA, Yang SY, Newcomb CE, Sumpter JA, Lane J, et al. Evaluation of mitochondrial DNA copy number estimation techniques. PLOS ONE. Public Library of Science; 2020;15:e0228166.
Lee H-C, Wei Y-H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol. 2005;37:822–34.
Lee HC, Yin PH, Lu CY, Chi CW, Wei YH. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem J. 2000;348:425–32.
• Picard M. Blood mitochondrial DNA copy number: what are we counting? Mitochondrion. 2021;60:1–11. This review outlines important considerations when using mtDNAcn to measure mitochondrial dysfunction.
Stefano GB, Bjenning C, Wang F, Wang N. Kream RM. Mitochondrial heteroplasmy: Mitochondrial dynamics in cardiovascular medicine. Springer International Publishing; 2017:577–94.
Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–42.
•• Cowell W, Brunst K, Colicino E, Zhang L, Zhang X, Bloomquist TR, et al. Placental mitochondrial DNA mutational load and perinatal outcomes: findings from a multi-ethnic pregnancy cohort. Mitochondrion. 2021;59:267–75. This study demonstrates an association between heteroplasmy in placental mtDNA and disparate birth outcomes.
Vaz Fragoso CA, Manini TM, Kairalla JA, Buford TW, Hsu F-C, Gill TM, et al. Mitochondrial DNA variants and pulmonary function in older persons. Exp Gerontol. 2019;115:96–103.
Tranah GJ, Maglione JE, Yaffe K, Katzman SM, Manini TM, Kritchevsky S, et al. Mitochondrial DNA m.13514G>A heteroplasmy is associated with depressive symptoms in the elderly. Int J Geriatr Psychiatry. 2018;33:1319–26.
Buford TW, Manini TM, Kairalla JA, McDermott MM, Vaz Fragoso CA, Chen H, et al. Mitochondrial DNA sequence variants associated with blood pressure among 2 cohorts of older adults. J Am Heart Assoc. 2018;7:e010009.
Sosa MX, Sivakumar IKA, Maragh S, Veeramachaneni V, Hariharan R, Parulekar M, et al. Next-generation sequencing of human mitochondrial reference genomes uncovers high heteroplasmy frequency. PLOS Comput Biol. Public Library of Science; 2012;8:e1002737.
Sobenin IA, Mitrofanov KY, Zhelankin AV, Sazonova MA, Postnov AY, Revin VV, et al. Quantitative assessment of heteroplasmy of mitochondrial genome: perspectives in diagnostics and methodological pitfalls. Biomed Res Int. 2014;2014:292017.
Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312.
Sakamuru S, Attene-Ramos MS, Xia M. Mitochondrial membrane potential assay. Methods Mol Biol. 2016;1473:17–22.
Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, et al. Mitochondrial membrane potential. Anal Biochem. 2018;552:50–9.
Logan A, Pell VR, Shaffer KJ, Evans C, Stanley NJ, Robb EL, et al. Assessing the mitochondrial membrane potential in cells and in vivo using targeted click chemistry and mass spectrometry. Cell Metab. 2016;23:379–85.
Tian Y, Tian W, Li T, Xu J. Commentary: the effects of different 787 fluorescent indicators in observing the changes of the mitochondrial membrane potential during oxidative stress-induced mitochondrial injury of cardiac H9c2 cells. J Cardiol Cardiovasc Sci. 2021;5(1):1–3.
Horan MP, Pichaud N, Ballard JWO. Review: Quantifying mitochondrial dysfunction in complex diseases of aging. J Gerontol: Series A. 2012;67:1022–35.
•• Acin-Perez R, Benincá C, Shabane B, Shirihai OS, Stiles L. Utilization of human samples for assessment of mitochondrial bioenergetics: gold standards, limitations, and future perspectives. Life (Basel). 2021;11:949. This review evaluates the different human samples used to measure mitochondrial bioenergetics.
Ziech D, Franco R, Georgakilas AG, Georgakila S, Malamou-Mitsi V, Schoneveld O, et al. The role of reactive oxygen species and oxidative stress in environmental carcinogenesis and biomarker development. Chem Biol Interact. 2010;188:334–9.
Valavanidis A, Vlachogianni T, Fiotakis C. 8-Hydroxy-2′-deoxyguanosine (8-OHdG): a critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health, Part C. Taylor & Francis; 2009;27:120–39.
Fernandez-Sanz C, De la Fuente S, Sheu S-S. Mitochondrial Ca2+ concentrations in live cells: quantification methods and discrepancies. FEBS Lett. 2019;593:1528–41.
Toward consensus in the analysis of urinary 8-oxo-7,8-dihydro-2′-deoxyguanosine as a noninvasive biomarker of oxidative stress. FASEB J. 2010;24:1249–60.
Giorgi C, Marchi S, Pinton P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol. 2018;19:713–30.
Xu Z, Zhang D, He X, Huang Y, Shao H. Transport of calcium ions into mitochondria. Curr Genomics. 2016;17:215–9.
Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529:57–68.
Dudek J. Role of cardiolipin in mitochondrial signaling pathways. Front Cell Dev Biol. 2017;5:90.
• Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Role of cardiolipin in mitochondrial function and dynamics in health and disease: molecular and pharmacological aspects. Cells. 2019;8:728. This review outlines the importance of cardiolipin in mitochondrial function.
Paradies G, Paradies V, Ruggiero FM, Petrosillo G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20:14205–18.
• Al Amir Dache Z, Otandault A, Tanos R, Pastor B, Meddeb R, Sanchez C, et al. Blood contains circulating cell-free respiratory competent mitochondria. The FASEB J. 2020;34:3616–30. This study indicates the presence of circulating cell-free mitochondria in human blood samples.
Stier A. Human blood contains circulating cell-free mitochondria, but are they really functional? American Journal of Physiology-Endocrinology and Metabolism. Am Physiol Soc. 2021;320:E859–63.
• Trumpff C, Michelson J, Lagranha CJ, Taleon V, Karan KR, Sturm G, et al. Stress and circulating cell-free mitochondrial DNA: a systematic review of human studies, physiological considerations, and technical recommendations. Mitochondrion. 2021;59:225–45. This review establishes the associations between physiological stressors and circulating cell-free mtDNA.
Li L, Hann H-W, Wan S, Hann RS, Wang C, Lai Y, et al. Cell-free circulating mitochondrial DNA content and risk of hepatocellular carcinoma in patients with chronic HBV infection. Sci Rep. 2016;6:23992.
Dwivedi DJ, Toltl LJ, Swystun LL, Pogue J, Liaw K-L, Weitz JI, et al. Prognostic utility and characterization of cell-free DNA in patients with severe sepsis. Crit Care. 2012;16:R151.
Khailova LS, Rokitskaya TI, Kotova EA, Antonenko YN. Effect of cyanide on mitochondrial membrane depolarization induced by uncouplers. Biochemistry Moscow. 2017;82:1140–6.
Caito SW, Aschner M. Mitochondrial redox dysfunction and environmental exposures. Antioxidants & Redox Signaling. Mary Ann Liebert, Inc., publishers; 2015;23:578–95.
Heinz S, Freyberger A, Lawrenz B, Schladt L, Schmuck G, Ellinger-Ziegelbauer H. Mechanistic investigations of the mitochondrial complex I inhibitor rotenone in the context of pharmacological and safety evaluation. Sci Rep. 2017;7:45465.
Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production *. J Biol Chem. 2003;278:8516–25.
Nomura R, Sato T, Sato Y, Medin JA, Kushimoto S, Yanagisawa T. Azidothymidine-triphosphate impairs mitochondrial dynamics by disrupting the quality control system. Redox Biol. 2017;13:407–17.
Samuels DC. Mitochondrial AZT metabolism. IUBMB Life. 2006;58:403–8.
Abdullah CS, Alam S, Aishwarya R, Miriyala S, Bhuiyan MAN, Panchatcharam M, et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci Rep. 2019;9:2002.
Wallace KB, Sardão VA, Oliveira PJ. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ Res. American Heart Association; 2020;126:926–41.
Rothman N, Vermeulen R, Zhang L, Hu W, Yin S, Rappapor SM, et al. Metabolome-wide association study of occupational exposure to benzene. Carcinogenesis. 2021;42(11):1326–36.
Carugno M, Pesatori AC, Dioni L, Hoxha M, Bollati V, Albetti B, et al. Increased mitochondrial DNA copy number in occupations associated with low-dose benzene exposure. Environ Health Perspect. 2012;120:210–5.
McHale CM, Zhang L, Smith MT. Current understanding of the mechanism of benzene-induced leukemia in humans: implications for risk assessment. Carcinogenesis. 2012;33:240–52.
Shen M, Zhang L, Bonner MR, Liu C-S, Li G, Vermeulen R, et al. Association between mitochondrial DNA copy number, blood cell counts, and occupational benzene exposure. Environ Mol Mutagen. 2008;49:453–7.
Patel AB, Shaikh S, Jain KR, Desai C, Madamwar D. Polycyclic aromatic hydrocarbons: sources, toxicity, and remediation approaches. Frontiers in Microbiology [Internet]. 2020 [cited 2022 Jan 25];11. Available from: https://www.frontiersin.org/article/https://doi.org/10.3389/fmicb.2020.562813
Backer JM, Weinstein IB. Mitochondrial DNA is a major cellular target for a dihydrodiol-epoxide derivative of benzo[a]pyrene. Science. 198075.
Allen JA, Coombs MM. Covalent binding of polycyclic aromatic compounds to mitochondrial and nuclear DNA. Nature. 1980;287:244–5.
Cohen BH. Pharmacologic effects on mitochondrial function. Dev Disabil Res Rev. 2010;16:189–99.
Pavanello S, Dioni L, Hoxha M, Fedeli U, Mielzynska-Švach D, Baccarelli AA. Mitochondrial DNA copy number and exposure to polycyclic aromatic hydrocarbons. Cancer Epidemiol Biomarkers Prev. American Association for Cancer Research; 2013;22:1722–9.
Bhargava A, Kumari R, Khare S, Shandilya R, Gupta PK, Tiwari R, et al. Mapping the mitochondrial regulation of epigenetic modifications in association with carcinogenic and noncarcinogenic polycyclic aromatic hydrocarbon exposure. Int J Toxicol. SAGE Publications Inc; 2020;39:465–76.
Hori A, Yoshida M, Shibata T, Ling F. Reactive oxygen species regulate DNA copy number in isolated yeast mitochondria by triggering recombination-mediated replication. Nucleic Acids Res. 2009;37:749–61.
Xu Y, Lindh CH, Jönsson BAG, Broberg K, Albin M. Occupational exposure to asphalt mixture during road paving is related to increased mitochondria DNA copy number: a cross-sectional study. Environ Health. 2018;17:29.
Hou J, Yin W, Li P, Hu C, Zhang Y, Wang X, et al. Seasonal modification of the associations of exposure to polycyclic aromatic hydrocarbons or phthalates of cellular aging. Ecotoxicol Environ Saf. 2019;182:109384.
Cao X, Li J, Cheng L, Deng Y, Li Y, Yan Z, et al. The associations between prenatal exposure to polycyclic aromatic hydrocarbon metabolites, umbilical cord blood mitochondrial DNA copy number, and children’s neurobehavioral development. Environ Pollut. 2020;265:114594.
Ling X, Zhang G, Sun L, Wang Z, Zou P, Gao J, et al. Polycyclic aromatic hydrocarbons exposure decreased sperm mitochondrial DNA copy number: a cross-sectional study (MARHCS) in Chongqing. China Environmental Pollution. 2017;220:680–7.
Wong JY, Hu W, Downward GS, Seow WJ, Bassig BA, Ji B-T, et al. Personal exposure to fine particulate matter and benzo[a]pyrene from indoor air pollution and leukocyte mitochondrial DNA copy number in rural China. Carcinogenesis. 2017;38:893–9.
Du J, Pan B, Cao X, Li J, Yang J, Nie J. Urinary polycyclic aromatic hydrocarbon metabolites, peripheral blood mitochondrial DNA copy number, and neurobehavioral function in coke oven workers. Chemosphere. 2020;261:127628.
• Zhao X, Yang A, Fu Y, Zhang B, Li X, Pan B, et al. Reduction of mitochondrial DNA copy number in peripheral blood is related to polycyclic aromatic hydrocarbons exposure in coke oven workers: Bayesian kernel machine regression. Environ Pollut. 2020;260:114026. This study identifies a negative association between a PAH mixture and mtDNAcn using a novel mixture approach.
Duan X, Yang Y, Zhang H, Liu B, Wei W, Wang L, et al. Polycyclic aromatic hydrocarbon exposure, miRNA genetic variations, and associated leukocyte mitochondrial DNA copy number: a cross-sectional study in China. Chemosphere. 2020;246:125773.
Pieters N, Koppen G, Smeets K, Napierska D, Plusquin M, Prins SD, et al. Decreased mitochondrial DNA content in association with exposure to polycyclic aromatic hydrocarbons in house dust during wintertime: from a population enquiry to cell culture. PLOS ONE. Public Library of Science; 2013;8:e63208.
Fetterman JL, Sammy MJ, Ballinger SW. Mitochondrial toxicity of tobacco smoke and air pollution. Toxicology. 2017;391:18–33.
Chew S, Lampinen R, Saveleva L, Korhonen P, Mikhailov N, Grubman A, et al. Urban air particulate matter induces mitochondrial dysfunction in human olfactory mucosal cells. Part Fibre Toxicol. 2020;17:18.
Shang Y, Xue W, Kong J, Chen Y, Qiu X, An X, et al. Ultrafine black carbon caused mitochondrial oxidative stress, mitochon- drial dysfunction and mitophagy in SH-SY5Y cells. Sci Total Environ. 2022;813:151899.
Breton CV, Song AY, Xiao J, Kim S-J, Mehta HH, Wan J, et al. Effects of air pollution on mitochondrial function, mitochondrial DNA methylation, and mitochondrial peptide expression. Mitochondrion. 2019;46:22–9.
Ku T, Ji X, Zhang Y, Li G, Sang N. PM2.5, SO2 and NO2 co-exposure impairs neurobehavior and induces mitochondrial injuries in the mouse brain. Chemosphere. 2016;163:27–34.
Grevendonk L, Janssen BG, Vanpoucke C, Lefebvre W, Hoxha M, Bollati V, et al. Mitochondrial oxidative DNA damage and exposure to particulate air pollution in mother-newborn pairs. Environ Health. 2016;15:10.
Lin W, Zhu T, Xue T, Peng W, Brunekreef B, Gehring U, et al. Association between changes in exposure to air pollution and biomarkers of oxidative stress in children before and during the Beijing Olympics. Am J Epidemiol. 2015;181:575–83.
Hou L, Zhang X, Dioni L, Barretta F, Dou C, Zheng Y, et al. Inhalable particulate matter and mitochondrial DNA copy number in highly exposed individuals in Beijing, China: a repeated-measure study. Part Fibre Toxicol. 2013;10:17.
Pieters N, Janssen BG, Dewitte H, Cox B, Cuypers A, Lefebvre W, et al. Biomolecular markers within the core axis of aging and particulate air pollution exposure in the elderly: a cross-sectional study. Environ Health Perspect. 2016;124:943–50.
Bai Y, Casas L, Scheers H, Janssen BG, Nemery B, Nawrot TS. Mitochondrial DNA content in blood and carbon load in airway macrophages. A panel study in elderly subjects. Environ Int. 2018;119:47–53.
Clemente DBP, Casas M, Vilahur N, Begiristain H, Bustamante M, Carsin A-E, et al. Prenatal ambient air pollution, placental mitochondrial DNA content, and birth weight in the INMA (Spain) and ENVIRONAGE (Belgium) birth cohorts. Environ Health Perspect. 2016;124:659–65.
Peng C, Cayir A, Sanchez-Guerra M, Di Q, Wilson A, Zhong J, et al. Associations of annual ambient fine particulate matter mass and components with mitochondrial DNA abundance. Epidemiology. 2017;28:763–70.
Rosa MJ, Just AC, Guerra MS, Kloog I, Hsu H-HL, Brennan KJ, et al. Identifying sensitive windows for prenatal particulate air pollution exposure and mitochondrial DNA content in cord blood. Environ Int. 2017;98:198–203.
Brunst KJ, Sanchez-Guerra M, Chiu Y-HM, Wilson A, Coull BA, Kloog I, et al. Prenatal particulate matter exposure and mitochondrial dysfunction at the maternal-fetal interface: effect modification by maternal lifetime trauma and child sex. Environ Int. 2018;112:49–58.
Hou L, Zhu Z-Z, Zhang X, Nordio F, Bonzini M, Schwartz J, et al. Airborne particulate matter and mitochondrial damage: a cross-sectional study. Environ Health. 2010;9:48.
Byun H-M, Panni T, Motta V, Hou L, Nordio F, Apostoli P, et al. Effects of airborne pollutants on mitochondrial DNA methylation. Part Fibre Toxicol. 2013;10:18.
Zhong J, Cayir A, Trevisi L, Sanchez-Guerra M, Lin X, Peng C, et al. Traffic-related air pollution, blood pressure, and adaptive response of mitochondrial abundance. Circulation. 2016;133:378–87.
Janssen BG, Byun H-M, Gyselaers W, Lefebvre W, Baccarelli AA, Nawrot TS. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: an ENVIRONAGE birth cohort study. Epigenetics. Taylor & Francis; 2015;10:536–44.
• Frye RE, Cakir J, Rose S, Delhey L, Bennuri SC, Tippett M, et al. Prenatal air pollution influences neurodevelopment and behavior in autism spectrum disorder by modulating mitochondrial physiology. Mol Psychiatry. 2021;26:1561–77. This study demonstrates that associations between prenatal particulate matter exposure and child neurodevelopment may be mediated through long-term changes in mitochondrial respiration.
Gruzieva O, Xu CJ, Breton CV, Annesi-Maesano I, Antó JM, Auffray C, et al. Epigenome-wide meta-analysis of methylation in children related to prenatal NO2 air pollution exposure. Environ Health Perspec. 2017;125:104–10.
Brunst KJ, Hsu H-HL, Zhang L, Zhang X, Carroll KN, Just A, et al. Prenatal particulate matter exposure and mitochondrial mutational load at the maternal-fetal interface: effect modification by genetic ancestry. Mitochondrion. 2022;62:102–10.
Winckelmans E, Nawrot TS, Tsamou M, Den Hond E, Baeyens W, Kleinjans J, et al. Transcriptome-wide analyses indicate mitochondrial responses to particulate air pollution exposure. Environ Health. 2017;16:87.
Belyaeva EA, Sokolova TV, Emelyanova LV, Zakharova IO. Mitochondrial electron transport chain in heavy metal-induced neurotoxicity: effects of cadmium, mercury, and copper. Sci World J. Hindawi; 2012;2012:e136063.
Prakash C, Soni M, Kumar V. Mitochondrial oxidative stress and dysfunction in arsenic neurotoxicity: a review. J Appl Toxicol. 2016;36:179–88.
Sanchez-Guerra M, Peng C, Trevisi L, Cardenas A, Wilson A, Osorio-Yáñez C, et al. Altered cord blood mitochondrial DNA content and pregnancy lead exposure in the PROGRESS cohort. Environ Int. 2019;125:437–44.
• Kupsco A, Sanchez-Guerra M, Amarasiriwardena C, Brennan KJM, Estrada-Gutierrez G, Svensson K, et al. Prenatal manganese and cord blood mitochondrial DNA copy number: effect modification by maternal anemic status. Environ Int. 2019;126:484–93. This study identifies that exposure to manganese is associated with higher mtDNAcn in mother-child pairs, but the relationship was inversed in anemic mothers.
Liu B, Song L, Zhang L, Wu M, Wang L, Cao Z, et al. Prenatal aluminum exposure is associated with increased newborn mitochondrial DNA copy number. Environ Pollut. 2019;252:330–5.
• Smith AR, Lin P-ID, Rifas-Shiman SL, Rahman ML, Gold DR, Baccarelli AA, et al. Prospective associations of early pregnancy metal mixtures with mitochondria DNA copy number and telomere length in maternal and cord blood. Environ Health Perspect. 2021;129:117007. This study examines metal exposures and mtDNAcn in both child and maternal blood while exploring nonlinearities as well as metal mixtures.
Wu M, Shu Y, Song L, Liu B, Zhang L, Wang L, et al. Prenatal exposure to thallium is associated with decreased mitochondrial DNA copy number in newborns: evidence from a birth cohort study. Environ Int. 2019;129:470–7.
Song L, Liu B, Wang L, Wu M, Zhang L, Liu Y, et al. Exposure to arsenic during pregnancy and newborn mitochondrial DNA copy number: a birth cohort study in Wuhan. China Chemosphere. 2020;243:125335.
Thomas C, Mackey MM, Diaz AA, Cox DP. Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: implications for diseases associated with iron accumulation. Redox Report Taylor & Francis. 2009;14:102–8.
Li X, Hao S, Han A, Yang Y, Fang G, Liu J, et al. Intracellular Fenton reaction based on mitochondria-targeted copper(II)–peptide complex for induced apoptosis. J Mater Chem B. The Royal Society of Chemistry; 2019;7:4008–16.
Wang Y, Fang J, Leonard SS, Krishna Rao KM. Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radical Biol Med. 2004;36:1434–43.
Monteiro C, Ferreira de Oliveira JMP, Pinho F, Bastos V, Oliveira H, Peixoto F, et al. Biochemical and transcriptional analyses of cadmium-induced mitochondrial dysfunction and oxidative stress in human osteoblasts. J Toxicol Environ Health, Part A. Taylor & Francis; 2018;81:705–17.
Branca JJV, Pacini A, Gulisano M, Taddei N, Fiorillo C, Becatti M. Cadmium-induced cytotoxicity: effects on mitochondrial electron transport chain. Front Cell Dev Biol. 2020;8:604377.
Kang Z, Qiao N, Liu G, Chen H, Tang Z, Li Y. Copper-induced apoptosis and autophagy through oxidative stress-mediated mitochondrial dysfunction in male germ cells. Toxicol In Vitro. 2019;61:104639.
Gottipolu RR, Davuljigari CB. Perinatal exposure to lead: reduction in alterations of brain mitochondrial antioxidant system with calcium supplement. Biol Trace Elem Res. 2014;162:270–7.
Mattalloni MS, Deza-Ponzio R, Albrecht PA, Fernandez-Hubeid LE, Cancela LM, Virgolini MB. Brain ethanol-metabolizing enzymes are differentially expressed in lead-exposed animals after voluntary ethanol consumption: pharmacological approaches. Neurotoxicology. 2019;75:174–85.
Jia G, Aroor AR, Martinez-Lemus LA, Sowers JR. Mitochondrial functional impairment in response to environmental toxins in the cardiorenal metabolic syndrome. Arch Toxicol. 2015;89:147–53.
Keshavarz-Bahaghighat H, Sepand MR, Ghahremani MH, Aghsami M, Sanadgol N, Omidi A, et al. Acetyl-l-carnitine attenuates arsenic-induced oxidative stress and hippocampal mitochondrial dysfunction. Biol Trace Elem Res. 2018;184:422–35.
Iranpak F, Saberzadeh J, Vessal M, Takhshid MA. Sodium valproate ameliorates aluminum-induced oxidative stress and apoptosis of PC12 cells. Iran J Basic Med Sci. 2019;22:1353–8.
Ellis JK, Athersuch TJ, Thomas LD, Teichert F, Pérez-Trujillo M, Svendsen C, et al. Metabolic profiling detects early effects of environmental and lifestyle exposure to cadmium in a human population. BMC Med. 2012;10:61.
Borchard S, Bork F, Rieder T, Eberhagen C, Popper B, Lichtmannegger J, et al. The exceptional sensitivity of brain mitochondria to copper. Toxicol In Vitro. 2018;51:11–22.
Behzadfar L, Abdollahi M, Sabzevari O, Hosseini R, Salimi A, Naserzadeh P, et al. Potentiating role of copper on spatial memory deficit induced by beta amyloid and evaluation of mitochondrial function markers in the hippocampus of rats. Metallomics. 2017;9:969–80.
Belyaeva EA, Glazunov VV, Korotkov SM. Cyclosporin A-sensitive permeability transition pore is involved in Cd2+-induced dysfunction of isolated rat liver mitochondria: doubts no more. Arch Biochem Biophys. 2002;405:252–64.
Gobe G, Crane D. Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol Lett. 2010;198:49–55.
Al-Nasser IA, Al-Nasser I. Cadmium hepatotoxicity and alterations of the mitochondrial function. J Toxicol: Clin Toxicol. Taylor & Francis; 2000;38:407–13.
Sousa CA, Soares EV. Mitochondria are the main source and one of the targets of Pb (lead)-induced oxidative stress in the yeast Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2014;98:5153–60.
Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR Jr, Lee D-H, et al. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev. 2012;33:378–455.
Larsson K, Ljung Björklund K, Palm B, Wennberg M, Kaj L, Lindh CH, et al. Exposure determinants of phthalates, parabens, bisphenol A and triclosan in Swedish mothers and their children. Environ Int. 2014;73:323–33.
Marroqui L, Tudurí E, Alonso-Magdalena P, Quesada I, Nadal Á, Santos RS dos. Mitochondria as target of endocrine-disrupting chemicals: implications for type 2 diabetes. J Endocrinol. Bioscientifica Ltd; 2018;239:R27–45.
• Zhou Z, Goodrich JM, Strakovsky RS. Mitochondrial epigenetics and environmental health: making a case for endocrine disrupting chemicals. Toxicol Sci. 2020;178:16–25. This review highlights the impacts of endocrine-disrupting chemicals on mtDNA methylation.
Zhang Q, Zhao Y, Talukder M, Han Y, Zhang C, Li X-N, et al. Di(2-ethylhexyl) phthalate induced hepatotoxicity in quail (Coturnix japonica) via modulating the mitochondrial unfolded protein response and NRF2 mediated antioxidant defense. Sci Total Environ. 2019;651:885–94.
Hornos Carneiro MF, Shin N, Karthikraj R, Barbosa F, Kannan K, Colaiácovo MP. Antioxidant CoQ10 restores fertility by rescuing bisphenol A-induced oxidative DNA damage in the Caenorhabditis elegans germline. Genetics. 2020;214:381–95.
Savchuk I, Söder O, Svechnikov K. Mono-2-ethylhexyl phthalate stimulates androgen production but suppresses mitochondrial function in mouse Leydig cells with different steroidogenic potential. Toxicol Sci. 2015;145:149–56.
Li X, Zhou L, Ni Y, Wang A, Hu M, Lin Y, et al. Nonylphenol induces pancreatic damage in rats through mitochondrial dysfunction and oxidative stress. Toxicol Res Camb. 2017;6:353–60. https://doi.org/10.1039/c6tx00450d.
Lan T, Jie Y, Jie X. The effects of environmental endocrine disruptors on myocardial mitochondrial: a review. Asian J Ecotoxicol. 2020;4:123–8.
Kaur K, Chauhan V, Gu F, Chauhan A. Bisphenol A induces oxidative stress and mitochondrial dysfunction in lymphoblasts from children with autism and unaffected siblings. Free Radical Biol Med. 2014;76:25–33.
Huffman AM, Wu H, Rosati A, Rahil T, Sites CK, Whitcomb BW, et al. Associations of urinary phthalate metabolites and lipid peroxidation with sperm mitochondrial DNA copy number and deletions. Environ Res. 2018;163:10–5.
• Wang L, Song L, Liu B, Wu M, Liu Y, Bi J, et al. Prenatal exposure to bisphenol S and altered newborn mitochondrial DNA copy number in a baby cohort study: sex-specific associations. Chemosphere. 2021;263:128019. This study identifies that bisphenol S has a sex-specific effect on mtDNAcn.
Jiang Y, Xia W, Zhu Y, Li X, Wang D, Liu J, et al. Mitochondrial dysfunction in early life resulted from perinatal bisphenol A exposure contributes to hepatic steatosis in rat offspring. Toxicol Lett. 2014;228:85–92.
Jiang Y, Xia W, Yang J, Zhu Y, Chang H, Liu J, et al. BPA-induced DNA hypermethylation of the master mitochondrial gene PGC-1α contributes to cardiomyopathy in male rats. Toxicology. 2015;329:21–31.
Azevedo LF, Hornos Carneiro MF, Dechandt CRP, Cassoli JS, Alberici LC, Barbosa F. Global liver proteomic analysis of Wistar rats chronically exposed to low-levels of bisphenol A and S. Environ Res. 2020;182:109080.
Leung MCK, Meyer JN. Mitochondria as a target of organophosphate and carbamate pesticides: revisiting common mechanisms of action with new approach methodologies. Reprod Toxicol. 2019;89:83–92.
Karami-Mohajeri S, Ahmadipour A, Rahimi H-R, Abdollahi M. Adverse effects of organophosphorus pesticides on the liver: a brief summary of four decades of research. Arch Ind Hyg Toxicol. 2017;68:261–75.
Farkhondeh T, Mehrpour O, Forouzanfar F, Roshanravan B, Samarghandian S. Oxidative stress and mitochondrial dysfunction in organophosphate pesticide-induced neurotoxicity and its amelioration: a review. Environ Sci Pollut Res. 2020;27:24799–814.
Park C-M, Kim K-T, Rhyu D-Y. Exposure to a low concentration of mixed organochlorine pesticides impairs glucose metabolism and mitochondrial function in L6 myotubes and zebrafish. J Hazard Mater. 2021;414:125437.
Liu Q, Wang Q, Xu C, Shao W, Zhang C, Liu H, et al. Organochloride pesticides impaired mitochondrial function in hepatocytes and aggravated disorders of fatty acid metabolism. Sci Rep. 2017;7:46339.
Budnik LT, Kloth S, Baur X, Preisser AM, Schwarzenbach H. Circulating mitochondrial DNA as biomarker linking environmental chemical exposure to early preclinical lesions elevation of mtDNA in human serum after exposure to carcinogenic halo-alkane-based pesticides. PLOS ONE. Public Library of Science; 2013;8:e64413.
Chen X, Zhou Y, Hu C, Xia W, Xu S, Cai Z, et al. Prenatal exposure to benzotriazoles and benzothiazoles and cord blood mitochondrial DNA copy number: a prospective investigation. Environ Int. 2020;143:105920.
Mohammadi-Bardbori A, Ghazi-Khansari M. Alternative electron acceptors: proposed mechanism of paraquat mitochondrial toxicity. Environ Toxicol Pharmacol. 2008;26:1–5.
Yuan B, Liang S, Jin Y-X, Zhang M-J, Zhang J-B, Kim N-H. Toxic effects of atrazine on porcine oocytes and possible mechanisms of action. PLOS ONE. Public Library of Science; 2017;12:e0179861.
Drechsel DA, Patel M. Chapter 21 Paraquat‐induced production of reactive oxygen species in brain mitochondria. Methods in enzymology [Internet]. Academic Press; 2009 [cited 2021 Oct 13]. p. 381–93. Available from: https://www.sciencedirect.com/science/article/pii/S0076687908044212
Tatjana V, Domitille S, Jean-Charles S. Paraquat-induced cholesterol biosynthesis proteins dysregulation in human brain microvascular endothelial cells. Sci Rep. 2021;11:18137.
Zhou R, Liu R, Li W, Wang Y, Wan X, Song N, et al. The use of different sublethal endpoints to monitor atrazine toxicity in nematode Caenorhabditis elegans. Chemosphere. 2021;274:129845.
Wang Q, Yang L, Hua Y, Nair S, Xu X, Ren J. AMP-activated protein kinase deficiency rescues paraquat-induced cardiac contractile dysfunction through an autophagy-dependent mechanism. Toxicol Sci. 2014;142:6–20.
Foley S, Crowley C, Smaihi M, Bonfils C, Erlanger BF, Seta P, et al. Cellular localisation of a water-soluble fullerene derivative. Biochem Biophys Res Commun. 2002;294:116–9.
Maurer LL, Meyer JN. A systematic review of evidence for silver nanoparticle-induced mitochondrial toxicity. Environ Sci: Nano. The Royal Society of Chemistry; 2016;3:311–22.
• Wu D, Ma Y, Cao Y, Zhang T. Mitochondrial toxicity of nanomaterials. Sci Total Environ. 2020;702:134994. This review highlights the various mechanisms in which different nanomaterials induce mitochondrial dysfunction.
Teodoro JS, Simões AM, Duarte FV, Rolo AP, Murdoch RC, Hussain SM, et al. Assessment of the toxicity of silver nanoparticles in vitro: a mitochondrial perspective. Toxicol In Vitro. 2011;25:664–70.
Yuan X, Zhang X, Sun L, Wei Y, Wei X. Cellular toxicity and immunological effects of carbon-based nanomaterials. Part Fibre Toxicol. 2019;16:18.
Xu Y-M, Tan HW, Zheng W, Liang Z-L, Yu F-Y, Wu D-D, et al. Cadmium telluride quantum dot-exposed human bronchial epithelial cells: a further study of the cellular response by proteomics. Toxicol Res (Camb). 2019;8:994–1001.
Nguyen KC, Rippstein P, Tayabali AF, Willmore WG. Mitochondrial toxicity of cadmium telluride quantum dot nanoparticles in mammalian hepatocytes. Toxicol Sci. 2015;146:31–42.
Jaworski S, Strojny B, Sawosz E, Wierzbicki M, Grodzik M, Kutwin M, et al. Degradation of mitochondria and oxidative stress as the main mechanism of toxicity of pristine graphene on U87 glioblastoma cells and tumors and HS-5 cells. Int J Mol Sci. 2019;20:650.
Jayaram DT, Runa S, Kemp ML, Payne CK. Nanoparticle-induced oxidation of corona proteins initiates an oxidative stress response in cells. Nanoscale. The Royal Society of Chemistry; 2017;9:7595–601.
Xue Y, Chen Q, Sun J. Hydroxyapatite nanoparticle-induced mitochondrial energy metabolism impairment in liver cells: in vitro and in vivo studies. J Appl Toxicol. 2017;37:1004–16.
Maher BA, González-Maciel A, Reynoso-Robles R, Torres-Jardón R, Calderón-Garcidueñas L. Iron-rich air pollution nanoparticles: an unrecognised environmental risk factor for myocardial mitochondrial dysfunction and cardiac oxidative stress. Environ Res. 2020;188:109816.
Ren C, Hu X, Li X, Zhou Q. Ultra-trace graphene oxide in a water environment triggers Parkinson’s disease-like symptoms and metabolic disturbance in zebrafish larvae. Biomaterials. 2016;93:83–94.
Rottenberg H, Hoek JB. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell. 2017;16:943–55.
Ly LD, Xu S, Choi S-K, Ha C-M, Thoudam T, Cha S-K, et al. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp Mol Med. 2017;49:e291–e291.
Bertero E, Maack C. Calcium signaling and reactive oxygen species in mitochondria. Cir Res. American Heart Association; 2018;122:1460–78.
Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909–50.
Angelova PR, Abramov AY. Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. 2018;592:692–702.
Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med. 2019;51:1–13.
Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry. 2012;17:389–401.
Sharma C, Kim S, Nam Y, Jung UJ, Kim SR. Mitochondrial dysfunction as a driver of cognitive impairment in Alzheimer’s disease. Int J Mol Sci. 2021;22:4850.
de Mello AH, Costa AB, Engel JDG, Rezin GT. Mitochondrial dysfunction in obesity. Life Sci. 2018;192:26–32.
Roshanravan B, Kestenbaum B, Gamboa J, Jubrias SA, Ayers E, Curtin L, et al. CKD and muscle mitochondrial energetics. Am J Kidney Dis. 2016;68:658–9.
Kohlhaas M, Nickel AG, Maack C. Mitochondrial energetics and calcium coupling in the heart. J Physiol. 2017;595:3753–63.
Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW, Kitsis RN, et al. Mitochondrial function, biology, and role in disease. Cir Res. American Heart Association; 2016;118:1960–91.
Visioli F, Rodríguez-Pérez M, Gómez-Torres Ó, Pintado-Losa C, Burgos-Ramos E. Hydroxytyrosol improves mitochondrial energetics of a cellular model of Alzheimer’s disease. Nutr Neurosci. Taylor & Francis; 2020;0:1–11.
Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000;23:298–304.
Requejo-Aguilar R, Bolaños JP. Mitochondrial control of cell bioenergetics in Parkinson’s disease. Free Radical Biol Med. 2016;100:123–37.
Sunny NE, Bril F, Cusi K. Mitochondrial adaptation in nonalcoholic fatty liver disease: novel mechanisms and treatment strategies. Trends Endocrinol Metab. 2017;28:250–60.
Kanaan GN, Patten DA, Redpath CJ, Harper M-E. Atrial fibrillation is associated with impaired atrial mitochondrial energetics and supercomplex formation in adults with type 2 diabetes. Can J Diabetes. 2019;43:67-75.e1.
Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018;592:728–42.
Buneeva O, Fedchenko V, Kopylov A, Medvedev A. Mitochondrial dysfunction in Parkinson’s disease: focus on mitochondrial DNA. Biomedicines. Multidisciplinary Digital Publishing Institute; 2020;8:591.
Fetterman JL, Holbrook M, Westbrook DG, Brown JA, Feeley KP, Bretón-Romero R, et al. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovasc Diabetol. 2016;15:53.
Hu H, Lin Y, Xu X, Lin S, Chen X, Wang S. The alterations of mitochondrial DNA in coronary heart disease. Exp Mol Pathol. 2020;114:104412.
Pirola CJ, Garaycoechea M, Flichman D, Castaño GO, Sookoian S. Liver mitochondrial DNA damage and genetic variability of cytochrome b – a key component of the respirasome – drive the severity of fatty liver disease. J Intern Med. 2021;289:84–96.
Boyapati RK, Tamborska A, Dorward DA, Ho G-T. Advances in the understanding of mitochondrial DNA as a pathogenic factor in inflammatory diseases. F1000Res. 2017;6:169.
Huang Y, Chi J, Wei F, Zhou Y, Cao Y, Wang Y. Mitochondrial DNA: a new predictor of diabetic kidney disease. Int J Endocrinol. Hindawi; 2020;2020:e3650937.
Govers LP, Toka HR, Hariri A, Walsh SB, Bockenhauer D. Mitochondrial DNA mutations in renal disease: an overview. Pediatr Nephrol. 2021;36:9–17.
Włodarczyk M, Nowicka G. Obesity, DNA damage, and development of obesity-related diseases. Int J Mol Sci. 2019;20:1146.
Cameron RB, Beeson CC, Schnellmann RG. Development of therapeutics that induce mitochondrial biogenesis for the treatment of acute and chronic degenerative diseases. J Med Chem American Chemical Society. 2016;59:10411–34.
Singh A, Faccenda D, Campanella M. Pharmacological advances in mitochondrial therapy. EBioMedicine. 2021;65:103244.
Nie S, Lu J, Wang L, Gao M. Pro-inflammatory role of cell-free mitochondrial DNA in cardiovascular diseases. IUBMB Life. 2020;72:1879–90.
El-Hafidi M, Correa F, Zazueta C. Mitochondrial dysfunction in metabolic and cardiovascular diseases associated with cardiolipin remodeling. Biochimica et Biophysica Acta (BBA)-Mol Basis Dis. 2020;1866:165744.
Acknowledgements
Figures 1 and 2 were adapted from “Mitochondria (2D, outer membrane)” and “Mitochondria,” respectively, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates
Funding
Research support was provided by a National Institutes of Environmental Health Sciences grant (R00ES030749) to A.K.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Environmental Epigenetics.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Reddam, A., McLarnan, S. & Kupsco, A. Environmental Chemical Exposures and Mitochondrial Dysfunction: a Review of Recent Literature. Curr Envir Health Rpt 9, 631–649 (2022). https://doi.org/10.1007/s40572-022-00371-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-022-00371-7