
Abstract
Fibrosis is defined as an excessive accumulation of extracellular matrix components that lead to the destruction of organ architecture and impairment of organ function. Moreover, fibrosis is an intricate process attributable to a variety of interlaced fibrogenic signals and intrinsic mechanisms of activation of myofibroblasts. Being the dominant matrix-producing cells in organ fibrosis, myofibroblasts may be differentiated from various types of precursor cells. Identification of the signal pathways that play a key role in the pathogenesis of fibrotic diseases may suggest potential therapeutic targets. Here, we emphasize several intracellular signaling pathways that control the activation of myofibroblasts and matrix production.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
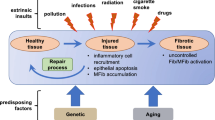
Fibrosis, characterized by excessive accumulation of extracellular matrix (ECM) in and around inflamed or damaged tissue, leads to tissue destruction,permanent scarring, and organ malfunction [1••]. Although fibrotic conditions may result from diverse causes, it is generally thought that an initial injury activates a repair process that aims to restore the original tissue structure, and that a failure to delicately regulate this process results in sustained fibroblast activation, matrix deposition, and tissue devastation [2]. Fibrosis is part of progressively chronic diseases in parenchymal organs throughout the body, and the fibrotic process plays an essential role in the deterioration of these organs [1••, 3••, 4]. Few effective therapies to halt tissue fibrosis, or to reverse it, are available in clinical practice [5••]. Consequently, it is important to thoroughly understand the cellular and molecular mechanisms of fibrogenesis, not only to acquire novel insights into the pathogenesis of the fibrotic process, but also to further exploit efficient strategies to treat patients with fibrotic disorders.
Fibrogenesis is a dynamic and progressive process in which nonresolving inflammation following a persistent injury sets the stage for fibrosis and triggers the activation of matrix-producing myofibroblasts differentiated from a variety of precursor cell types through different mechanisms, including activation of resident fibroblasts and pericytes, phenotypic transition of epithelial and endothelial cells, phenotypic modulation of vascular smooth muscle cells, and recruitment of circulating multipotent monocytes and fibrocytes [5••]. The mechanisms governing the fibrotic process have been studied in great detail in the past years. Regardless of the initiating event, a feature common to all fibrotic diseases is the activation of myofibroblasts, key mediators in fibrotic tissue remodeling. Once activated, myofibroblasts produce and secrete components of ECM, such as collagen and fibronectin [5••, 6•, 7•]. Because fibrogenesis may be evoked by a variety of stimuli, it is conceivable that a diverse array of signaling networks and mediators might be involved in regulating the progression of fibrosis. The scope of this review is limited to several key intracellular signal transduction pathways that are essential in controlling a host of transcription regulators and signaling mediators that are necessary for fibrogenesis. It is hoped that a precise delineation of these fibrotic signaling cascades will yield further insights into the pathogenesis of fibrosis and lead to the identification of novel targets for therapy.
Smad Pathway: Canonical Pathway of Transforming Growth Factor-β Signaling
Transforming growth factor-β (TGF-β) is a profibrotic factor and a central mediator of fibrogenesis. The classical Smad-dependent pathway for TGF-β signaling occurs when TGF-β receptor type 2, which is constitutively active, transphosphorylates and forms a complex with the TGF-β-bound TGF-β receptor type 1. This complex then phosphorylates serine residues of cytoplasmic receptor-activated Smad (R-Smad), a complex of Smad2 and Smad3. These two heterodimerize and bind to the common mediator Smad (Co-Smad) Smad4, and the whole complex translocates across the nuclear membrane to interact with specific cis-acting elements in the regulatory regions of its target genes [8••], recruiting coactivators such as p300 and CBP; corepressors such as c-Ski, SnoN, transforming growth-inhibiting factor, and Smad nuclear-interacting protein 1; or transcription factors such as AP-1 and Sp1 to modulate gene expression [4]. Inhibitory Smad (I-Smad) Smad6 or Smad7, acting as negative regulators, not only antagonizes the TGF-β/Smad pathway by binding to TGF-β1 or competing with activated R-Smad for binding to Co-Smad, but also recruits the E3 ubiquitin–protein ligases Smurf1 and Smurf2, which target Smad proteins for proteasomal degradation, thereby blocking Smad2/3 activation, facilitating receptor degradation, and eventually terminating Smad-mediated signaling [4, 8••, 9–11].
A large amount of evidence shows that Smad signaling plays a crucial role in governing TGF-β-induced fibrosis. A variety of genes for mediating myofibroblast activation and epithelial–mesenchymal transition (EMT) are the downstream targets of TGF-β/Smad signaling, including connective tissue growth factor (CTGF), Snail, α-smooth muscle actin (α-SMA), collagen IA2, inhibitor of differentiation 1 (Id1), Wnt, matrix metalloproteinase 2 (MMP-2), integrin-linked kinase (ILK), β1-integrin, and PINCH-1 [12–15, 16•]. Mounting studies indicate that the canonical Smad2/3 pathway induces transition of the increased number of fibroblasts into active myofibroblasts and EMT, and acts on the latter cells as transcriptional activators of multiple ECM proteins as well as regulators of matrix accumulation [17].
The necessity of Smad signaling in fibrogenesis is illustrated clearly in Smad3 knockout mice [4, 16•]. Mice lacking Smad3 are protected from bleomycin-induced skin and lung fibrosis, dimethylnitrosamine-induced liver fibrosis, renal interstitial fibrosis due to unilateral ureteral obstruction, skin fibrosis following irradiation, and cardiac fibrosis [18–23]. In addition, Smad4-deficient mice also are protected from renal fibrosis after obstructive injury [8••, 24]. Conversely, I-Smad, such as Smad7, counteracts TGF-β-induced fibrosis. Overexpression of Smad7 blocks bleomycin-induced lung fibrosis and bile duct ligation-induced liver fibrosis [25, 26]. Dysregulation of SnoN, Ski, or Smurf2 may give rise to deviant TGF-β/Smad signaling in the pathogenesis of kidney fibrosis [27, 28].
Mitogen-Activated Protein Kinase Pathway: Erk, p38, and Jnk
Besides activating the Smad-dependent pathway, TGF-β also can signal in a noncanonical manner. One of the Smad-independent pathways is the mitogen-activated protein kinase (MAPK) family. As the ubiquitous intracellular serine/threonine kinases, MAPK family members can transmit extracellular signals from cell surface receptors to intracellular targets, ultimately activating or inhibiting nuclear transcription factors and determining the cell’s fate [4]. MAPKs contain three major subfamilies: the extracellular signal-regulated kinases (Erk1 and Erk2), the p38/MAP kinases (α, β, γ, and δ), and the stress-activated protein kinases known as c-Jun N-terminal kinases (Jnk1, Jnk2, and Jnk3). All three MAPK pathways may be activated by TGF-β, and signaling through these cascades can further regulate the expression of Smad proteins and mediate Smad-independent TGF-β responses [9]. These three MAPK pathways are all involved in TGF-β-induced fibrosis [8••, 9, 29, 30].
TGF-β induces phosphorylation on TGF-β receptors 1 and 2 and/or Shc, which recruit Grb2/Sos to activate Erk through membrane-anchored Ras and downstream MAPK cascades [8••, 31]. Depending on the cell type studied, the activation of Erk can up- or down-regulate Smad signaling activity, whereas Jnk or p38 generally promote TGF-β-stimulated responses [4]. The TGF-β-Erk axis can mediate the transcription of genes controlling the EMT process, CTGF expression, and collagen I production by working with the Smad-dependent pathway, whereas Erk also can repress R-Smad activity through phosphorylation [4, 8••, 32, 33]. Together with Smads, the activated JnK/p38 pathways regulate TGF-β-induced fibroblast differentiation into myofibroblasts [8••, 30].
Phosphoinositide 3-Kinase Pathway: Akt/mTOR and PAK2/c-Abl
The phosphatidylinositol 3-kinase (PI3K) pathway is another non-Smad pathway contributing to TGF-β-induced fibrosis. It induces two profibrotic pathways: Akt–mammalian target of rapamycin (mTOR) and p21-activated kinase 2 (PAK2)/Abelson kinase (c-Abl) [4, 8••, 9, 34].
Akt is a serine/threonine kinase that can engage multiple downstream signaling substrates and pathways. One of its downstream targets is Tuberin [tuberous sclerosis complex 2 (TSC2)], which binds to hamartin (TSC1). TSC1 contributes stability and prolongs the half-life of TSC2 within this complex, and TSC2 becomes phosphorylated and thereby inactivated. TSC2 functions as a GTPase-activating protein (GAP) specifically for Ras homolog enriched in brain (Rheb) [17, 35]. Rheb exists in either an active or inactive GDP-bound state, and Rheb–GTP activates target of rapamycin complex 1 (TORC1) [36–38]. TORC1 consists of several protein components, including mTOR itself, the regulatory-associated protein of mTOR (Raptor), and mLST [39, 40]. Active (nonphosphorylated) TSC2 converts mTORC1-activating Rheb–GTP to Rheb–GDP. Thus, active TSC2 is an inhibitor of mTORC1, and loss of TSC2 activity by phosphorylation increases mTORC1 activity. This activates downstream substrates, including p70 S6 kinase (S6K), a translational activator of many proteins, including cell cycle proteins, and hence, proliferation [17]. The Akt–mTORC1–P70S6K branch pathway contributes to fibroblast proliferation and myofibroblast differentiation. Abnormal activation of mTORC1 is involved in the pathogenesis of fibrotic disorders. Activation of mTORC1 induced by TGF-β is essential for its profibrotic effects on collagen production, whereas pharmacologic and genetic manipulation that decreases mTORC1 activity prevents fibrotic changes [17, 26, 41]. Rheb/mTORC1 signaling may promote activation of kidney fibroblasts and contributes to the development of interstitial fibrosis [38].
PI3K also acts as a branch point in response to TGF-β, leading to activation of PAK2/c-Abl. The latter not only stimulates collagen gene expression in normal fibroblasts, but also induces fibroblast proliferation, thereby increasing the number of myofibroblast precursors [42]. PAK2/c-Abl promotes fibrosis through its downstream mediators, including PKCδ/Fli-1 and early growth response (Egr)-1, -2, and -3 [8••, 43•, 44]. Unrestrained TGF-β activity might be associated with aberrantly sustained c-Abl activation, leading to Erk1/2-dependent upregulation and persistent expression of Egr-1 in fibroblasts [45]. Sustained Egr-1 expression in target tissues in turn would induce or perpetuate fibrotic responses. Activation of the c-Abl–Egr-1 pathway, presumably through PI3K and PAK2, represents an important novel mechanism for mediating TGF-β responses in fibroblasts [45].
Rho GTPase Signaling Pathway
Rho GTPases, a subfamily of small GTP-binding proteins belonging to the Ras superfamily, modulate the actin cytoskeleton. Their activity is regulated by Rho guanine nucleotide exchange factors. The latter can interact directly with Rho proteins, allowing exchange of GDP for GTP [8••]. Rho is activated due to GTP-bound state and it can interact with downstream effector proteins, most notably Rho-associated, coiled-coil containing protein kinase (ROCK) and mouse diaphanous-related formin 1 (mDia1), which together initiate and stabilize actin stress fibers. Mechanistically, mDia1 induces F-actin filament nucleation, whereas phosphorylated ROCK regulates stabilization of F-actin through multiple downstream target genes [8••]. Hence, the amount of F-actin is increased, resulting in a decrease in G-actin monomer-free myocardin-related transcription factor (MRTF) to travel into the nucleus, where it cooperates with serum response factor (SRF) to induce gene transcription [8••].
Multiple target genes for MRTF/SRF are known drivers of fibrosis, including CTGF, α-SMA, and collagen [46, 47]. SRF-mediated gene transcription is crucial for the induction and maintenance of myofibroblast differentiation [48–50]. Of interest, Rho GTPase signaling with its downstream gene transcription mechanism MRTF/SRF seems to play a convergent role in pathways downstream of TGF-β, lysophosphatidic acid (LPA), endothelin 1 (ET-1), and integrins in fibrosis [8••]. It suggests that the Rho/MRTF/SRF transcriptional pathway may be a therapeutic target for fibrotic diseases [8••].
Canonical Wnt/β-Catenin Signaling
Wnt proteins deliver their signal across the plasma membrane by interacting with Fizzled receptors and coreceptors, members of the LDL receptor-related protein 5/6. Once Wnts bind to their receptors/coreceptors, they initiate a chain of downstream signaling events implicating Disheveled, axin, adenomatosis polyposis coli (APC), casein kinase 1 (CK-1), and glycogen synthase kinase 3β (GSK-3β), leading to dephosphorylation of β-catenin [16•]. Escaping from degradation mediated by the ubiquitin/proteasome system stabilized β-catenin accumulates in the cytoplasm and translocates into the nuclei, where it interacts with its DNA-binding partner, known as T cell factor (TCF)/lymphocyte enhancer-binding factor 1 (LEF1), to stimulate the transcription of Wnt target genes [2, 16•, 51].
Wnt/β-catenin is an evolutionarily conserved cellular signaling system that plays an essential role in a diverse array of biologic processes, such as organogenesis, tissue homeostasis, and the pathogenesis of many human diseases [16•]. The contribution of Wnt/β-catenin signaling to fibrogenesis is becoming increasingly clear. Several studies suggest that the aberration of components of this pathway that fine-tunes the signaling is an essential driver of β-catenin-mediated fibrosis in fibrotic diseases. Activation of Wnt/β-catenin signaling has been reported in skin, kidney, liver, lung, and cardiac fibrosis [1••, 2].
With regard to cellular targets, several in vitro studies show that activation of Wnt/β-catenin signaling enhances proliferation, migration, and matrix production in human dermal fibroblasts, suggesting a key role for this signaling pathway in fibroproliferation and differentiation of fibroblasts into myofibroblasts [52, 53]. In addition to fibroblasts, epithelium is a notable target of Wnt/β-catenin signaling. In vivo, β-catenin is upregulated predominantly in renal tubular epithelium in fibrotic kidneys, and in vitro, activation of β-catenin in tubular epithelial cells induces EMT as well as the expression of several fibrosis-related genes, such as Snail1, plasminogen activator inhibitor 1 (PAI-1), MMP-7, and fibronectin [54, 55•, 56–58]. Likewise, activation of Wnt/β-catenin signaling is involved in mediating podocyte EMT, podocyte dysfunction, and glomerulosclerosis [59•]. Furthermore, many β-catenin target genes are key EMT regulatory transcription factors and mediators, including Snail, Twist, LEF1, and Jagged1 [16•]. Interestingly, recent studies demonstrate that tubular β-catenin can control the fate of interstitial fibroblasts via MMP-7-mediated epithelial–mesenchymal communication in renal fibrosis after obstructive injury [58]. Besides the aforementioned cell types, several studies indicate that a multipotent adipogenic progenitor that can be changed toward a fibrotic phenotype may be a critical target of Wnt/β-catenin signaling in fibrotic diseases [2]. By inhibiting adipogenic transcription factors CCAAT/enhancer-binding protein α and peroxisome proliferator-activated receptor γ [60], Wnt/β-catenin signaling antagonizes adipogenic gene expression and promotes dedifferentiation toward a phenotype of myofibroblast in both hepatic lipofibroblasts [61] and 3T3-L1 cells [2, 62].
Increasing evidence exists regarding crosstalk between Wnt/β-catenin and TGF-β. Wnt/β-catenin signaling can upregulate expression of TGF-β1 [52, 63], and TGF-β1 can activate β-catenin [56, 63, 64]. Smad3 knockout mice display less β-catenin activation, whereas a lack of β-catenin attenuates the ability of TGF-β to promote proliferation in fibroblasts [63]. β-Catenin and TGF-β can synergize to coregulate the same genes via Smad and TCF-binding sites within the promoter [65]. Although it is unclear whether Wnt/β-catenin signaling simply collaborates with the profibrotic factor TGF-β to promote matrix synthesis and assembly or it primarily governs cell fate decisions leading to abundant myofibroblasts through differentiation or transition from various other cell types during fibrogenesis, it is exciting to see that pharmacologic inhibition of Wnt/β-catenin signaling by different approaches is protective, resulting in amelioration of tissue fibrosis in models of fibrotic disease [2, 54, 58, 66•, 67, 68, 69•].
Sonic Hedgehog Signaling
Hedgehog signaling is a highly conserved signaling pathway that orchestrates multiple aspects of embryogenesis, development, and tissue remodeling in a wide spectrum of systems [70, 71]. Hedgehog transmits its signaling by binding to the plasma membrane receptor, Patched (Ptc). In the absence of hedgehog, Ptc keeps the coreceptor Smoothened (Smo) in its inactive form and silences the Smo-dependent downstream intracellular signaling. When the extracellular microenvironment is enriched with soluble hedgehog, the interaction of hedgehog and Ptc leads to Ptc internalization and degradation, resulting in the derepression of Smo. Activated Smo moves from an intracellular vesicle to the cell membrane [72•], leading to an intracellular signaling cascade that ultimately drives the activation and nuclear translocation of glioblastoma (Gli) family zinc-finger transcription factors. The binding of Gli proteins to their cognate cis-acting elements regulates the expression of hedgehog target genes [73]. Gli1 and Gli2 mostly are responsible for providing prolonged cellular responses to hedgehog, whereas Gli3 primarily acts as a signaling repressor [74, 75]. Direct targets of hedgehog signaling contain several components in its own pathway, such as Ptc, Smo, Glis, and hedgehog-interacting protein 1, thereby providing both positive and negative feedback to ensure the delicate regulation of the pathway [72•, 76].
Among the three vertebrate hedgehogs, sonic hedgehog (Shh) is the best characterized and most widely studied. Shh signaling has been implicated in the regulation of injury repair and wound healing after tissue damage [72•, 77•]. Shh expression is upregulated in chronic fibrotic lung diseases, and the Shh signaling pathway may be involved in the remodelling of damaged pulmonary epithelium [78]. In mouse cholangiocytes, coculture with myofibroblastic hepatic stellate cells, a source of Shh, promotes EMT and cell migration, whereas addition of Shh-neutralizing antibodies to cocultures blocks these effects. Moreover, mice haploinsufficient for the Shh inhibitor Ptc exhibit increased Shh signaling activity, and their livers show enhanced fibrogenesis after bile duct injury and elevated expression of Gli2 and several mesenchymal markers [74, 79]. Thus, activation of Shh signaling promotes EMT and contributes to the evolution of biliary fibrosis during chronic cholestasis [74, 79]. In a mouse model of obstructive nephropathy, Shh is induced predominantly in renal tubular epithelium but targets interstitial fibroblasts. Either genetic Gli1 ablation or pharmacologic inhibition of Smo attenuates matrix gene expression and mitigates renal fibrotic lesions. This epithelial–mesenchymal communication, mediated by Shh/Gli1 signaling, probably plays a crucial role in the pathogenesis of renal fibrosis [72•].
Shh signaling elicits its action by regulating the transcription of its target genes [72•, 76]. The best-characterized direct target and downstream mediator of Shh is Gli1; another is Snail1, a key transcription factor for mediating EMT and fibroblast migration. Besides regulating its target genes, Shh might directly control the expression of a battery of fibrogenic genes, such as α-SMA, fibronectin, collagen I, and desmin, genes directly involved in myofibroblast activation and matrix production [72•]. It is worthwhile to note that through crosstalk with Wnt/β-catenin signaling and Notch signaling, Shh signaling can act in concert with other fibrogenic signaling pathways as well [76, 80], and the expression of Snail1 may be induced by both Wnt/β-catenin and Shh signaling [54, 59•, 72•]. Consequently, targeting Shh signaling might be a promising strategy for therapeutic intervention in a variety of fibrotic diseases.
Notch Signaling
Notch proteins are single-pass transmembrane receptors with conserved expression among animal species during evolution. Their principal function is the regulation of many developmental processes, including proliferation, differentiation, and apoptosis. Mammals possess four different Notch receptors, referred to as Notch1–4. The Notch receptor consists of an extracellular domain, which is involved in ligand binding, and an intracellular domain that works in signal transduction. Notch ligands also are single-pass transmembrane proteins named Jagged (Jag1 and 2) and Delta (Dll1, 3, and 4) [81, 82]. Activation of this signaling pathway requires cell–cell contact. Interaction of ligands with the Notch receptors triggers a series of proteolytic cleavages, by a metalloprotease of the ADAM family (TACE; tumor necrosis factor-α-converting enzyme) and finally by the γ-secretase complex. The final cleavage leads to the release of Notch intracellular domain (NICD), which travels to the nucleus and binds to other transcriptional regulators (mainly of the CBF1/RBP-Jκ, SU(H), Lag1 family) to trigger the transcription of the target genes, classically belonging to the Hes and Hey family. This core signal transduction pathway is used in most Notch-dependent processes and is known as the canonical pathway [81, 83••, 84].
During the past few years, activation of Notch signaling has shown fibrogenic effects in a wide spectrum of diseases, including systemic sclerosis (SSc) [85•, 86], scleroderma, idiopathic pulmonary fibrosis (IPF) [87], kidney fibrosis [81, 88, 89], and cardiac fibrosis [83••].
Activated Notch1 and an elevated level of NICD are found in the lesional skin of SSc patients and in the skin and lungs of mouse models with SSc. Blocking the release of NICD with a γ-secretase inhibitor or treating SSc mice with Notch siRNA may reduce the collagen content in the skin and lungs [85•, 86, 90]. Activation of Notch signaling has major implications on fibroblast activation in SSc. Stimulation of SSc fibroblasts with a recombinant Jag-1-Fc chimera results in their differentiation into myofibroblasts overexpressing α-SMA and producing large amounts of ECM, whereas pharmacologic blockade of Notch signaling normalizes the proliferation rate of dermal fibroblasts extracted from lesional skin [85•, 90].
Activation of Notch signaling is associated with abnormal differentiation of respiratory epithelial cells in progressive IPF or secondary pulmonary fibrosis as observed in SSc. Hes-1, a Notch target gene, is upregulated in lung mucus cells from patients with chronic obstructive pulmonary disease, idiopathic pulmonary arterial hypertension, and IPF [87]. Activation of Notch signaling is critical for lung fibroblasts to differentiate into myofibroblasts. In bleomycin-induced lung fibrosis, Notch is indispensable in the upregulation of α-SMA induced by FIZZ1 (found in the inflammatory zone), a cysteine-rich secreted protein with fibrogenic properties, thus triggering the differentiation of fibroblasts into myofibroblasts [91]. Activation of Notch signaling induces expression of α-SMA, collagen I, and vimentin in alveolar epithelial cells in rats with lung fibrosis, which suggests that this signaling is involved in the EMT process, and Notch-mediated induction of Snail1 is required for TGF-β1-induced EMT in human alveolar epithelial cells [92, 93].
The participation of Notch in chronic kidney disease (CKD) has been studied in detail. Elevated levels of Notch ligands and receptors are detected in several glomerular diseases and tubulointerstitial fibrosis [81, 88, 89]. Either genetic deletion of a Notch transcriptional partner (CBF1), specifically in podocytes and tubular cells, or pharmacologic blockade of the Notch pathway with a γ-secretase inhibitor protects against glomerular injury and tubulointerstitial fibrosis in vivo [81]. Overexpression of Notch in renal tubular cells is necessary and sufficient for tubulointerstitial fibrosis development, and upregulation of Notch1 NICD (NICD1) increases ECM production in cultured tubular cells [94•]. NICD1 promotes tubulointerstitial fibrosis and glomerulosclerosis when overexpressed conditionally in tubular cells and podocytes in vivo, respectively [88, 95].
Integrin-Linked Kinase Signaling
Abundant studies demonstrate that integrin signaling plays a critical role in the production and assembly of matrix proteins. Integrins are a family of heterodimeric transmembrane receptors containing α and β subunits. Integrins integrate signals between cells and their extracellular environment by connecting the cytoskeleton to the ECM. Because they have no enzymatic or actin-binding activity, integrins transmit their signals by activating their downstream effector kinases, focal adhesion kinase (FAK), and integrin-linked kinase (ILK) [5••, 16•, 96].
ILK can integrate and mediate several different intracellular signals associated with fibrogenesis, mainly because it is both a scaffolding protein and a serine/threonine protein kinase. Ectopic expression of ILK directly phosphorylates GSK-3β, then the inactivation of GSK-3β leads to stabilization of Snail1 and β-catenin. Snail1 regulates EMT and fibroblast motility, whereas β-catenin directly governs the transcription of fibronectin and PAI-1. On the other hand, as a scaffolding protein, ILK assembles a multicomponent protein complex that contains ILK, PINCH, and parvin [5••, 14, 97]. These characteristics of ILK enable it to bridge the integrins, actin cytoskeleton, and other growth factor signal cascades [5••]. It is worth noting that the integrity and activity of this complex are necessary for ECM production, because either inhibition of ILK activity or destruction of this complex attenuates ECM expression, deposition, and extracellular assembly [14, 96, 98, 99]. Several critical fibrogenic signals induce ECM production by regulating this complex. For instance, TGF-β1 promotes induction of β1-integrin, ILK, and PINCH1, as well as their assembly. CTGF binds to both integrins and LDL receptor-related protein 1 (LRP1), resulting in the activation of ILK and production of matrix components. PDGF and FGF2 signaling are potentially linked to this complex via PINCH1. Tissue-type plasminogen activator promotes collagen I expression by triggering LRP1-mediated β1-integrin recruitment and ILK activation. Angiotensin II induces TGF-β1 expression, activates Smad signaling, and induces β1-integrin and ILK expression. Therefore, this molecular complex functions as a platform that integrates various fibrogenic signals and controls ECM production [5••].
Conclusion
Given the diversity of known fibrogenic mediators, the intracellular signal cascades involved might be immensely complex, with almost immeasurable crosstalk and feedback. In essence, the activation of major fibrogenic signaling mentioned earlier is characteristic of various fibrotic diseases. During the past decade, our understanding of the profibrotic mechanisms of these signaling pathways has evolved and improved considerably. It seems intriguing and meaningful to identify some of the converging molecular machineries that integrate various signal inputs and control the transcriptional program for myofibroblast activation and matrix production. Because several strategies to target key profibrotic mediators and signaling pathways to block the process of fibrosis are effective in different animal models and clinical trials [1••, 5••, 100••, 101••], therapeutic strategies based on these observations will bring new hope to clinicians and patients in the future.
References
Papers of particular importance, published from 2009 to present, have been highlighted as: • Of importance •• Of major importance
•• Wynn TA, Ramalingam TR (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18:1028–1040. The authors discuss how key components of the innate and adaptive immune response contribute to the pathogenesis of fibrosis
Lam AP, Gottardi CJ (2011) Beta-catenin signaling: a novel mediator of fibrosis and potential therapeutic target. Curr Opin Rheumatol 23:562–567
•• Zeisberg M, Kalluri R (2013) Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol 304:C216–C225. This review focuses on common and organ-specific pathways of tissue fibrosis
Biernacka A, Dobaczewski M, Frangogiannis NG (2011) TGF-beta signaling in fibrosis. Growth Factors 29:196–202
•• Liu Y (2011) Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol 7:684–696. Renal fibrogenesis is a dynamic process that can be divided into four overlapping phases—priming, activation, execution, and progression—in which many of these events occur concomitantly
• Kendall RT, Feghali-Bostwick CA (2014) Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol 5:123. The authors illustrate a central role for fibroblasts in the pathology of fibrosis, including ECM production in relation to fibrosis, and provide some examples of critical chemical signaling and myofibroblast differentiation
• LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R (2013) Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19:1047–1053. The authors use multiple genetic mouse models to track, fate map and ablate cells to determine the source and function of myofibroblasts in kidney fibrosis
•• Tsou PS, Haak AJ, Khanna D, Neubig RR (2014) Cellular mechanisms of tissue fibrosis. 8. Current and future drug targets in fibrosis: focus on Rho GTPase-regulated gene transcription. Am J Physiol Cell Physiol 307:C2–C13. The authors discuss the various molecular pathways involved in fibrosis and highlights the Rho GTPase signaling pathway and its downstream gene transcription output through MRTF/SRF as a convergence point for targeting this complex set of diseases
Kamato D, Burch ML, Piva TJ, Rezaei HB, Rostam MA, Xu S, Zheng W, Little PJ, Osman N (2013) Transforming growth factor-beta signalling: role and consequences of Smad linker region phosphorylation. Cell Signal 25:2017–2024
Massague J, Gomis RR (2006) The logic of TGFbeta signaling. FEBS Lett 580:2811–2820
Feng XH, Derynck R (2005) Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol 21:659–693
Phanish MK, Wahab NA, Colville-Nash P, Hendry BM, Dockrell ME (2006) The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem J 393:601–607
Li Y, Yang J, Dai C, Wu C, Liu Y (2003) Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J Clin Invest 112:503–516
Li Y, Dai C, Wu C, Liu Y (2007) PINCH-1 promotes tubular epithelial-to-mesenchymal transition by interacting with integrin-linked kinase. J Am Soc Nephrol 18:2534–2543
Li Y, Yang J, Luo JH, Dedhar S, Liu Y (2007) Tubular epithelial cell dedifferentiation is driven by the helix-loop-helix transcriptional inhibitor Id1. J Am Soc Nephrol 18:449–460
• Liu Y (2010) New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol 21:212–222. This review highlights the current understanding of EMT and its underlying mechanisms to stimulate further discussion on its role, both in the pathogenesis of renal interstitial fibrosis and in the onset of podocyte dysfunction, proteinuria, and glomerulosclerosis
Wang S, Wilkes MC, Leof EB, Hirschberg R (2010) Noncanonical TGF-beta pathways, mTORC1 and Abl, in renal interstitial fibrogenesis. Am J Physiol Renal Physiol 298:F142–F149
Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, Frangogiannis NG (2010) Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res 107:418–428
Flanders KC, Sullivan CD, Fujii M, Sowers A, Anzano MA, Arabshahi A, Major C, Deng C, Russo A, Mitchell JB, Roberts AB (2002) Mice lacking Smad3 are protected against cutaneous injury induced by ionizing radiation. Am J Pathol 160:1057–1068
Lakos G, Takagawa S, Chen SJ, Ferreira AM, Han G, Masuda K, Wang XJ, DiPietro LA, Varga J (2004) Targeted disruption of TGF-beta/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am J Pathol 165:203–217
Latella G, Vetuschi A, Sferra R, Catitti V, D’Angelo A, Zanninelli G, Flanders KC, Gaudio E (2009) Targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice. Liver Int 29:997–1009
Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A (2003) Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest 112:1486–1494
Zhao J, Shi W, Wang YL, Chen H, Bringas P Jr, Datto MB, Frederick JP, Wang XF, Warburton D (2002) Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol 282:L585–L593
Meng XM, Huang XR, Xiao J, Chung AC, Qin W, Chen HY, Lan HY (2012) Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int 81:266–279
Dooley S, Hamzavi J, Breitkopf K, Wiercinska E, Said HM, Lorenzen J, Ten Dijke P, Gressner AM (2003) Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 125:178–191
Nakao A, Fujii M, Matsumura R, Kumano K, Saito Y, Miyazono K, Iwamoto I (1999) Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J Clin Invest 104:5–11
Tan R, He W, Lin X, Kiss LP, Liu Y (2008) Smad ubiquitination regulatory factor-2 in the fibrotic kidney: regulation, target specificity, and functional implication. Am J Physiol Renal Physiol 294:F1076–F1083
Yang J, Zhang X, Li Y, Liu Y (2003) Downregulation of Smad transcriptional corepressors SnoN and Ski in the fibrotic kidney: an amplification mechanism for TGF-beta1 signaling. J Am Soc Nephrol 14:3167–3177
Pannu J, Nakerakanti S, Smith E, ten Dijke P, Trojanowska M (2007) Transforming growth factor-beta receptor type I-dependent fibrogenic gene program is mediated via activation of Smad1 and ERK1/2 pathways. J Biol Chem 282:10405–10413
Yu L, Hebert MC, Zhang YE (2002) TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J 21:3749–3759
Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM, Derynck R (2007) TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J 26:3957–3967
Leask A, Holmes A, Black CM, Abraham DJ (2003) Connective tissue growth factor gene regulation. Requirements for its induction by transforming growth factor-beta 2 in fibroblasts. J Biol Chem 278:13008–13015
Liu X, Sun SQ, Hassid A, Ostrom RS (2006) cAMP inhibits transforming growth factor-beta-stimulated collagen synthesis via inhibition of extracellular signal-regulated kinase 1/2 and Smad signaling in cardiac fibroblasts. Mol Pharmacol 70:1992–2003
Wilkes MC, Mitchell H, Penheiter SG, Dore JJ, Suzuki K, Edens M, Sharma DK, Pagano RE, Leof EB (2005) Transforming growth factor-beta activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res 65:10431–10440
Inoki K, Li Y, Xu T, Guan KL (2003) Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17:1829–1834
Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, Tavare S, Inoki K, Shimizu S, Narita M (2011) Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 332:966–970
Goorden SM, Hoogeveen-Westerveld M, Cheng C, van Woerden GM, Mozaffari M, Post L, Duckers HJ, Nellist M, Elgersma Y (2011) Rheb is essential for murine development. Mol Cell Biol 31:1672–1678
Jiang L, Xu L, Mao J, Li J, Fang L, Zhou Y, Liu W, He W, Zhao AZ, Yang J, Dai C (2013) Rheb/mTORC1 signaling promotes kidney fibroblast activation and fibrosis. J Am Soc Nephrol 24:1114–1126
McMahon G, Weir MR, Li XC, Mandelbrot DA (2011) The evolving role of mTOR inhibition in transplantation tolerance. J Am Soc Nephrol 22:408–415
Proud CG (2011) mTOR signalling in health and disease. Biochem Soc Trans 39:431–436
Wang S, Wilkes MC, Leof EB, Hirschberg R (2005) Imatinib mesylate blocks a non-Smad TGF-beta pathway and reduces renal fibrogenesis in vivo. FASEB J 19:1–11
Wilkes MC, Leof EB (2006) Transforming growth factor beta activation of c-Abl is independent of receptor internalization and regulated by phosphatidylinositol 3-kinase and PAK2 in mesenchymal cultures. J Biol Chem 281:27846–27854
• Bhattacharyya S, Fang F, Tourtellotte W, Varga J (2013) Egr-1: new conductor for the tissue repair orchestra directs harmony (regeneration) or cacophony (fibrosis). J Pathol 229:286–297. This observation highlights a previously unsuspected fundamental physiologic function for the Egr-1–Nab2 signalling axis in regulating fibrogenesis
Fang F, Shangguan AJ, Kelly K, Wei J, Gruner K, Ye B, Wang W, Bhattacharyya S, Hinchcliff ME, Tourtellotte WG, Varga J (2013) Early growth response 3 (Egr-3) is induced by transforming growth factor-beta and regulates fibrogenic responses. Am J Pathol 183:1197–1208
Bhattacharyya S, Ishida W, Wu M, Wilkes M, Mori Y, Hinchcliff M, Leof E, Varga J (2009) A non-Smad mechanism of fibroblast activation by transforming growth factor-beta via c-Abl and Egr-1: selective modulation by imatinib mesylate. Oncogene 28:1285–1297
Luchsinger LL, Patenaude CA, Smith BD, Layne MD (2011) Myocardin-related transcription factor-A complexes activate type I collagen expression in lung fibroblasts. J Biol Chem 286:44116–44125
Selvaraj A, Prywes R (2004) Expression profiling of serum inducible genes identifies a subset of SRF target genes that are MKL dependent. BMC Mol Biol 5:13
Akhmetshina A, Dees C, Pileckyte M, Szucs G, Spriewald BM, Zwerina J, Distler O, Schett G, Distler JH (2008) Rho-associated kinases are crucial for myofibroblast differentiation and production of extracellular matrix in scleroderma fibroblasts. Arthritis Rheum 58:2553–2564
Chai J, Norng M, Tarnawski AS, Chow J (2007) A critical role of serum response factor in myofibroblast differentiation during experimental oesophageal ulcer healing in rats. Gut 56:621–630
Sandbo N, Kregel S, Taurin S, Bhorade S, Dulin NO (2009) Critical role of serum response factor in pulmonary myofibroblast differentiation induced by TGF-beta. Am J Respir Cell Mol Biol 41:332–338
Huang H, He X (2008) Wnt/beta-catenin signaling: new (and old) players and new insights. Curr Opin Cell Biol 20:119–125
Carre AL, James AW, MacLeod L, Kong W, Kawai K, Longaker MT, Lorenz HP (2010) Interaction of wingless protein (Wnt), transforming growth factor-beta1, and hyaluronan production in fetal and postnatal fibroblasts. Plast Reconstr Surg 125:74–88
Poon R, Nik SA, Ahn J, Slade L, Alman BA (2009) Beta-catenin and transforming growth factor beta have distinct roles regulating fibroblast cell motility and the induction of collagen lattice contraction. BMC Cell Biol 10:38
He W, Kang YS, Dai C, Liu Y (2011) Blockade of Wnt/beta-catenin signaling by paricalcitol ameliorates proteinuria and kidney injury. J Am Soc Nephrol 22:90–103
• He W, Dai C, Li Y, Zeng G, Monga SP, Liu Y (2009) Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol 20:765–776. This study establishes a critical role for hyperactive Wnt/β-catenin signaling in the pathogenesis of renal fibrosis and presents a novel target for therapeutic intervention in fibrotic kidney diseases
He W, Tan R, Dai C, Li Y, Wang D, Hao S, Kahn M, Liu Y (2010) Plasminogen activator inhibitor-1 is a transcriptional target of the canonical pathway of Wnt/beta-catenin signaling. J Biol Chem 285:24665–24675
He W, Tan RJ, Li Y, Wang D, Nie J, Hou FF, Liu Y (2012) Matrix metalloproteinase-7 as a surrogate marker predicts renal Wnt/beta-catenin activity in CKD. J Am Soc Nephrol 23:294–304
Zhou D, Tan RJ, Zhou L, Li Y, Liu Y (2013) Kidney tubular beta-catenin signaling controls interstitial fibroblast fate via epithelial-mesenchymal communication. Sci Rep 3:1878
• Dai C, Stolz DB, Kiss LP, Monga SP, Holzman LB, Liu Y (2009) Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J Am Soc Nephrol 20:1997–2008. This study provides clear evidence of a pivotal role for hyperactive Wnt/β-catenin signaling in mediating podocytopathy
Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, MacDougald OA (2000) Inhibition of adipogenesis by Wnt signaling. Science 289:950–953
Cheng JH, She H, Han YP, Wang J, Xiong S, Asahina K, Tsukamoto H (2008) Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am J Physiol Gastrointest Liver Physiol 294:G39–G49
Gustafson B, Eliasson B, Smith U (2010) Thiazolidinediones increase the wingless-type MMTV integration site family (WNT) inhibitor Dickkopf-1 in adipocytes: a link with osteogenesis. Diabetologia 53:536–540
Cheon SS, Wei Q, Gurung A, Youn A, Bright T, Poon R, Whetstone H, Guha A, Alman BA (2006) Beta-catenin regulates wound size and mediates the effect of TGF-beta in cutaneous healing. FASEB J 20:692–701
Sato M (2006) Upregulation of the Wnt/beta-catenin pathway induced by transforming growth factor-beta in hypertrophic scars and keloids. Acta Derm Venereol 86:300–307
Labbe E, Letamendia A, Attisano L (2000) Association of Smads with lymphoid enhancer binding factor 1/T cell-specific factor mediates cooperative signaling by the transforming growth factor-beta and wnt pathways. Proc Natl Acad Sci USA 97:8358–8363
• Hao S, He W, Li Y, Ding H, Hou Y, Nie J, Hou FF, Kahn M, Liu Y (2011) Targeted inhibition of beta-catenin/CBP signaling ameliorates renal interstitial fibrosis. J Am Soc Nephrol 22:1642–1653. The therapeutic efficacy of ICG-001 in obstructive nephropathy is evaluated, and the results suggest that targeting β-catenin/CBP signaling by this small-molecule inhibitor is a novel and effective approach for treating fibrotic CKD
Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, Jernigan KK, Meyers KC, Hang BI, Waterson AG, Kim K, Melancon B, Ghidu VP, Sulikowski GA, LaFleur B, Salic A, Lee LA, Miller DM 3rd, Lee E (2010) Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat Chem Biol 6:829–836
Gonsalves FC, Klein K, Carson BB, Katz S, Ekas LA, Evans S, Nagourney R, Cardozo T, Brown AM, DasGupta R (2011) An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc Natl Acad Sci USA 108:5954–5963
• Zhou L, Li Y, Zhou D, Tan RJ, Liu Y (2013) Loss of Klotho contributes to kidney injury by derepression of Wnt/beta-catenin signaling. J Am Soc Nephrol 24:771–785. This study demonstrates that Klotho is an antagonist of endogenous Wnt/β-catenin activity
Briscoe J (2009) Making a grade: sonic hedgehog signalling and the control of neural cell fate. EMBO J 28:457–465
Jenkins D (2009) Hedgehog signalling: emerging evidence for non-canonical pathways. Cell Signal 21:1023–1034
• Ding H, Zhou D, Hao S, Zhou L, He W, Nie J, Hou FF, Liu Y (2012) Sonic hedgehog signaling mediates epithelial-mesenchymal communication and promotes renal fibrosis. J Am Soc Nephrol 23:801–813. This study demonstrates that Shh/Gli1 signaling plays a critical role in promoting fibroblast activation, ECM production, and the development of renal interstitial fibrosis
Nagoshi S (2013) Osteopontin: versatile modulator of liver diseases. Hepatol Res 44:22–30
Omenetti A, Porrello A, Jung Y, Yang L, Popov Y, Choi SS, Witek RP, Alpini G, Venter J, Vandongen HM, Syn WK, Baroni GS, Benedetti A, Schuppan D, Diehl AM (2008) Hedgehog signaling regulates epithelial-mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest 118:3331–3342
Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL (2002) Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 129:4753–4761
Katoh Y, Katoh M (2009) Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med 9:873–886
• Choi SS, Omenetti A, Syn WK, Diehl AM (2011) The role of Hedgehog signaling in fibrogenic liver repair. Int J Biochem Cell Biol 43:238–244. This study suggests that injury-related activation of the hedgehog pathway modulates several important aspects of repair, containing hepatic accumulation of myofibroblasts and liver fibrosis
Stewart GA, Hoyne GF, Ahmad SA, Jarman E, Wallace WA, Harrison DJ, Haslett C, Lamb JR, Howie SE (2003) Expression of the developmental sonic hedgehog (Shh) signalling pathway is up-regulated in chronic lung fibrosis and the Shh receptor patched 1 is present in circulating T lymphocytes. J Pathol 199:488–495
Greenbaum LE (2008) Hedgehog signaling in biliary fibrosis. J Clin Invest 118:3263–3265
Katoh Y, Katoh M (2008) Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (review). Int J Mol Med 22:271–275
Sharma S, Sirin Y, Susztak K (2011) The story of Notch and chronic kidney disease. Curr Opin Nephrol Hypertens 20:56–61
Bray SJ (2006) Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 7:678–689
•• Kavian N, Servettaz A, Weill B, Batteux F (2012) New insights into the mechanism of notch signalling in fibrosis. Open Rheumatol J 6:96–102. This review summarizes the structure and activation of the Notch family members, and focuses on recent findings regarding the role of Notch in organ fibrogenesis, in humans and in animal models
Fortini ME (2009) Notch signaling: the core pathway and its posttranslational regulation. Dev Cell 16:633–647
• Dees C, Tomcik M, Zerr P, Akhmetshina A, Horn A, Palumbo K, Beyer C, Zwerina J, Distler O, Schett G, Distler JH (2011) Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Ann Rheum Dis 70:1304–1310. Notch signaling is activated in SSc and plays an important role in fibroblast activation and collagen release
Kavian N, Servettaz A, Mongaret C, Wang A, Nicco C, Chereau C, Grange P, Vuiblet V, Birembaut P, Diebold MD, Weill B, Dupin N, Batteux F (2010) Targeting ADAM-17/notch signaling abrogates the development of systemic sclerosis in a murine model. Arthritis Rheum 62:3477–3487
Plantier L, Crestani B, Wert SE, Dehoux M, Zweytick B, Guenther A, Whitsett JA (2011) Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 66:651–657
Niranjan T, Bielesz B, Gruenwald A, Ponda MP, Kopp JB, Thomas DB, Susztak K (2008) The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med 14:290–298
Murea M, Park JK, Sharma S, Kato H, Gruenwald A, Niranjan T, Si H, Thomas DB, Pullman JM, Melamed ML, Susztak K (2010) Expression of Notch pathway proteins correlates with albuminuria, glomerulosclerosis, and renal function. Kidney Int 78:514–522
Dees C, Zerr P, Tomcik M, Beyer C, Horn A, Akhmetshina A, Palumbo K, Reich N, Zwerina J, Sticherling M, Mattson MP, Distler O, Schett G, Distler JH (2011) Inhibition of Notch signaling prevents experimental fibrosis and induces regression of established fibrosis. Arthritis Rheum 63:1396–1404
Liu T, Hu B, Choi YY, Chung M, Ullenbruch M, Yu H, Lowe JB, Phan SH (2009) Notch1 signaling in FIZZ1 induction of myofibroblast differentiation. Am J Pathol 174:1745–1755
Matsuno Y, Coelho AL, Jarai G, Westwick J, Hogaboam CM (2012) Notch signaling mediates TGF-beta1-induced epithelial-mesenchymal transition through the induction of Snai1. Int J Biochem Cell Biol 44:776–789
Saad S, Stanners SR, Yong R, Tang O, Pollock CA (2010) Notch mediated epithelial to mesenchymal transformation is associated with increased expression of the Snail transcription factor. Int J Biochem Cell Biol 42:1115–1122
• Bielesz B, Sirin Y, Si H, Niranjan T, Gruenwald A, Ahn S, Kato H, Pullman J, Gessler M, Haase VH, Susztak K (2010) Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest 120:4040–4054. This study suggests that Notch activation is associated with EMT of cultured tubular cells and is sufficient to induce severe tubulointerstitial fibrosis in vivo
Waters AM, Wu MY, Onay T, Scutaru J, Liu J, Lobe CG, Quaggin SE, Piscione TD (2008) Ectopic notch activation in developing podocytes causes glomerulosclerosis. J Am Soc Nephrol 19:1139–1157
Li Y, Tan X, Dai C, Stolz DB, Wang D, Liu Y (2009) Inhibition of integrin-linked kinase attenuates renal interstitial fibrosis. J Am Soc Nephrol 20:1907–1918
Legate KR, Montanez E, Kudlacek O, Fassler R (2006) ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol 7:20–31
Margadant C, Sonnenberg A (2010) Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep 11:97–105
Yeh YC, Wei WC, Wang YK, Lin SC, Sung JM, Tang MJ (2010) Transforming growth factor-{beta}1 induces Smad3-dependent {beta}1 integrin gene expression in epithelial-to-mesenchymal transition during chronic tubulointerstitial fibrosis. Am J Pathol 177:1743–1754
•• Decleves AE, Sharma K (2014) Novel targets of antifibrotic and anti-inflammatory treatment in CKD. Nat Rev Nephrol 10:257–267. This review highlights new insights into the mechanisms that contribute to CKD and approaches that might facilitate the development of disease-arresting therapies for CKD
•• Tampe D, Zeisberg M (2014) Potential approaches to reverse or repair renal fibrosis. Nat Rev Nephrol 10:226–237. This review summarizes strategies targeting various components of the fibrotic pathway, from signaling molecules including TGF-β, PI3K, and chemokines to microRNAs
Author information
Authors and Affiliations
Corresponding authors
Additional information
This article is part of the Topical Collection on Matrix Pathobiology.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
He, W., Dai, C. Key Fibrogenic Signaling. Curr Pathobiol Rep 3, 183–192 (2015). https://doi.org/10.1007/s40139-015-0077-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-015-0077-z