
Abstract
Ba0.5Sr0.5Co0.8Fe0.2O3–δ (BSCF) powders were prepared by solution combustion synthesis using single and double fuels. The effect of the fuel mixture on the main properties of this well-known solid oxide fuel cell cathode material with high oxygen ion and electronic conduction was investigated in detail. Results showed that the fuel mixture significantly affected the area-specific resistance of the BSCF cathode materials, by controlling the oxygen deficiency and stabilizing the Co2+ oxidation state. It was demonstrated that high fuel-to-metal cations molar ratios and high reducing power of the combustion fuel mixture are mainly responsible for the decreasing of the area-specific resistance of BSCF cathode materials. Moreover, a new metastable monoclinic phase with Ba0.5Sr0.5CO3 composition was discovered in the as-burned BSCF powders, enlarging the existing information on the BSCF phase formation mechanism.
Similar content being viewed by others
Introduction
The increasing worldwide demand of efficient, low-cost, and eco-friendly energy technologies has been leading in the last years toward an intense research on intermediate temperature (450–700 °C) solid oxide fuel cell (IT-SOFC) devices [1]. In particular, cathode materials to be used in IT-SOFCs should meet some essential requirements, like high oxygen and electronic transport properties (easy oxygen reduction reaction, surface exchange, and diffusion) at intermediate temperatures and high compatibility with the electrolyte material [1, 2]. To meet these requirements, two main strategies have been adopted in the literature: looking for new chemical compositions [3] and developing new preparation routes [4] (or improving the existing preparation and processing procedures [5]) to increase the active area where the cathode reaction can take place.
Ba1–xSr x Co1–yFe y O3–δ (BSCF) perovskite-type compounds, already known as oxygen conducting membrane materials [6], have attracted the attention of the scientific community as IT-SOFC cathode materials owing to their very good oxygen conducting properties at intermediate temperatures [7–10]. Nevertheless, their chemical instability [6, 11, 12] and reactivity to CO2 [13] in the intermediate temperature range have so far limited their industrial applicability. Despite the described limits, BSCF is still a very interesting material to investigate, since the information obtained about the factors governing its high conductivity may help the selection of more stable cathode materials [9, 10, 14–16]. BSCF has been usually prepared by solid-state reaction synthesis [9, 10, 17–19] or by EDTA–citric acid complexing method [5–7, 18, 20, 21], although few other preparation routes have been described as well in the literature, for example the co-precipitation method [22] and the solution combustion synthesis (SCS) [17, 18]. SCS is based on both the concepts of sol–gel and propellant chemistry [23, 24] and it has been successfully used in the literature to obtain powders at lower temperatures, with smaller crystal size and larger surface area or with metastable structures, compared to other preparation methods [23, 25, 26]. The properties of the materials prepared by SCS are essentially governed by the fuel type and by the reducer/oxidizer molar ratio, the so called “Φ” ratio [24, 26, 27]. The effect of the combustion fuel on the properties of heterometallic oxides, and especially of SOFC materials prepared by SCS, has been investigated recently, for both single fuels and fuel mixtures [28–30]. In particular, it has been found that the fuel type affects the microstructural properties of the powders [29–31], the phase formation mechanism [28] and the electrical conducting properties [29, 31, 32] of the final material. Nevertheless, only very general explanations about the effect of the combustion fuel have been so far proposed. Moreover, the use of a mixture of fuels has been rarely reported for the preparation of such materials.
In this paper, fuels such as sucrose, cellulose, citric acid and polyethylene glycol (PEG) were used, for the first time alone or in combination, for the preparation of BSCF powders of Ba0.5Sr0.5Co0.8Fe0.2O3–δ composition. Moreover, the effect of the fuel mixture composition on the phase formation process, structure, microstructure and conductivity of BSCF prepared by SCS was investigated in detail, going a step further toward the control of the IT-SOFC materials properties.
If the use of citric acid is already well known in the literature [25] the cellulose templating method [33, 34] and sucrose method [35, 36] are less common and they have been not yet used for the preparation of BSCF powders, to the best of our knowledge. Moreover, the experimental conditions used in this work for the synthesis from cellulose and sucrose-containing fuel mixtures were quite different from the literature and need to be commented. Sucrose is usually accompanied by nitric acid at very acid pH, in order to form saccharic acid, which is able to polymerize alone or in the presence of PVA [37, 38]. In this work, sucrose was used without any nitric acid, alone or in combination with PEG, and the resultant pH was around 5.
Experimental procedure
Powder preparation
The same general procedure was used for the preparation of BSCF powders of Ba0.5Sr0.5Co0.8Fe0.2O3–δ composition, irrespective of the fuel used. Ba(NO3)2 (99.1 %, VWR Prolabo), Sr(NO3)2 (≥99 %, Merck), Co(NO3)2·6H2O (≥99 %, Fluka), and Fe(NO3)3·9H2O (≥98 %, Carlo Erba) were carefully weighted in the required proportions to obtain 3 g of final product (unless a different amount was specified), poured in a 1.2-L stainless steel beaker and dissolved in distilled water at 60 °C under magnetic stirring. Ammonium nitrate and the fuel were then added to the metal precursor solution as oxidant additive and propellant–template–complexant, respectively. Citric acid (anhydrous, Fluka, 99.9 %), cellulose (powder DS-O for thin layer chromatography, Fluka Analytical), sucrose (Eridania), polyethylene glycol (PEG, MW 20000, Fluka Chemika) were used as fuels, alone or in combination. For one of the samples, ammonia solution (NH4OH 30 vol %) was added to the citric acid fuel as a pH regulator.
Taking into account only the primary fuel, the fuel/metal cation molar ratio was set to 2, unless otherwise specified, while the reducers/oxidizers ratio Φ was set to the stoichiometric value for the specific combustion reaction, following the method described by Jain et al. [24]. The water was left to evaporate at 80 °C (a vertex was inserted in the precursor solution) under constant magnetic stirring until the formation of a homogeneous gel. Initiation of the combustion reaction was performed by putting the stainless steel beaker on a hotplate and setting the temperature to around 250 °C.
The as-burnt powders were fired at higher temperatures (600–950 °C) for the crystallization of the perovskite-type phase and to eliminate any carbon residues derived from the combustion process.
Temperature/time profiles of the combustion process
A K-type thermocouple 1.5 mm in diameter, which covered a temperature range from ambient temperature up to 1,100 °C, was used to measure the temperature variations during the combustion reaction. Concurrently, the signals from the thermocouple were stored and processed using a data acquisition system: a data logger (PICO technology) with a sampling velocity of 20 bit/s and a computer with Picolog software. The thermocouple was carefully inserted into the combustion medium and the temperature was registered from the time the beaker was put into contact with the hotplate until the end of the combustion process. The duration of the process was calculated from the full width half maximum (FWHM) of the peak profile.
Powder characterization
X-ray diffraction analysis
The same general X-ray diffraction (XRD) measurements were carried out on a SIEMENS D5000 X-ray powder diffractometer equipped with a Kristalloflex 760 X-ray generator and with a curved graphite monochromator that made possible the selection of the Cu Kα radiation (40 kV/30 mA). The 2θ step size was 0.03° for the as-burned powders and the powders fired at 600 °C, whereas a 0.02° step was used for the powders fired at higher temperatures. The integration time was 3 s per step, and the 2θ scan range was from 18° to 90°. The diffraction patterns were analyzed by Rietveld refinement using the GSAS package [37] Chebyschev polynomials and Pearson VII functions were chosen for the background and for the peak profile fitting, respectively. In the structure refinement, lattice constants, zero offset, scale factors, FWHM, Debye–Waller factors and microstrain were considered as variable parameters. In those cases where the XRD patterns were complex and with overlapping peaks, Debye–Waller factors and microstrain were set to a default value (0.025 and 0, respectively). From fitting results, the structural parameters of the investigated compounds and, in particular, the cell edge lengths and the relative phase composition were obtained. When possible, an estimation of the crystal size values was obtained from Scherrer equation, in agreement with the GSAS package procedure. The agreement factors (“R values”) [38] were generally acceptable and they were reported along with the graphical Rietveld fits showed in the figures. A standard deviation (SD) of ±0.003 Å for the refined cell parameters was estimated in the experimental conditions used in this work.
Simultaneous thermal analysis
Thermo-gravimetric and differential thermal analysis (TG–DTA) measurements were performed using a Perkin Elmer STA 6000 equipment, in the 50–1,000 °C temperature range (heating rate = 10 °C/min) in static air or in nitrogen flow (ca. 20 ml/min), on both as-burned and fired powders.
Nitrogen adsorption
Specific surface area (BET method) [39] and mesopore size distributions (BJH method) [40] were evaluated performing nitrogen adsorption/desorption measurements at −196 °C, using a Sorptomatic 1900 Carlo Erba equipment. All the samples were pre-treated under vacuum at 250 °C for 2 h prior to the measurements.
Temperature-programmed reaction techniques
Reduction properties of the perovskite oxides were studied by temperature-programmed reduction (TPR) measurements. Experiments were carried out with a Micromeritics Autochem 2910 apparatus, equipped with a thermal conductivity detector (TCD) and a mass quadrupole spectrometer (QMS) (Thermostar™, Balzers). Hydrogen consumption (mmol/g) was calculated through TCD calibration experiments. Before TPR, the samples (0.17 g) were pre-treated in a quartz reactor in O2 (5 % in He) at 500 °C for 30 min, followed by purging and cooling to room temperature in Ar flow. Then, the flow was switched to the H2 mixture (5 % in Ar, 50 mL/min) and the temperature was increased with a heating rate of 10 °C/min up to 950 °C. The same instrument was used to study the reactivity towards oxygen of the perovskite oxides, performing temperature-programmed oxidation (TPO) measurements. The fired samples (0.17 g), without any pre-treatment, were flowed under O2 (5 % in He, 50 mL/min) by increasing linearly the temperature from 25 to 950 °C. The temperature was kept at 950 °C for 2 h under oxygen atmosphere, and then the samples were cooled down to room temperature. Finally, to study the oxygen desorption properties of the perovskite oxides, He-TPD experiments were performed directly on the calcined samples (0.17 g) by flowing He (50 mL/min) from 25 to 950 °C. The temperature was kept at 950 °C for 2 h under He stream, and then cooling down under the same atmosphere. The evolution of gaseous species was monitored by on-line TCD and QMS.
SEM-EDX
Typical micrographs and energy-dispersive X-ray (EDX) analysis of the powder samples and fractured cross section of the co-sintered electrode/electrolyte interfaces were acquired using a FEI Quanta 200 SEM microscope operating in high vacuum at 20 keV, after deposition by sputtering of a thin gold layer.
Electrochemical characterization of symmetric cells
Dense electrolyte pellets, 13 mm in diameter, were obtained by uniaxially pressing Sm0.2Ce0.8O2 (SDC, AGC Seimi Chemical Co., Ltd., Japan) powders at 250 MPa and sintering at 1,500 °C for 5 h. Cathode inks were prepared mixing the BSCF powders with an organic vehicle (α-terpineol as a solvent, di-n-butyl phthalate as a plasticizer, and polyvinylbutyral as a binder) until an appropriate viscosity for brushing was achieved. Symmetric cells were prepared by brush painting the cathode slurry to each side of the SDC pellet, drying at 150 °C for 3 h, and firing at 1,000 °C for 2 h.
Electrochemical impedance spectroscopy (EIS) measurements were performed using a multichannel potentiostat (model VMP3, Biologic Science Instruments) in the frequency range between 100 mHz and 500 kHz, with an AC voltage amplitude ranging between 10 and 50 mV, at temperatures ranging between 500 and 800 °C. Silver mesh current collectors and four silver lead wires were pressed against the samples in a quartz reactor by the use of a ceramic screw-and-bolt assembly. The quartz reactor was placed into a temperature-controlled furnace, and dry air was fed through the reactor at a rate of 50 cm3 min−1. For the measurements at different oxygen partial pressures, the atmosphere in the quartz reactor was changed to different pO2 values using various O2–Ar and air–Ar gas mixtures, and monitoring the pO2 of the outlet gas with a zirconia oxygen sensor.
In the acquired complex impedance plane plots, the intercept with the real axis at high frequencies was considered as the Ohmic resistance (ROhmic) composed of the electrolyte resistance, possibly electrode-sheet resistance, and lead-contact resistance. The difference between the high frequency and low-frequency intercepts with the real axis, in some cases composed of the sum of two contributions, was considered as the electrode polarization resistance (Rp). The different contributions in the impedance plots were calculated by fitting the experimental data with a Zview software. An equivalent circuits made of LR0(R1Q1)(R2Q2) was used to fit the complex impedance plane plots. In the selected equivalent circuits, L represents the set-up inductance, R0 is the total ohmic resistance, R1 and R2 are the resistance values due to the electrode processes, whereas Q1 and Q2 are the constant phase elements associated with R1 and R2, respectively.
The area-specific resistance (ASR) of the electrode was calculated from the electrode resistance (Rp) by
where A is the geometrical electrode area and the factor \( \frac{1}{2} \) takes into account that symmetrical cells were used.
Results and discussion
Combustion process and formation mechanism of BSCF cathode materials
BSCF powders were prepared by SCS using two single fuels and four fuel mixtures. Fuels such as sucrose, cellulose, citric acid, ammonia and polyethylene glycol were used alone or in combination. Table 1 describes the fuel mixture composition for all the samples studied in this work.
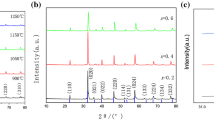
BSCF perovskite-type compounds with cubic structure were obtained from the A1–A6 fuel mixtures by using a solution combustion synthesis followed by a thermal treatment in static air. Figure 1 shows the XRD patterns before (as-burned powders) and after heating to 600, 750 and 950 °C for 5 h of a representative sample, A4, prepared from sucrose and PEG. The XRD patterns of the as-burned samples, regardless of the fuel (single fuel or mixture), showed the formation of two types of Ba–Sr carbonates, one being the already known orthorhombic aragonite-type carbonate (ar-Ba0.5Sr0.5CO3), the other a new metastable δ phase with monoclinic structure (δ-Ba0.5Sr0.5CO3). After heating to 600 °C, δ-Ba0.5Sr0.5CO3 was totally converted into ar-Ba0.5Sr0.5CO3 (Fig. 1). A Fe–Co spinel was also detected in all the powders heated to 600 °C (Fig. 1). After heating to 750 °C, no more carbonates were present in the XRD patterns and two intermediate phases were formed, P1 hexagonal and P′ cubic, of Ba1–xSr x Co1–yFe y O3–δ composition, with x (and y) content different from 0.5 (and 0.2) (Fig. 1). Such two phases (P1 and P′) merged into a cubic P phase after thermal treatment at 950 °C. The heating temperature for the formation of a pure BSCF phase reported by other authors in the literature [17, 18, 41] was 50–100 °C lower than the value obtained in this work, though using slightly different experimental conditions.

Evolution of the phase composition with the temperature for a representative BSCF sample (A4). The main peaks of stable aragonite-type Ba0.5Sr0.5CO3 and metastable δ-type Ba0.5Sr0.5CO3 phases are indicated by ar and δ, respectively; the desired BSCF cubic phase is indicated by a P, while the other intermediate BSCF hexagonal and cubic phases are indicated by P1 and P′, respectively. Peaks belonging to the Fe–Co spinel-type phase are indicated by sp
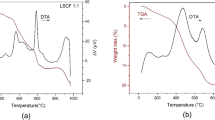
TG–DTA measurements were performed under static air on both as-burned and calcined BSCF powder samples. The TG–DTA of a representative as-burned sample (A3) is reported in Fig. 2a. Figure 2b shows TG–DTA profiles of the same sample after calcination at 950 °C/5 h.

TG and DTA curves under static air for a representative as-burned sample (a); TG curve under static air for the A3 fired sample (b)
In the as-burned sample (Fig. 2a), after the initial water loss, a sharp step ascribable to the residual carbon decomposition was visible at around 400 °C. At around 850 °C a substantial weight loss occurred, which was related to the formation of the final BSCF cubic phase from ar-Ba0.5Sr0.5CO3 and Fe-Co spinel. The BSCF formation was completed at 900 °C. Above that temperature, the weight continued to decrease. The absence of a definite plateau in the TGA curves hindered a proper determination of the exact formation temperature and it was ascribed to the peculiar property of the BSCF powders to lose oxygen even under oxidative environment [19, 42]. According to this hypothesis, an evident weight loss was observed in the TGA of A3 after calcination at 950 °C/5 h (Fig. 2b). Such weight loss was around 1.5 wt % in the 200–900 °C temperature range, in agreement with the literature [21]. Following the considerations of Li et al. [43], three different regions were identified in the TGA of A3 sample (Fig. 2b). The oxidation of Co3+ and Fe3+ to the IV state took place between 200 and 350 °C with a slight weight gain. Above 350 °C the reduction of Co4+ and Fe4+ to the (III) state was more favored. Then, the temperature-driven reduction of Co3+ to Co2+ occurred above 800 °C. These redox changes were accompanied by oxygen content variations for charge compensation.
The evolution of the BSCF phase composition with the heating temperature in the 600–950 °C range showed in Fig. 1 was in agreement with the literature [20], whereas the phase composition of the as-burned-powders needs to be commented, since it enlarges the actual picture on the BSCF phase formation mechanism.
Figure 3 shows the XRD pattern and Rietveld refinement for a representative as-burned sample, A2, which showed the largest percentage of the δ-Ba0.5Sr0.5CO3. Table 2 provides the phase composition of such sample together with the structural information about the new metastable phase. A considerable amount of metallic cobalt was also found in the as-burned powders (Table 2). The formation of metallic Co is not surprising since it is known to form, eventually with CoO, in the as-burned powders prepared by SCS, depending on the flame temperature and on the nature of the combustion process [44]. It can be noticed that Fe was not mentioned in Table 2, although it is probably still hidden as amorphous phase, since it appeared as Co–Fe spinel after heating to 600 °C.

Graphical results of the Rietveld refinement for a typical as-burned BSCF sample (A2), where the new metastable δ-Ba0.5Sr0.5CO3 monoclinic phase represents the main phase; the two main peaks belonging to metallic cobalt are indicated by Co°. Rietveld reliability factors are as well reported in the figure
The formation of the new metastable δ-Ba0.5Sr0.5CO3 phase deserves a more detailed discussion. This is very similar to the δ-BaCO3 found in the literature [45, 46], although it has a much smaller lattice volume (145 Å3) with respect to the δ-BaCO3 phases previously described in the literature (155 and 158 Å3) (Table 2), in agreement with the smaller ionic radius of Sr2+ with respect to that of Ba2+. The XRD pattern of the new δ-Ba0.5Sr0.5CO3 phase identified in this work resembles the “OP1” Ba–Sr carbonate phase observed by Yi et al. [47] on the surface layer of a BSCF membrane at the beginning of the thermal treatment in CO2 [45], although they indexed it with an orthorhombic Pmma group. In our work, any attempt made to fit the δ-Ba0.5Sr0.5CO3 phase according to an orthorhombic symmetry with the cell parameters suggested by Yi et al. was unsuccessful. Like its δ-BaCO3 analogue, which is known to be stabilized by the incorporation of SO4− anions [46] or by an abrupt temperature change [45], it is likely that the new metastable δ-Ba0.5Sr0.5CO3 phase was formed at the beginning of the combustion process due to the fast combustion. Figure 4 shows the temperature/time profiles of sample A2, prepared by citric acid-assisted SCS, for 1, 1.5 and 3 g of sample produced. All the fuel mixtures used in this work displayed a single combustion peak similar to those of A2, with a maximum temperature at around 600–700 °C, where the combustion involved the whole mass (visual observation) in few seconds. The temperature/time profiles were reproducible for the same experimental conditions used in the synthesis, including the type of combustion beaker used (geometry and material) and the final mass of the produced samples. Both the duration and the maximum temperature of the combustion increased with the BSCF powder mass produced in the synthesis. Moreover, the amount of δ-Ba0.5Sr0.5CO3 phase, determined by Rietveld analysis of the XRD data, decreased, whereas the ar-Ba0.5Sr0.5CO3 phase and metallic cobalt amounts proportionally increased by increasing the sample mass of the same as-burned sample (table inset of Fig. 4). A larger duration of the combustion process favors the conversion of the metastable δ-Ba0.5Sr0.5CO3 into the more stable ar-Ba0.5Sr0.5CO3.

Temperature–time profiles for the A2 sample, for increasing sample mass of powder prepared in the same batch; the included table shows the corresponding variation of the ar-Ba0.5Sr0.5CO3, δ-Ba0.5Sr0.5CO3, and Co° wt. % with the combustion process duration and combustion profile maximum temperature
It should be emphasized that, although the phase composition of the as-burned samples was dependent on the sample weight, such dependence was not observed after heating to 600 °C/5 h, where only the ar-Ba0.5Sr0.5CO3 and Fe–Co spinel were present, irrespective of the amount of powder produced in one batch, in agreement with the existing literature [20].
Structural parameters and B-site cation stability of BSCF cathode materials
Figure 5 shows the Rietveld refinement of a representative sample, A4, after firing at 950 °C/5 h. A closer look by Rietveld refinement of the XRD patterns of the powders heated to 950 °C revealed the presence of a mixture of BSCF phases with the same cubic symmetry and minor amounts (below 5 wt %) of a rhombohedral phase (a = b = 5.67 Å, c = 35.2 Å). This phase can be well described with a Ba3Co10O17-like structure oriented parallel to the (111)cubic plane, as already observed by Efimov et al. [48], who stated that the probable composition of this phase is Ba1–xSr x Co2–yFe y O5–δ. The mixture of cubic phases was observed in all the samples, although some clear differences were present in terms of cell parameter and/or respective weight percentage. The two phases have different cell parameters and oxygen deficiency. The BSCF cubic phase with the smaller cell parameter (phase 1) was the typical BSCF cubic phase (3.986 Å for A4) in agreement with the literature [49]. The other cubic phase (phase 2) had a larger lattice parameter value (3.994 Å for A4), demonstrating a larger oxygen deficiency due to the increased formation of Co2+ ions with larger ionic radius. The formation of an oxygen-deficient BSCF with cubic structure can be explained by the fact that BSCF, with respect to other perovskite-type oxides, is able to keep very high oxygen deficiency without forming the ordered brownmillerite structure [50]. From the refined cell parameters and phase composition, the total percentage of Co2+ was calculated, taking into account the relationship found by Kriegel et al. [19] between the BSCF cell parameter and the oxygen deficiency, hypothesizing that BSCF contains more Co3+ and/or Co2+ than Co4+ and more Fe3+ than Fe4+ [51]. It was found that Co2+ changed with the type of fuel mixture, ranging between 5 wt % (A2) and 32 wt% (A6). Figure 6 compares temperature-programmed experiments under reductive (TPR) and inert (He-TPD) environments for the A2 and A6 samples, performed by using the same weight amounts. For both the investigated samples, the shape of TPR curves was very similar: a main reduction peak was evident at around 500 °C, together with another low-intensity peak at around 250 °C (Fig. 6a). The main peak was ascribed to the Co3+, Co2+ step reduction to Co0, in agreement with the hydrogen consumption (5.3–5.5 mmol H2/g), while the low temperature peak (2.3 mmol H2/g) was likely related to both the reduction of Fe4+ to Fe3+ and of Co4+ to Co3+ [52]. The lower hydrogen consumption of this peak indicates that both samples contained more Co3+/Co2+ than Co4+, in agreement with the above discussion. Two additional peaks with very low intensity and total hydrogen consumption of 1.4 mmol H2/g were also visible above 700 °C, which could be ascribed to the subsequent reduction of Fe3+ and Fe2+ species into the metallic state [53]. It is important to note that the maximum temperature of the first two peaks, below 600 °C, is distinctly higher for the A6 sample than for the A2 sample (Fig. 6a). A different reduction temperature suggests that the two samples probably contain cobalt ions with a different surrounding and a different oxidation state (Co3+/Co2+). This is supported by the slightly lower hydrogen consumption registered for the main peak of the A6 sample (5.3 mmol H2/g) with respect to the A2 sample (5.5 mmol H2/g). The higher Co2+ content calculated from XRD results for A6 is in agreement with the higher content of Co2+ suggested by TPR results for the same sample.

Graphical results of the Rietveld refinement of the XRD patterns of the A4 sample heated to 950 °C for 5 h. (h k l) planes are also indicated for the BSCF cubic phase. Rietveld reliability factors are reported in the figure as well. The inset is a magnification of the (110) XRD peak at around 32°2θ, which evidences the contribution from the three phases

TPR (a) and He-TPD (b) profiles for the A2 and A6 samples
Figure 6b describes the oxygen desorption from the A2 and A6 samples after heating in static air to 950 °C/5 h. One strong desorption peak was detected at around 400 °C; a second less intense desorption peak was present at around 800 °C. Figure 6b clearly shows that the overall oxygen desorption of sample A6 was smaller than that shown by sample A2, while the desorption peak maximum temperature was almost the same, in agreement with the fact that removal of oxygen was easier and higher for the less oxygen-deficient sample. XRD patterns were registered after the He-TPD experiments, without oxygen, and it was found that the cell parameters of both the cubic phases increased and phase 2 reached a value of 4.005 Å. This phase, whose composition is Ba0.5Sr0.5Co0.8Fe0.2O2 with maximal Co2+ content, it is described in the literature as the most deficient cubic phase with disordered oxygen vacancies [50].
TPO experiments, with 5 vol.% O2, were also performed in order to check for any O2 consumption or CO2 evolution. No O2 consumption was observed, while CO2 evolution peaks were detected between 500 and 800 °C in both TPO and TPD profiles, probably due to the decomposition of residual carbonate-like species. XRD patterns after TPO were very similar to the XRD patterns of the samples obtained after heating in static air, although the calculated Co2+ total amount was slightly larger after TPO, in agreement with the lower O2 content in the gas (5 vol.% O2).
Figure 7 shows the TG–DTA curves measured under nitrogen flow for the A1 and A6 samples, after annealing at 950 °C/5 h, where A1 has a smaller total Co2+ content (10 wt%) than A6 (32 wt%). At temperatures below 500 °C, where no significant CO2 evolution was detected by TPD experiments, the weight changes could be easily ascribed to oxygen depletion. The TG–DTA under nitrogen were very similar in shape to those obtained in air: the Co3+ to Co4+ oxidation step and the two Co4+ to Co3+ and Co3+ to Co2+ reduction steps were visible, although the weight loss in the 200–500 °C range was generally larger than that for the samples treated in air. It is worth noticing that the Co3+ to Co4+ oxidation step was much more evident in the A1 sample, with structural parameters and Co2+ content close to A2, confirming a larger Co3+ percentage, in agreement with the results previously discussed. DTA results evidenced a difference in the temperature for the Co3+ to Co2+ reduction, which occurred at 890 °C and at 850 °C for the A1 and the A6 sample, respectively, indicating that the Co2+ reduced state was better stabilized by using the sucrose–PEG fuel mixture than the cellulose–citric acid one. Both the structural and redox results pointed out that the fuel mixture was able to control the structural parameters of the two BSCF cubic phases through the stabilization of the cobalt (II) oxidation state.

TG and DTA curves under N2 flow for A1 and A6 fired samples
Electrochemical and microstructural properties of BSCF cathode materials
Figure 8a shows the Arrhenius plot of the area-specific resistance (ASR) for the symmetrical cells with BSCF electrodes annealed at 1,000 °C for 2 h on both sides of dense SDC pellets. It is worth noticing that, for a given temperature, the difference in the ASR between the A1 and A6 samples was as large as one order of magnitude, for the same nominal material composition (Fig. 8a). The A6 cathode showed the smallest ASR in the whole tested temperature range (500–800 °C), achieving 0.016 Ω cm2 at 800 °C and 0.10 Ω cm2 at 600 °C. These ASR values were comparable with values reported in the literature for BSCF cathodes measured in similar electrochemical conditions [13, 21, 54]. The activation energy values, albeit slightly smaller, were in agreement with literature data [21].

ASR versus the reciprocal of temperature for BSCF cathodes prepared using different combustion fuel mixtures (a); variation with the oxygen partial pressure of the ASR derived from the first and second semicircles for the A6 sample, measured at 650 °C. In the inset of this figure, a typical EIS spectrum of A6 cathode showing the two semicircles is shown (b)
A one order of magnitude difference in the ASR values at a given temperature of BSCF with the same chemical composition could be caused by several factors which could be related to differences in microstructure [4, 55–58], oxygen deficiency [2, 9, 10, 58–61] or surface chemical composition (cation enrichment and/or impurity presence) [47]. Other factors could involve a difference in the extent of the adhesion of the powder to the electrolyte, or a difference in the thickness of the cathode layer.
N2-adsorption experiments conducted on all the investigated BSCF calcined powders, revealed a very low specific surface area (below 5 m2/g) and porosity (cumulative pore volume around 0.15 cm3/g and mean pore diameter around 4 nm). Figure 9 shows SEM micrographs (with low magnification images in the insets) for the A1, A2, A3 and A6 powders heated to 950 °C/5 h. Higher porosity and lower mean grain size (around 1 μm) were characteristic of sample A1 (Fig. 9). On the other hand, A2, A3, and A6 were less porous, showed a mean grain size around 3 μm and very small differences could be observed among the three samples (Fig. 9). For example, the A3 sample appeared slightly denser than the A6 sample, both before (Fig. 9) and after (Fig. 10) depositing the powders on the SDC electrolyte. The cathode thickness was very close for all the investigated samples (20 μm) and the cathode–electrolyte adhesion in the half-cells, as observed by SEM, was good (Fig. 10). Peculiar nanoscaled features were well evident in all the investigated BSCF samples (Fig. 9). Some authors ascribed the high oxygen conductivity of BSCF powders to peculiar nanoscaled features on the surface of BSCF powders prepared by coprecipitation [22]. Although this hypothesis cannot be completely ruled out, no evident correlation was found in this work between size/shape/distribution of the nanoscaled features and the observed ASR trend. These features were, for example, almost identical for the A2 and A6 samples (Fig. 8a), which showed very different ASR values at the same temperature (Fig. 8a). On the other hand, A1 and A2, which have very similar ASR values, showed very different morphologies. It can be reasonably postulated that the minor wt% of (111)-oriented rhombohedral phase detected by XRD in all the investigated samples (Fig. 5) was somewhat related to the nanoscaled features observed by SEM.

SEM micrographs of four powders—A1, A2, A3 and A6—at two different magnifications (lower in the insets)

SEM images of the half cell sections for two representative cathode powders, A3 and A6, after deposition on the SDC pellet
EDX results for three of the six samples are reported in Table 3, as atomic ratios of Ba, Sr and Co with respect to Fe. The EDX results were very similar for all the samples investigated. A slight Sr depletion (ca. 25 at%) was noticeable for all the investigated samples (Table 3) and this is in agreement with the observations of Efimov et al. [48] about the chemical composition of the (111) oriented hexagonal phase.
In the present case, it appears that the observed differences in the ASR values of the BSCF cathodes at a given temperature (Fig. 8a) cannot be explained solely by microstructural differences. For example, the very slight microstructural differences between A3 and A6 samples (Figs. 9, 10) cannot explain a difference of half order of magnitude in the ASR (Fig. 8a). On the other hand, the more evident microstructural differences between A1 and A2 samples (Fig. 9), apparently did not affect their electrochemical properties (Fig. 8a).
Microstructural properties are mainly responsible for differences in the electrochemical properties of LSCF cathode materials [62], which was not the case here for BSCF. To understand the difference, one should consider the rate determining steps responsible for the overall oxygen reduction reaction in the case of BSCF and LSCF cathodes. Figure 8b shows the ASR associated to each of the two distinct semicircles observed in the low-frequency region of the Nyquist plots as a function of the oxygen partial pressure, measured at 650 °C. ASR is proportional to (pO2)−n, where the n value gives information about the electrode reaction involved. The n value of 0.2 (Fig. 8b), corresponding to the first semicircle, was indeed indicative of a rate-limiting step related to the charge transfer at the electrode/electrolyte interfaces, while the n value of 1 corresponding to the second semicircle indicated a rate-limiting step due to bulk and surface diffusion of oxygen species. As confirmed in Fig. 8b, the rate determining step of a BSCF cathode is governed by gas diffusion and charge transfer at the electrode/electrolyte interface [63]. On the other hand, in the case of LSCF cathodes the dissociative oxygen adsorption is identified as the main factor governing the oxygen reduction reaction [58, 62, 64]. Therefore, it is reasonable to assume that BSCF electrochemical properties are less affected by the cathode microstructure than LSCF compounds.
Bulk and surface oxygen vacancy diffusion is improved in the presence of disordered oxygen vacancies that enhance the oxygen incorporation in the BSCF [63]. It is worth noticing that the sample A6, with more oxygen vacancies and Co2+ content, showed a smaller ASR value than the sample A2, at the same temperature (Fig. 8a). The combustion fuel mixture might affect indirectly the ASR values by governing the oxygen deficiency of the BSCF powders. We searched to correlate the measured ASR values with the most important SCS processing parameters, connected with the fuel mixture. In order to consider both the fuel-to-metal cations molar ratio and the reducing power of each different fuel in the combustion mixture, a new parameter was introduced here. The WF/M ratio, or the fuel-to-metal cations molar ratio weighted for the reducing power of the specific fuel in the mixture, is equivalent to the fuel-to-metal cations ratio, but it can be used for fuel mixtures, where each fuel has a specific reducing power [24]. Figure 11 shows the relationship of both the log ASR measured at a fixed temperature (730 °C) and the Co2+ total content in the sample with the WF/M ratio. An inverse correlation was found between the log ASR and the WF/M (Fig. 11). On the contrary, the Co2+ content increased with increasing WF/M (Fig. 11). It is here argued that, during the combustion process and with the other parameters fixed, the larger the reducing power of the fuel mixture, the larger the tendency of the fuel mixture to stabilize the atoms that will form later the BSCF structure in an oxygen vacancy-rich configuration. It can be further argued that, for a given fuel mixture, this tendency is also positively influenced by larger fuel-to-metal cations ratios, where a higher local concentration of fuels with high reducing power exists around the metal cations.

Correlation between log ASR (at 730 °C), the Co2+ in each sample calculated from XRD results and the fuels/metal cation ratio weighted for the reducing power of each fuel in the combustion mixture (WF/M)
Conclusions
BSCF powders were produced by solution combustion synthesis using various single and double fuels. We showed that the fuel mixture has an explicit influence on the structural parameters, the oxidation state stability at the B-site, the microstructure and the electrochemical properties of the BSCF cathode materials. Moreover, it was observed that such improvement could not be solely ascribed to an improvement in the microstructural features, although a further optimization of the BSCF electrochemical properties by obtaining nanometer-sized powders still remains a challenge due to processing problems. Nonetheless, some important guidelines for the improvement of the electrochemical properties of BSCF cathode powders are proposed in this work. The main conclusion of this work is that BSCF cathode materials with optimized conductivity and high oxygen deficiency can be easily obtained by solution combustion synthesis, making use of high-interacting fuel mixtures having high fuel-to-metal cations molar ratio and high reducing power. In particular, the best electrochemical properties were observed for the A6 sample, where a mixture of sucrose and PEG20000 in a 1:2.5 molar ratio was used. Moreover, a new metastable monoclinic phase with Ba0.5Sr0.5CO3 composition was discovered in the as-burned BSCF powders, extending the existing information on the phase formation mechanism of BSCF compounds. It is expected that most of the information acquired in this work can be successfully used for other chemical compositions. All these results confirmed the importance of the combustion process itself in tailoring the final materials properties, focusing the attention at the fuel mixture–metal cations interaction, contributing to fill an important gap between materials synthesis and materials properties.
References
Brett, D.J.L., Atkinson, A., Brandon, N.P., Skinner, S.J.: Intermediate temperature solid oxide fuel cells. Chem. Soc. Rev. 37, 1568–78 (2008)
Sun, C., Hui, R., Roller, J.: Cathode materials for solid oxide fuel cells: a review. J. Solid State Electrochem. 14, 1125–1144 (2010)
Tarancón, A., Burriel, M., Santiso, J., Skinner, S.J., Kilner, J.A.: Advances in layered oxide cathodes for intermediate temperature solid oxide fuel cells. J. Mater. Chem. 20, 3799–3813 (2010)
Kim, J.H., Park, Y.M., Kim, H.: Nano-structured cathodes based on La0.6Sr0.4Co0.2Fe0.8O3–δ for solid oxide fuel cells. J. Power Sources 196, 3544–3547 (2011)
Patra, H., Rout, S.K., Pratihar, S.K., Bhattacharya, S.: Effect of process parameters on combined EDTA–citrate synthesis of Ba0.5Sr0.5Co0.8Fe0.2O3–δ perovskite. Powder Technol. 209, 98–104 (2011)
Liang, F., Jiang, H., Luo, H., Caro, J., Feldhoff, A.: Phase stability and permeation behavior of a dead-end Ba0.5Sr0.5Co0.8Fe0.2O3–δ tube membrane in high-purity oxygen production. Chem. Mater. 23, 4765–4772 (2011)
Shao, Z., Haile, S.M.: A high-performance cathode for the next generation of solid-oxide fuel cells. Nature 431, 170–173 (2004)
Zhou, W., Ran, R., Shao, Z.: Progress in understanding and development of Ba0.5Sr0.5Co0.8Fe0.2O3–δ-based cathodes for intermediate-temperature solid-oxide fuel cells: A review. J. Power Sources 192, 231–246 (2009)
Yáng, Z., Harvey, A.S., Infortuna, A., Schoonman, J., Gauckler, L.J.: Electrical conductivity and defect chemistry of Ba x Sr1−xCo y Fe1−yO3−δ perovskites. J. Solid State Electrochem. 15, 277–284 (2011)
Yang, Z., Martynczuk, J., Efimov, K., Harvey, A.S., Infortuna, A., Kocher, P., Gauckler, L.J.: Oxygen-vacancy-related structural phase transition of Ba0.8Sr0.2Co0.8Fe0.2O3–δ. Chem. Mater. 23, 3169–3175 (2011)
Mueller, D.N., De Souza, R.A., Weirich, T.E., Roehrens, D., Mayer, J., Martin, M.: A kinetic study of the decomposition of the cubic perovskite-type oxide Ba x Sr1–xCo0.8Fe0.2O3–δ (BSCF) (x = 0.1 and 0.5). Phys. Chem. Chem. Phys. 12, 10320–10328 (2010)
Efimov, K., Halfer, T., Kuhn, A., Heitjans, P., Caro, J., Feldhoff, A.: Novel cobalt-free oxygen-permeable perovskite-type membrane. Chem. Mater. 22, 1540–1544 (2010)
Yan, A., Yang, M., Hou, Z., Dong, Y., Cheng, M.: Investigation of Ba1–xSr x Co0.8Fe0.2O3–δ as cathodes for low-temperature solid oxide fuel cells both in the absence and presence of CO2. J. Power Sources 185, 76–84 (2008)
Hung, I.-M., Liang, C.-Y., Ciou, C.-J., Lee, Y.-C.: Conductivity and electrochemical performance of (Ba0.5Sr0.5)0.8La0.2CoO3–δ cathode for intermediate-temperature solid oxide fuel cell. Ceram. Int. 36, 1937–1943 (2010)
Kotomin, E.A., Mastrikov, Y.A., Kuklja, M.M., Merkle, R., Roytburd, A., Maier, J.: First principles calculations of oxygen vacancy formation and migration in mixed conducting Ba0.5Sr0.5Co1–yFe y O3–δ perovskites. Solid State Ionics 188, 1–5 (2011)
Ding, X., Kong, X., Jiang, J., Cui, C., Guo, X.: Characterization and electrochemical performance of (Ba0.6Sr0.4)1–xLa x Co0.6Fe0.4O3–δ (x = 0, 0.1) cathode for intermediate temperature solid oxide fuel cells. Mater. Res. Bull. 45, 1271–1277 (2010)
Tan, L., Gu, X., Yang, L., Jin, W., Zhang, L., Xu, N.: Influence of powder synthesis methods on microstructure and oxygen permeation performance of Ba0.5Sr0.5Co0.8Fe0.2O3–δ perovskite-type membranes. J. Membr. Sci. 212, 157–165 (2003)
Liu, B., Zhang, Y.: Ba0.5Sr0.5Co0.8Fe0.2O3 nanopowders prepared by glycine–nitrate process for solid oxide fuel cell cathode. J. Alloys Compd. 453, 418–422 (2008)
Kriegel, R., Kircheisen, R., Toepfer, J.: Oxygen stoichiometry and expansion behavior of Ba0.5Sr0.5Co0.8Fe0.2O3–δ. Solid State Ionics 181, 64–70 (2010)
Martynczuk, J., Arnold, M., Wang, H., Caro, J., Feldhoff, A.: How (Ba0.5Sr0.5)(Fe0.8Zn0.2)O3–δ and (Ba0.5Sr0.5)(Co0.8Fe0.2)O3–δ perovskites form via an edta/citric acid complexing method. Adv. Mater. 19, 2134–2140 (2007)
Chen, Z., Ran, R., Zhou, W., Shao, Z., Liu, S.: Assessment of Ba0.5Sr0.5Co1–yFe y O3–δ (y = 0.0–1.0) for prospective application as cathode for IT-SOFCs or oxygen permeating membrane. Electrochim. Acta 52, 7343–7351 (2007)
Toprak, M.S., Darab, M., Syvertsen, G.E., Muhammed, M.: Synthesis of nanostructured BSCF by oxalate co-precipitation—As potential cathode material for solid oxide fuels cells. Int. J. Hydrogen En. 35, 9448–9454 (2010)
Specchia, S., Galletti, C., Specchia, V.: Solution combustion synthesis as intriguing technique to quickly produce performing catalysts for specific applications. Stud. Surf. Sci. Catal. 175, 59–67 (2010)
Jain, S.R.: Energetics of propellants, fuels and explosives: A chemical valence approach. Propellants, Explos. Pyrotech. 12, 188–195 (1987)
Deganello, F., Marcì, G., Deganello, G.: Citrate–nitrate auto-combustion synthesis of perovskite-type nanopowders: A systematic approach. J. Eur. Ceram. Soc. 29, 439–450 (2009)
Mukasyan, A.S., Epstein, P., Dinka, P.: Solution combustion synthesis of nanomaterials. Proc. Combust. Inst. 31, 1789–1795 (2007)
Jain, S.R., Adiga, K.C., Verneker, V.R.P.: A new approach to thermochemical calculations of condensed fuel-oxidizer mixtures. Combust. Flame 40, 71–79 (1981)
Öncel, Ç., Gülgün, M.A.: Chemical Synthesis of Mixed-Oxide Powders for Solid Oxide Fuel Cell (SOFC) Electrolyte and Electrodes. In: Sheffield, J.W., Sheffield, Ç. (eds.) Assessment of hydrogen energy for sustainable development, pp. 147–159. Springer, Dordrecht (2007)
da Conceição, L., Silva, A.M., Ribeiro, N.F.P., Souza, M.M.V.M.: Combustion synthesis of La0.7Sr0.3Co0.5Fe0.5O3 (LSCF) porous materials for application as cathode in IT-SOFC. Mater. Res. Bull. 46, 308–314 (2011)
da Silva, A.L.A., Castro, G.G.G., Souza, M.M.V.M.: Synthesis of Sr-doped LaCrO3 powders by combustion method. Influence of the fuel agent. J. Therm. Anal. Calorim. 109, 33–38 (2011)
Vijaya Lakshmi, V., Bauri, R., Paul, S.: Effect of fuel type on microstructure and electrical property of combustion synthesized nanocrystalline scandia stabilized zirconia. Mater. Chem. Phys. 126, 741–746 (2011)
Park, K., Nam, S.W.: Thermoelectric properties of Ca2.8Cu0.2Co4O9 ceramics fabricated by solution combustion method. Curr. Appl. Phys. 11, 939–944 (2011)
Shigapov, A.N., Graham, G.W., McCabe, R.W., Plummer Jr, H.K.: The preparation of high-surface area, thermally-stable, metal-oxide catalysts and supports by a cellulose templating approach. Appl. Catal. A: Gen. 210, 287–300 (2001)
Zhou, W., Shao, Z.P., Ran, R., Jin, W.Q., Xu, N.P.: Functional nano-composite oxides synthesized by environmental-friendly auto-combustion within a micro-bioreactor. Mater. Res. Bull. 43, 2248–2259 (2008)
Das, R.N.: Nanocrystalline ceramics from sucrose process. Mater. Lett. 47, 344–350 (2001)
Alinejada, B., Sarpoolaky, H., Beitollahi, A., Saberi, A., Afshar, S.: Synthesis and characterization of nanocrystalline MgAl2O4 spinel via sucrose process. Mater. Res. Bull. 43, 1188–1194 (2008)
Larson, A.C., Von Dreele, R.B.: GSAS general structure analysis system. LANSCE, MS-H 805, Los Alamos National Laboratory, Los Alamos, NM 87545, USA (1998)
McCusker, L.B., Von Dreele, R.B., Coxd, D.E., Louer, D., Scardi, P.: Rietveld refinement guidelines. J. Appl. Crystallogr. 32, 36–50 (1999)
Brunauer, S., Emmett, P.H., Teller, E.: Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 60, 309–319 (1938)
Barret, E.P., Joyner, L.G., Halenda, P.P.: The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J. Am. Chem. Soc. 73, 373–380 (1951)
Bo, Y., Wenqiang, Z., Jingming, X., Jing, C.: Microstructure characterization and electrochemical properties of Ba0.5Sr0.5Co0.5FeO3–δ and its application for anode of SOEC. Int. J. Hydrog. Energy 33, 6873–6877 (2008)
Magnone, E., Miyayama, M., Traversa, E.: Structural properties and electrochemical characteristics of Ba0.5Sr0.5Co1–xFe x O3–δ phases in different atmospheres. J. Electrochem. Soc. 159, B1059–B1066 (2009)
Li, Z., Wei, B., Lü, Z., Huang, X., Su, W.: Structure, electrical and thermal properties of (Ba0.5Sr0.5)1–xGd x Co0.8Fe0.2O3–δ perovskite as a solid-oxide fuel cell cathode. Solid State Ionics 207, 38–43 (2012)
Toniolo, J.C., Takimi, A.S., Bergmann, C.P.: Nanostructured cobalt oxides (Co3O4 and CoO) and metallic Co powders synthesized by the solution combustion method. Mater. Res. Bull. 45, 672–676 (2010)
Strobel, R., Maciejewski, M., Pratsinis, S.E., Baiker, A.: Unprecedented formation of metastable monoclinic BaCO3 nanoparticles. Thermochim. Acta 445, 23–26 (2006)
Nishino, T.: Characterization of γ-BaCO3. Solid State Ionics 32/33, 394–397 (1989)
Yi, J., Schroeder, M.: High temperature degradation of Ba0.5Sr0.5Co0.8Fe0.2O3–δ membranes in atmospheres containing concentrated carbon dioxide. J. Membrane Sci. 378, 163–170 (2011)
Efimov, K., Xu, Q., Feldhoff, A.: Transmission electron microscopy study of Ba0.5Sr0.5Co0.8Fe0.2O3–δ perovskite decomposition at intermediate temperatures. Chem. Mater. 22, 5866–5875 (2010)
Koster, H., Mertins, F.H.B.: Powder diffraction of the cubic perovskite Ba0.5Sr0.5Co0.8Fe0.2O3–δ. Powder Diffr. 18, 56–59 (2003)
Mueller, D.N., De Souza, R.A., Yoo, H.-I., Martin, M.: Phase stability and oxygen nonstoichiometry of highly oxygen-deficient perovskite-type oxides: A case study of (Ba, Sr)(Co, Fe)O3–δ. Chem. Mater. 24, 269–274 (2012)
Harvey, A.S.: Jochen Litterst, F., Yang, Z., Rupp, J.L.M., Infortuna, A., Gauckler, L.J.: Oxidation states of Co and Fe in Ba1–xSr x Co1–yFe y O3–δ (x, y = 0.2–0.8) and oxygen desorption in the temperature range 300–1273 K. Phys. Chem. Chem. Phys. 11, 3090–3098 (2009)
Crapanzano, S., Babich, I.V., Lefferts, L.: Selection of mixed conducting oxides for oxidative dehydrogenation of propane with pulse experiments. Appl. Catal. A Gen. 391, 70–77 (2011)
Khan, A., Smirniotis, P.G.: Relationship between temperature-programmed reduction profile and activity of modified ferrite-based catalysts for WGS reaction. J. Mol. Catal. A Chem. 280, 43–51 (2008)
Zhang, Y., Liu, J., Huang, X., Lu, Z., Su, W.: Low temperature solid oxide fuel cell with Ba0.5Sr0.5Co0.8Fe0.2O3 cathode prepared by screen printing. Solid State Ionics 179, 250–255 (2008)
Deganello, F., Esposito, V., Miyayama, M., Traversa, E.: Cathode performance of nanostructured La1–aSr a Co1–bFe b O3–x on a Ce0.8Sm0.2O2. Electrolyte prepared by citrate–nitrate autocombustion. J. Electrochem. Soc. 154, A89–A96 (2007)
Baqué, L., Caneiro, A., Moreno, M.S., Serquis, A.: High performance nanostructured IT-SOFC cathodes prepared by novel chemical method. Electrochem. Commun. 10, 1905–1908 (2008)
Hieu, N.T., Parka, J., Tae, B.: Synthesis and characterization of nanofiber-structured Ba0.5Sr0.5Co0.8Fe0.2O3–δ perovskite oxide used as a cathode material for low-temperature solid oxide fuel cells. Mater. Sci. Eng. B 177, 205–209 (2012)
Adler, S.B.: Factors governing oxygen reduction in solid oxide fuel cell cathodes. Chem. Rev. 104, 4791–4843 (2004)
Molenda, J., Świerczek, K., Zając, W.: Functional materials for the IT-SOFC. J. Power Sources 173, 657–670 (2007)
Deganello, F., Liotta, L.F., Longo, A., Casaletto, M.P., Scopelliti, M.: Cerium effect on the phase structure, phase stability and redox properties of Ce-doped strontium ferrates. J. Solid State Chem. 179, 3406–3419 (2006)
Neagu, D., Irvine, J.T.S.: Structure and properties of La0.4Sr0.4TiO3 ceramics for use as anode materials in solid oxide Fuel cells. Chem. Mater. 22, 5042–5053 (2010)
Yin. Q., Yang, Z., Lin, Y.S.: Effects of microstructure on oxygen transport in perovskite-type oxides. J. Mater. Sci. 41, 4865–4870 (2006)
Wang, L., Merkle, R., Maier, J.: Oxygen exchange on mixed conducting oxides: (Ba, Sr)(Co, Fe)O3–δ SOFC cathode materials. J. Electrochem. Soc. 157, B1802–B1808 (2010)
Zhao, H., Huo, L., Sun, L., Yu, L., Gao, S., Zhao, J.: Preparation, chemical stability and electrochemical properties of LSCF–CBO composite cathodes. J. Mater. Chem. Phys. 88, 160–166 (2004)
Feldhoff, A., Martynczuk, J., Wang, H. (2007) Advanced Ba0.5Sr0.5Zn0.2Fe0.8O3–δ perovskite-type ceramics as oxygen selective membranes: Evaluation of the synthetic process. Progress in Solid State Chem. 35, 339−353 (2007)
Acknowledgments
The authors are thankful to Dr. Francesco Giordano (ISMN-CNR UOS Palermo) for technical assistance in performing the XRD measurements and to Dr. Bartolomeo Megna (Laboratorio di Materiali per il Restauro e la Conservazione, Dipartimento di Ingegneria Civile, Ambientale e Aerospaziale, Università di Palermo) for technical assistance in performing the TG–DTA measurements. University of Palermo is also acknowledged for supporting in part this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Deganello, F., Liotta, L.F., Marcì, G. et al. Strontium and iron-doped barium cobaltite prepared by solution combustion synthesis: exploring a mixed-fuel approach for tailored intermediate temperature solid oxide fuel cell cathode materials. Mater Renew Sustain Energy 2, 8 (2013). https://doi.org/10.1007/s40243-013-0008-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-013-0008-z