
Abstract
Phytochemical investigation of the rhizomes of Paris polyphylla var. stenophylla led to the isolation of two new highly oxygenated spirostanol saponins, named paristenosides A (1) and B (2), together with seven known compounds. Their structures were established mainly on the base of NMR spectroscopic techniques and mass spectrometry, as well as chemical methods. In addition, the cytotoxicity of the two new saponins was tested.
Graphical Abstract
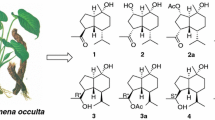
Two new highly oxygenated spirostanol saponins, paristenosides A (1) and B (2), were isolated from the rhizomes of Paris polyphylla var. stenophylla. Their structures were established mainly based on NMR spectroscopic techniques and mass spectrometry, as well as chemical methods.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The genus Paris (Liliaceae) comprises about 24 species, which grows as perennial rhizomatous herbs in the Eurasian continent from temperate zone to the tropics [1]. Rhizoma Paridis (‘Chonglou’ in Chinese), the dried rhizomes of P. polyphylla var. yunnanensis and P. polyphylla var. chinensis, have been recorded in the Pharmacopoeia of the People’s Republic of China as a traditional Chinese medicine for the treatment of traumatic injuries, insect and snake bites, and sore throat [2]. Plants of the genus Paris are known as a rich source of steroidal saponins, which have attracted great interests for their structural diversity and various bioactivities such as anti-tumor [3, 4], hemostatic [5], and antifungal [6] effects. In recent years, our group focuses the research on the chemical constituents, bioactivity, and sustainable utilization of resources of the genus [7–11].
P. polyphylla var. stenophylla Franch. is one of the variations of P. polyphylla and has been used as a herbal medicine to treat skin furuncle and skin tinea, stop bleeding, and move clean blood and bad blood out by the Yi nationality of Liangshan in Sichuan Province [12]. Due to having certain resources, its rhizomes have been as the main steam commodities of Rhizoma Paridis. However, there are a few reports about the research of its material basis [13]. In order to clarify its chemical quality, we performed a phytochemical investigation of the rhizomes of P. polyphylla var. stenophylla. As a result, two new polyhydroxylated steroidal saponis, named paristenosides A (1) and B (2), were isolated from the title species. Meanwhile, seven known compounds were obtained and identified as paris saponin H (3) [14], Tg (4) [15], Pb (5) [16], Th (6) [17], Methyl-Th (7) [18], parispseudoside A (8) [19] and β-ecdysone (9) [16]. In the current paper, we report the isolation, structural elucidation, and cytotoxicity of the two new compounds (Fig. 1).

Chemical structures of paristenosides A (1) and B (2)
2 Results and Discussion
Paristenoside A (1) was isolated as a white amorphous powder. Its molecular formula, C50H80O25, was assigned by the positive-ion HR-ESI-MS (m/z 1103.4881 [M + Na]+, calcd for 1103.4881) and 13C NMR data (Tables 1, 2). The IR spectrum displayed absorption bands ascribable to hydroxy (3426 cm−1) and olefinic bond (1631 cm−1) functionalities. The 13C NMR spectrum showed signals for a ketal carbon at δ C 111.5, a trisubstituted olefin carbons at δ C 139.4 (s) and 124.6 (d), and three methyls at δ C 17.0, 15.0, and 13.1, which were characteristic of a ∆5,6-spirostanol skeleton as an aglycone [7]. Its 1H NMR spectrum showed two singlet signals for two tertiary methyls at δ H 1.38 and 1.13, one doublet signal for a secondary methyl at δ H 1.03 (d, J = 6.8 Hz), an olefinic proton at δ H 5.55 (br d, J = 5.5 Hz), as well as four anomeric protons at δ H 6.41 (br s), 5.24 (d, J = 7.8 Hz), 4.91 (d, J = 7.6 Hz), and 4.78 (d, J = 7.7 Hz), which suggested the presence of four sugar units. In addition, one methyl at δ H 1.71 (d, J = 6.0 Hz) was the methyl of one 6-deoxyhexopyranose residue. Comparison of the 1H and 13C NMR spectra of 1 with those of padelaoside A [20] revealed that 1 possessed the same polyhydroxylated aglycone [(23S,24S,25S)-spirost-5-ene-1β,3β,21,23,24-pentol] as that of padelaoside A, which could be further verified by detailed 2D NMR data analysis (Figs. 2, 3). Furthermore, the result of acid hydrolysis and GC analysis of 1 with its corresponding trimethylsilated l-cysteine adducts afforded d-glucose, d-galactose, d-xylose, and l-rhamnose. The large coupling constants (3 J 1,2 > 7 Hz) were consistent with β-configuration for the glucose, galactose, and xylose, while the α-configuration for the rhamnose was deduced by comparing its 13C NMR spectroscopic data for C-3″ (δ C 72.4) and C-5″ (δ C 69.5) with those reported in the literature [21]. The sequence and binding sites of the sugar units were determined by following HMBC correlations: δ H 6.41 (H-1″ of Rha) with δ C 76.3 (C-2′ of Glc), δ H 4.91 (H-1″′ of Xyl) with δ C 88.3 (C-3′ of Glc), δ H 4.78 (H-1′ of Glc) with δ C 83.9 (C-1 of the aglycone), and δ H 5.24 (H-1″″ of Gal) with δ C 81.8 (C-24 of the aglycone). Assignment of all proton and carbon resonances was achieved by interpretation of 1H-1H COSY, HSQC, and HMBC spectra. Based on the above information, the structure of paristenoside A (1) was elucidated as 24-O-β-d-galactopyranosyl-(23S,24S,25S)-spirost-5-ene-1β,3β,21,23,24-pentol-1-O-α-l-rhamnopyranosyl-(1 → 2)-[β-d-xylopyranosyl-(1 → 3)]-β-d-glucopyranoside.

1H-1H COSY and Key HMBC correlations of paristenoside A (1)

Key ROESY correlations for the aglycone moiety of paristenoside A (1)
Paristenoside B (2) was obtained as a white amorphous powder with a molecular formula of C56H88O30, which was deduced from the positive HR-ESI-MS (m/z 1258.5689 [M + NH4]+, calcd for 1258.5699) and 13C NMR data (Tables 1, 2). The 1H and 13C NMR data suggested that compound 2 was also a highly oxygenated spirostanol glycoside. A comparison of the NMR spectra of 2 with those of parisyunnanoside I [22] disclosed that they both had the same aglycone and sugar chain linkages at C-1 and C-21, except for the replacement of the fucose by a galactose at C-24 in the latter. This was further supported by the HMBC correlation between δ H 5.82 (H-1″″′ of Gal) with δ C 82.0 (C-24 of the aglycone). The other parts of 2 were identical to those of parisyunnanoside I as confirmed by detailed analysis of its 2D NMR experiments. Therefore, the structure of paristenoside B (2) was established as 21-O-β-d-galactopyranosyl-24-O-β-d-galactopyranosyl-(23S,24S)-spirost-5,25(27)-diene-1β,3β,21,23,24-pentol-1-O-α-l-rhamnopyranosyl-(1 → 2)-[β-d-xylopyranosyl-(1 → 3)]-β-d-glucopyranoside.
The phytochemical investigation of the rhizomes of P. polyphylla var. stenophylla led to the identification of eight steroidal saponins and one ecdysone, including two new highly oxygenated spirostanol glycosides. Compounds 1 and 2 are a further addition to the diverse and complex steroidal saponins. Our results and the literature [13] suggested that P. polyphylla var. stenophylla contained the same bioactive components as those in the legally original plants of Rhizoma Paridis. However, whether the rhizomes of P. polyphylla var. stenophylla can be used as the original plant of Rhizoma Paridis are still valuable for us to further research. Considering cytotoxicity of steroidal glycosides, paristenosides A (1) and B (2) were evaluated for their cytotoxicity against two human cancer cell lines (HEK293 and HepG2) by using MTT method. Unfortunately, none of them showed significant activity at the concentration of 20 μM.
3 Experimental Section
3.1 General Experimental Procedures
Optical rotations were recorded on a JASCO P-1020 digital polarimeter. IR spectra were obtained on Bruker Tensor-27 infrared spectrophotometer with KBr pellets. ESI-MS spectra were recorded on a Bruker HCT/Esquire spectrometer. HR-ESI-MS were obtained on an Agilent 6200 Q-TOF MS system. 1D and 2D NMR spectra were obtained on a Bruker Avance III 600 MHz spectrometer in C5D5N; chemical shifts are given in δ (ppm), and coupling constants are reported in Hz. Column chromatography (CC) was performed on silica gel (200–300 mesh, Qingdao Puke Chemical Co. Ltd., China) and Rp-18 gel (40–63 μm, Merck, Darmstadt, Germany). GC analysis was performed on a HP5890 gas chromatograph equipped with an H2 flame ionization detector. Semi-preparative HPLC was run on Agilent 1100 liquid chromatograph equipped with a Zorbax SB-C18 column (5 μm, 25 cm × 9.4 mm) and a diode array detector (DAD). TLC was performed on HSGF254 (0.2 mm, Qingdao Puke Chemical Co. Ltd., China). Fractions were monitored by TLC and spots were visualized by heating silica gel plates sprayed with 10 % H2SO4 in EtOH.
3.2 Plant Material
The rhizomes of P. polyphylla var. stenophylla were collected in Octobor 2014 from Zhaoyang District, Zhaotong, Yunnan province, China. The plant material was authenticated by Dr. Yun-Heng Ji, Kunming Institute of Botany, Chinese Academy of Sciences. A voucher specimen (No. HY0023) was deposited at the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.
3.3 Extraction and Isolation
The air-dried and powdered rhizomes of P. polyphylla var. stenophylla (370 g) were extracted three times with 75 % EtOH under reflux and then the solvent was evaporated under reduced pressure to afford a crude extract (10 g). The crude extract was subjected to a silica gel column and eluted with a CHCl3–MeOH (15:1 → 1:2, v/v) gradient solvent system to yield seven fractions (A–G). Fr. C (1.1 g) was chromatographed through a silica gel column (CHCl3–MeOH, 15:1 → 1:1) and purified by semi-prep. HPLC (CH3CN-H2O, 40:60, v/v) to yield 3 (35 mg). Fr. E (1.5 g) was separated by an Rp-18 column (CH3OH–H2O, 50:50 → 70:30, v/v) to get 4 (9 mg) and 5 (30 mg). Fr. F (1.5 g) was separated by a silica gel column, eluting with CHCl3–MeOH (10:1 → 1:1) and was further purified by semi-prep. HPLC with the mobile phase of CH3CN–H2O (30:70, v/v) to obtain 1 (9 mg), 2 (5 mg), 6 (43 mg), 7 (50 mg), 8 (5 mg), and 9 (9 mg).
3.3.1 Paristenoside A (1)
White amorphous powder; \(\left[ \alpha \right]_{\text{D}}^{23}\) −76.8 (c = 0.05, MeOH); IR (KBr) v max: 3426, 1631, 1383, 1051 cm−1; 1H (600 MHz, C5D5N) and 13C NMR (150 MHz, C5D5N) data, see Tables 1 and 2; ESIMS m/z 1103 [M + Na]+; HRESIMS m/z 1103.4881 [M + Na]+ (cacld for C50H80O25Na, 1103.4881).
3.3.2 Paristenoside B (2)
White amorphous powder; \(\left[ \alpha \right]_{\text{D}}^{23}\) −55.8 (c = 0.09, MeOH); IR (KBr) v max: 3424, 1635, 1382, 1045 cm−1; 1H (600 MHz, C5D5N) and 13C NMR (150 MHz, C5D5N) data, see Tables 1 and 2; ESIMS m/z 1263 [M + Na]+; HRESIMS m/z 1258.5689 [M + NH4]+ (cacld for C56H92O30N, 1258.5699).
3.4 Sugar Analysis of 1 and 2
Compounds 1 and 2 (each 2 mg) were refluxed with 2 M HCl (1,4-dioxane/H2O 1:1, 2 mL) on water bath for 2 h. The mixture was concentrated in a vacuum, and the residue was suspended in H2O and then extracted with CHCl3 (5 mL × 3). The aqueous layer was neutralized with MeOH and then dried to give a mixture of sugars. Each mixture was dissolved in anhydrous pyridine (1 mL) and reacted with l-cysteine methyl ester hydrochloride (1.5 mg) stirred at 60 °C for 1.5 h. The trimethylsilylimidazole (1.0 mL) was subsequently added to the reaction mixtures, and they were kept at 60 °C for 30 min. The mixture (4 μL) was finally analyzed by an HP 5890 gas chromatograph with a 30QC2/AC-5 quartz capillary column (30 mm × 0.32 mm × 0.25 μm), respectively, under the following conditions: H2 flame ionization detector; carrier gas: N2 (1 mL/min); column temperature program: 180–280 °C with the rate of 3 °C/min; injector temperature: 250 °C; split ratio 1:50. Peaks of the hydrolysate were detected by comparison with retention times of authentic samples of d-glucose, d-galactose, d-xylose, and l-rhamnose after the aforementioned treatment in pyridine. The absolute configurations of the sugar moieties were identified as d-glucose (t R = 19.0 min), d-galactose (t R = 23.0 min), d-xylose (t R = 18.3 min), and l-rhamnose (t R = 15.4 min), respectively.
3.5 Cytotoxic Assay
Cytotoxic evaluations were performed for two human cancer cell lines (HEK293 and HepG2) using the MTT method described in the literature elsewhere [10]. (−)-OddC was used as the positive control and showed IC50 values for the two cancer cell lines with IC50 values of 0.30 and 0.17 µM, respectively. The experiments were conducted for three independent replicates, and IC50 > 20 μM was considered to be inactive.
References
H. Li, The Genus Paris (Science Press, Beijing, 1998), pp. 12–16
Chinese Pharmacopoeia Commission, Pharmacopoeia of the People’s Republic of China, vol. 1 (Chinese Medical Science and Technology Press, Beijing, 2015), pp. 243–244
Z.X. Cheng, B.R. Liu, X.P. Qian, Y.T. Ding, W.J. Hu, J. Sun, L.X. Yu, J. Ethnopharmacol. 120, 129–137 (2008)
S.L. Man, W.Y. Gao, Y.J. Zhang, L.L. Yan, C.Y. Ma, C.X. Liu, L.Q. Huang, Steroids 74, 1051–1056 (2009)
Y.L. Fu, Z.Y. Yu, X.M. Tang, Y. Zhao, X.L. Yuan, S. Wang, B.P. Ma, Y.W. Cong, J. Thromb. Haemost. 6, 524–533 (2007)
D.W. Deng, D.R. Lauren, J.M. Cooney, D.J. Jensen, K.V. Wurms, J.E. Upritchard, R.D. Cannon, M.Z. Wang, M.Z. Li, Planta Med. 74, 1397–1402 (2008)
X.J. Qin, D.J. Sun, W. Ni, C.X. Chen, Y. Hua, L. He, H.Y. Liu, Steroids 77, 1242–1248 (2012)
X.J. Qin, C.X. Chen, W. Ni, H. Yan, H.Y. Liu, Fitoterapia 84, 248–251 (2013)
C.L. Sun, W. Ni, H. Yan, Z.H. Liu, L. Yang, Y.A. Si, Y. Hua, C.X. Chen, L. He, J.H. Zhao, H.Y. Liu, Steroids 92, 90–95 (2014)
Y.S. Wen, W. Ni, X.J. Qin, H. Yan, C.X. Chen, Y. Hua, Y.C. Cheng, L. He, H.Y. Liu, Phytochem. Lett. 12, 31–34 (2015)
X.J. Qin, M.Y. Yu, W. Ni, H. Yan, C.X. Chen, Y.C. Cheng, L. He, H.Y. Liu, Phytochemistry 121, 20–29 (2016)
H.X. Yin, F.Y. Wen, H. Zhang, World Sci Technol. Mod. Trad. Chin. Med. Mat. Med. 16, 177–180 (2014)
H.X. Yin, D. Xue, N. Bai, C. Chen, Y. Chen, H. Zhang, J. Sichuan Univ. (Med. Sci. Edi.) 39, 485–488 (2008)
C.X. Chen, J. Zhou, Y.T. Zhang, C.K. Gao, Acta Bot. Yunnan. 5, 219–223 (1983)
C.X. Chen, Y.T. Zhang, J. Zhou, Acta Bot. Yunnan. 5, 91–97 (1983)
C.X. Chen, J. Zhou, Acta Bot. Yunnan. 3, 89–93 (1981)
T. Nohara, K. Migahara, T. Kawasaki, Chem. Pharm. Bull. 23, 872–885 (1975)
K. Nakano, K. Murakami, T. Nohara, T. Tomimatsu, T. Kawasaki, Chem. Pharm. Bull. 29, 1445–1451 (1981)
C.M. Xiao, J. Huang, X.M. Zhong, X.Y. Tan, P.C. Deng, Helv. Chim. Acta 92, 2587–2595 (2009)
T. Zhang, H. Liu, X.T. Liu, X.Q. Chen, Q. Wang, Steroids 74, 809–813 (2009)
R. Kasai, M. Okihara, J. Asakawa, K. Mizutani, O. Tanaka, Tetrahedron 35, 1427–1432 (1979)
L.P. Kang, Y.X. Liu, T. Eichhorn, E. Dapat, H.S. Yu, Y. Zhao, C.Q. Xiong, C. Liu, T. Efferth, B.P. Ma, J. Nat. Prod. 75, 1201–1205 (2012)
Acknowledgments
This work is funded by the National Natural Science Funding of China (Grand No. 31570363), the Key Research Program of the Chinese Academy of Sciences (Grand No. KSZD-EW-Z-004-03-4), and the Natural Science Foundation of Yunnan Province (Grand No. 2015FA031).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no conflict of interest.
Additional information
Ling-Yu Jin and Ting-Xiang Lu contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jin, LY., Lu, TX., Qin, XJ. et al. Two New Highly Oxygenated Spirostanol Saponins from Paris polyphylla var. stenophylla . Nat. Prod. Bioprospect. 6, 205–210 (2016). https://doi.org/10.1007/s13659-016-0102-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-016-0102-x