
Abstract
Several advances in fluid and tissue-based biomarkers for use in Parkinson’s disease (PD) and other synucleinopathies have been made in the last several years. While work continues on species of alpha-synuclein (aSyn) and other proteins which can be measured from spinal fluid and plasma samples, immunohistochemistry and immunofluorescence from peripheral tissue biopsies and alpha-synuclein seeding amplification assays (aSyn-SAA: including real-time quaking induced conversion (RT-QuIC) and protein misfolding cyclic amplification (PMCA)) now offer a crucial advancement in their ability to identify aSyn species in PD patients in a categorical fashion (i.e., of aSyn + vs aSyn −); to augment clinical diagnosis however, aSyn-specific assays that have quantitative relevance to pathological burden remain an unmet need. Alzheimer’s disease (AD) co-pathology is commonly found postmortem in PD, especially in those who develop dementia, and dementia with Lewy bodies (DLB). Biofluid biomarkers for tau and amyloid beta species can detect AD co-pathology in PD and DLB, which does have relevance for prognosis, but further work is needed to understand the interplay of aSyn tau, amyloid beta, and other pathological changes to generate comprehensive biomarker profiles for patients in a manner translatable to clinical trial design and individualized therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is pathologically characterized by inclusions of alpha-synuclein (aSyn) that compose Lewy bodies and Lewy neurites [1]. These inclusions are found in fairly stereotyped patterns that progress from brainstem nuclei, to limbic regions and lastly to neocortical areas [2]. At this time, a definitive diagnosis of PD can only be rendered after neuropathological assessments are performed, with levels of clinically established and clinically probable certainties being attainable during life [3]. Clinical diagnostic accuracy for PD has varied among studies over the last several decades and ranges from 50% to greater than 90% [4,5,6,7,8,9]. Factors that tend to relate to lower diagnostic accuracy are an older age at onset and a shorter degree of disease duration at time of assessment or a lower amount of clinical follow-up time [4, 9]. Thus, the diagnostic standard remains postmortem neuropathological diagnosis until a method to reliably detect aSyn in vivo is developed. Most biomarker studies rely on patients who have been clinically diagnosed with PD who do not go on to have autopsy validation. While this creates some uncertainty regarding the accuracy of diagnosis and this may be problematic in developing novel biomarkers, the current clinical criteria for PD are felt to have high specificity [9]. Furthermore, because there is no currently accepted quantitative aSyn biomarker, studies of these candidate biomarkers are compared to clinical metrics like motor severity or cognition which can be influenced by many factors and are fundamentally indirect measures of disease activity. While aSyn-specific biomarkers remain a critical unmet need for the field, they are especially needed for application in early disease when clinical diagnostic accuracy is at its lowest and also when disease-modifying interventions may have the greater utility.
Over the last decade, there has been considerable advancements in fluid and tissue-based assays in PD. Early work focused on CSF aSyn species including total aSyn, phosphorylated aSyn, and oligomeric aSyn species using immunoassays [10,11,12]. Plasma aSyn assays are under development as well [13, 14]. More recently, aSyn deposits have been noted in a variety of peripheral tissues of PD patients, including skin, submandibular glad, colon, and nasal mucosa and these observations have led to the development of methods to detect these deposits through immunohistochemistry or immunofluorescence methods [15, 16]. Additionally, the observations that pathologically misfolded aSyn species may induce sequential templating of normal monomeric aSyn in a prion-like fashion, has led to the development of aSyn-seeding amplification assays (aSyn-SAAs), which use these properties to identify patients who harbor pathogenic aSyn seeds in spinal fluid and peripheral tissues [17,18,19,20,21]. While some of these assays are still under development in the research setting, others are reaching levels of standardization and interlaboratory variability rapidly approaching possible acceptable levels for clinical use.
aSyn aggregates in Lewy bodies and Lewy neurites are the primary neuropathology and gold-standard for diagnosis of PD and their burden is roughly related to severity of disease and certain disease features like dementia [22,23,24,25,26]. However, multiple biological factors, even sex, can influence phenotypic expression of pathological burden [27, 28]. Additionally, it is exceedingly common in autopsy studies that other co-pathologies aside from aSyn are found; approximately 35–50% of PD patients with dementia with have moderate to high levels of AD neuropathologic change [29,30,31,32,33]. This number is considerably higher in DLB, where rates of moderate or severe AD co-pathology can reach 70% or greater [34, 35]. The presence of the AD co-pathology is well described to be related to older age of onset, faster time to dementia, decreased overall survival, greater likelihood of an akinetic-rigid motor phenotype, and specific cognitive features [31, 32, 36,37,38,39,40,41,42]. AD biomarkers are established using a framework for biological classification of AD based on positivity for amyloid-beta, tau, and neurodegeneration (A/T/N) [43]. A similar approach is understudied in PD and related synucleinopathies, but early work suggests that CSF AD biomarkers can be used to detect the presence of these AD co-pathologies in PD and predict cognitive and overall prognostic outcomes which could have utility in clinical care and trial design [44,45,46,47,48,49,50,51]. However, the application of AD CSF biomarkers in clinical care for PD is unclear, as biological factors related to aSyn may influence AD biomarkers in a manner independent from AD pathology, necessitating PD-specific diagnostic cut-points, but further studies with autopsy-confirmation are needed [46, 47, 52]. The presence of AD co-pathology is not universal, nor is it exclusively linked to worse prognosis; there are many cases of “pure” aSyn cases with fulminant presentations and precipitous clinical courses [38, 53, 54]. Finally, neuropathology of aging is complex and often includes additional mixed pathologies in PD and related disorders such as cerebrovascular disease, TDP-43 inclusions, age-related glial tau inclusions (ARTAG) and others which cannot be reliable measured through fluid assays at this time [55,56,57]. Here, we review the state of the science for aSyn related biomarkers in CSF, plasma, biopsy detection from peripheral tissues, and aSyn seeding amplification assays for PD with a focus on autopsy-confirmation which is a critical to help translate biomarker research into clinical practice.
CSF aSyn Assays
In vivo CSF aSyn levels have been studied extensively, but due to conflicting results and significant overlap in values with healthy controls and other disease states their current utility in PD is limited until further refinement occurs [11]. There are several technical considerations which make measuring CSF aSyn challenging. First, there is relatively little aSyn that is present in spinal fluid, on the orders ng/ml. There is high amounts in peripheral blood in red blood cells which can easily contaminate specimens [58]. In many studies, samples have had to be discarded if there are significant degrees of hemoglobin contamination in CSF samples [59, 60]. Polypropylene collection tubes are recommended for use in some assays to prevent loss of CSF aSyn, and there is variability even amongst different tube vendors in their effects on aSyn levels [61]. Time to storage, number of freeze–thaw cycles and other pre-analytic variables affects measured CSF aSyn levels [61, 62]. Most of these studies have used ELISA assays or the bead-based Luminex xMap platform which are calibrated against measurements made with recombinant aSyn (Table 1). ELISA assays may use different aSyn antibodies for capture and detection which may account for some degree of variability observed. For ELISA protocols, there tends to be relatively high intra-assay precision on repeated measurements (< 10%CV); however, there is less consistency between assays [62, 63]. Using these methods, several studies report that total CSF aSyn levels are lower in PD patients than normal controls or patients with other non-degenerative neurological diseases [45, 64,65,66]. In two meta-analyses, the sensitivity and specificity for distinguishing PD samples from normal controls were 72% and 88%, and 65% and 40% with modest positive predictive values and area under the curve values [67, 68]. The majority of these studies have been cross-sectional studies performed in early- to mid-stage, clinically defined PD (Table 1), although lower levels of CSF total aSyn is noted in prodromal PD as well in some patients with RBD and hyposmia [69]. While statistically significant group differences are observed, there is substantial overlap in individual values of PD patients and healthy controls, which limits the use of CSF total aSyn currently as a diagnostic tool. Furthermore, lower average levels of CSF total aSyn compared to controls are also found in DLB, progressive supranuclear palsy, and multiple systems atrophy as well which would make current assays of limited value in the differential diagnosis of parkinsonism [68, 70,71,72]. As PD progresses, aSyn levels in some patients rise and there is some correlation between these higher CSF aSyn levels and cognitive and motor dysfunction [46, 73,74,75,76,77], but this finding is not universal [60]. However, higher levels of CSF aSyn are also noted in Alzheimer’s disease, other neurodegenerative disease, such as Creutzfeldt Jakob disease [78,79,80,81,82], suggesting levels of this analyte degenerating synapses/neurons. Therefore, it is difficult to disentangle which processes may be PD-specific, and which may be due to non-specific neurodegeneration. As far as the association of CSF total aSyn and specific disease features, some studies have documented lower amounts of CSF total aSyn in non-tremor-dominant phenotypes than tremor dominant PD cases [45, 46, 60]. PD patients with RBD have higher CSF total aSyn than PD patients without RBD [83]. There are conflicting reports about whether lower [45] or higher [73, 75] CSF total aSyn relates to worse cognitive outcomes in PD. In summation, on average, CSF total aSyn is lower in PD than healthy controls, especially early in the disease course but there is significant overlap of CSF total aSyn levels with healthy controls and other neurological diseases and therefore current assays cannot acceptably function as a single test to aid in the diagnosis of PD. Ratios of CSF total aSyn and other analytes discussed below may offer some improvement in diagnostic utility [64, 80], but more work, especially longitudinal measurements are needed to clarify the utility of this biomarker in PD in relations to other conditions..
Other CSF aSyn species that have been studied in PD include phosphorylated and oligomeric forms of aSyn [75, 84]. Phosphorylated aSyn assays have focused on the pSer129 epitope which is a well described post-translational modification acidic tail near the C-terminal end of the protein in Lewy pathology [85]. In some studies, there is a U-shaped associated with disease severity with lower phosphorylated aSyn being associated with worse initial clinical presentations but later, higher levels being associated with worse motor and cognitive function [84, 86, 87].
The precise species of aSyn which contributes to neuronal dysfunction and neurodegeneration is not entirely clear, but the formation smaller oligomeric aggregations of aSyn may confer damage to synapses [88]. Higher levels of oligomeric aSyn from CSF samples compared to AD and healthy controls has been noted in PD and DLB [89]. Thus, there may be PD-specificity for oligomeric aSyn species detected in CSF as opposed to total aSyn measurements summarized above. Early studies suggest correlations of levels with CSF oligomeric aSyn with motor symptoms and may be useful as part of an oligomeric/total CSF aSyn ratio but replication in other laboratory settings is needed [90,91,92,93]. Early work in these studies use immunoassays which relay on epitope-specificity of the capture antibody used to detect oligomeric confirmations of aSyn. More recently, a study using a newer technique of single molecule counting technology was unable to detect pSer129 aSyn in CSF samples from PD patients, raising questions about epitopes specificity of immunoassays for different forms of aSyn in CSF [94]. Thus, these assays remain exploratory for PD research at the moment until further validation is performed.
Plasma aSyn Measurements
Plasma aSyn measurements in PD have yielded differing results, with most studies reporting higher plasma total aSyn levels than healthy controls [13, 95,96,97,98,99,100,101], but other report no difference [102, 103] and still others reporting lower amounts in PD patients compared to controls [14, 60, 104]. There also similarly remains substantial overlap in the ranges observed between PD patients and healthy controls, which make would make plasma aSyn difficult to use as a single diagnostic test for PD. Similar to CSF assays, these studies use different capture and detection antibodies and this and other sources of variability may influence results (aSyn levels in red blood cells from hemolysis, age variation, etc.). Initial studies largely have used ELISA-based assays but more sensitive assays like single molecule arrays or immunomagnetic reduction assays may provide improved clarity on these relationships with both diagnosis and clinical features [13, 14, 95,96,97, 101] (Table 2). There is conflicting evidence about whether increasing levels correlate with worse motor dysfunction [60, 95, 100, 102, 105, 106], but higher levels have been reported to be associated with worse cognitive function [13, 95, 101, 102]. One longitudinal study had noted an increase in plasma total aSyn over time in PD patients [106]. Heterogeneity in cohorts, sample size and methodological issues of sample collection and analysis could contribute to conflicting results across studies, necessitating further studies in large multicentered cohorts using standardized operating procedures. Future longitudinal studies in deeply phenotyped cohorts will provide further clarity on the use and evolution of plasma aSyn biomarkers although some likely changes over time can be surmised from the prior studies (Table 3).
ASyn Immunohistochemistry and Immunofluorescence from Tissue Samples
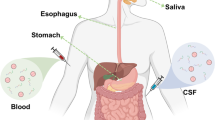
aSyn deposits are found in autonomic nerves that innervate a variety of peripheral tissues including, skin, olfactory mucosa, submandibular glands, and the colon in PD [16, 107,108,109,110]. Thus, the presence of these phosphorylated deposits in peripheral tissues could potentially aid the tissue diagnosis of living PD and other synucleinopathies.
Given that aSyn deposits likely occur early in the dorsal motor nucleus of the vagus nerve and the olfactory bulb, Heiko Braak and others posited that environmental factors could cause aSyn changes that could propagate to the central nervous system either from the gut via the vagus nerve or from the olfactory epithelium [111, 112]. Moreover, model systems find evidence to suggest pathological aSyn can propagate from the gut to the brain, which is abolished by vagotomy [113]. However, human autopsy studies do not find clear evidence of “incidental” peripheral aSyn in tissues (i.e., isolated aSyn in peripheral tissues without involvement of the brain), which argue against a peripheral origin of aSyn in PD and related synucleinopathies and instead peripheral aSyn may spread from early brainstem pathology [114]. It is very difficult to definitively define the epicenters or origins of neurodegenerative pathologies using cross-sectional autopsy tissue alone, but the findings of peripheral aSyn in PD and DLB offer an important minimally invasive method to obtain tissue diagnosis in living patients.
Given the regularity of colonoscopies as screening tests for colon cancer, investigations at assessing for aSyn pathology in colonic biopsies were performed but initial results were highly discordant with varying sensitivities in detecting pathological aSyn deposits in PD patients [115,116,117]. Multi-site studies were performed that showed good inter-rater reliability and helped to optimize methods, but results indicate that biopsies must include submucosal layers and be assessed by trained neuropathologists to determine if adequate neuronal elements and aSyn deposits are present [118, 119]. The submandibular glands of PD patients also demonstrate aSyn inclusions but there is a higher morbidity associated with needle biopsies compared to other peripheral tissue sampling and, because of inadequate sampling and immunohistochemical methods, sensitivity remains suboptimal in many studies [110, 116, 120,121,122]. aSyn deposits in skin biopsies are currently the most promising and least invasive tissue-based biomarker with optimization of methods that has occurred over the last several years. Initial studies discovered that different biopsy sites could yield different sensitivities in detecting phosphorylated aSyn deposits, with the abdomen and scalp showing lower rates of positivity but higher rates being shown in paracervical and lower leg sites in PD and DLB patients [16, 107,108,109, 123,124,125]. Different fixative methods influence results, with formalin fixed paraffin embedded tissue not performing as well as Zamboni fixation methods [15, 116, 124, 126,127,128,129,130,131,132]. The main reason for this may be that formalin may cause more extensive protein cross linking, making it more difficult for antibodies to attach to aSyn epitopes and the heat or chemically based retrieval methods may decrease aSyn signal if used too aggressively [116, 133, 134]. Depth of biopsy and section thickness affects yield as well [133]. Immunofluorescence using double labelling with antibodies against phosphorylated aSyn and neuron specific protein gene product (PGP) appears to perform better than bright field immunohistochemistry using diaminobenzamide chromagen (DAB) [15, 123, 127, 129,130,131, 135, 136]. The DAB chromagen is a staple of immunohistochemistry and creates a dark brown signal when detecting epitopes. However, in skin samples, it can be difficult to discern DAB signal from artifact and diffuse non-specific staining in small peripheral nerves and immunofluorescence facilitates double labelling to identify the overlap of small neurons innervating the skin and the presence of small phosphorylated synuclein inclusions simultaneously in the same tissue section [134, 137]. The most current methodologies using Zamboni fixative, cryosectioning, and immunofluorescence show 90% sensitivity and > 90% specificity for PD and DLB subjects in some studies [15, 125, 132, 138, 139]. Thus, standardization of pre-analytical factors, including sample handling are critical for the development of these tests for clinical use [140].
Aside from clinically manifest PD, skin aSyn deposits can be demonstrated in patients with REM sleep behavior disorder (RBD) and patients with pure-autonomic failure (pAF), both thought to be prodromal states that are highly likely to phenoconvert into PD or other synucleinopathy where presumably central nervous system pathology is more restricted [123, 126, 131, 137, 141,142,143,144]. It is not clear yet whether a positive skin biopsy predicts phenoconversion in RBD subjects but studies with longitudinal follow up are underway [137, 145]. Interestingly, there also may be differences in the characteristics of aSyn deposits between MSA and PD patients where MSA patients had phosphorylated aSyn deposits in somatic nerves whereas PD patients with orthostatic hypotension had deposits in autonomic nerve fibers in one study [146]. Furthermore, in PD patients, aSyn positive skin biopsies appear to have a rostro-caudal gradient, with more positive samples being noted from paracervical biopsy sites than limb sites; in MSA, however, there is a more uniform distribution of aSyn positivity in the different biopsy sites and higher density of phosphorylated aSyn in those biopsies [147]. Orthostatic hypotension is a common but not universal symptom of PD and its presence signifies autonomic involvement which may have relevance for skin biopsies in PD [148, 149]. One study found that PD patients with orthostatic hypotension had a more widespread and homogenous distribution of aSyn deposits whereas PD patients without orthostatic hypotension has aSyn pathology restricted to paracervical biopsy sites [150]. While these biopsies are likely useful in a categorical fashion, there are no features that correlate well with disease severity; however, one study of an MSA patient who underwent serial skin biopsies did note sequentially more skin structures affected, implying an evolution of skin aSyn deposits over time [146, 151]. aSyn skin deposits from PD patients with LRRK2, GBA, and SNCA mutations have also been demonstrated [152,153,154,155]. Given the pathological heterogeneity associated with LRRK2 mutations and the limited degree of central nervous system aSyn deposits in patient with PRKN mutations, further studies in autopsy validated subjects will be of interest [156, 157]. See Table 4 for selected studies of skin aSyn immunofluorescence in PD.
Peripheral biopsy testing for PD is nearing clinical use as there is a commercially available aSyn skin biopsy assay, the Syn-One test (CND Life Sciences). The SynOne test suggests obtaining samples using 3 mm punch biopsy tools, Zamboni fixative and requires double-immunostaining thick cryosection for neuronal elements (PGP 9.5) and phosphorylated aSyn (pSer129) using immunofluorescence [158]. Unpublished data using the Syn-One test has been presented at the American Academy of Neurology meeting in 2020 and the Lewy Body Disease association Biofluid/Tissue Biomarker symposium in 2021 reporting high sensitivity (74% from one biopsy site and 96% from three biopsy sites) and 99% accuracy of distinguishing synucleinopathies from controls [159]. This test is not FDA approved but is being further validated in a large multicentered clinical trial (NCT04700722) with a plan to enroll over 300 patients with synucleinopathies (PD: 105, MSA: 40, DLB: 90, pure autonomic failure: 65) and 200 healthy controls who will undergo three skin biopsies at the paracervical, distal thigh, and lower leg sites to determine sensitivity, specificity, accuracy, and precision of the current test [158].
Alpha Synuclein Seeding Amplification Assays
aSyn seeding amplification assays (aSyn-SAA) began as adaptations of prion disease assays and make use of the ability of aSyn seeds to template normal monomeric aSyn species to oligomeric and fibrillar forms in a prion-like fashion [160,161,162]. In these assays, a biological sample is added to a well containing monomeric aSyn with a fluorescent tag thioflavin-T. If a pathological aSyn seed is present, it will induce templating of the monomers and after a certain amount of time, the newly created fibrils will be broken down by shaking the plate allowing for more monomers to be recruited. After several hours, this creates an exponential rise in the fluorescence which can be detected. The standard diagnostic metrics collected is a binary positive or negative readout above a certain fluorescence threshold defined by the laboratory, but additional metrics including the time to positive signal (or lag time), maximum fluorescence, and the time to reach 50% of maximum fluorescence can also be reported if fluorescence measurements are captured at regular intervals (Fig. 1). In initial studies, remarkably high sensitivity and specificity (> 90%) was demonstrated in detecting aSyn seeding from CSF samples of patients with manifest PD and DLB [17, 163]. In the years since, multiple studies in independent laboratories have confirmed these findings [18, 20, 160, 163,164,165,166]. aSyn seeds are readily apparent in early PD when subjects within 2 years of diagnosis who had not started medications from the Parkinson’s Progression Marker Initiative were studied [166, 167], and high rates of positivity are also observed in prodromal patients with REM sleep behavior disorder and pure autonomic failure, conditions which have a high likelihood of underlying alpha-synuclein and phenoconverting into PD or DLB [165, 168,169,170]. In the case of REM sleep behavior disorder, it is not entirely clear if a positive aSyn-SAA results predicts phenoconversion to one of these syndromes. Some of the uncertainty is due to lack of longitudinal studies with serial sampling and differences in baseline rates of aSyn-SAA positivity in RBD cohorts studied [169, 170].

aSyn-SAA metrics. Tissue or fluid samples are analyzed with fluorescence measurements read at given intervals which can be used to establish curves shown in (a). From these curves, a variety of metrics can be derived including maximum fluorescence, time to threshold (or time lag), time to 50% of maximum fluorescence (T50) or area under the curve calculations; however, these metrics have not consistently been shown to relate to clinical characteristics or pathological burden within PD patients. If a sample undergoes serial dilution and is analyzed at these different dilutions as shown in (b), the dilution at which 50% of well remain positive can be used to estimate the SD50 which may have more relevance to disease activity in some studies. In this example the estimated -log(SD50) = 7. Created with Biorender.com
This body of work suggests that these assays are extremely sensitive and specific for detecting the categorical presence/absence of aSyn seeds in CSF of patients with manifest disease and prodromal states where presumably aSyn pathology is more restricted. It is less clear whether the quantitative metrics collected by these assays have quantitative value in relation to clinical variables in PD. Aside from MSA, there are no major differences in maximum fluorescence, time to positivity, or area under the curve between PD, DLB, pure autonomic failure or REM sleep behavior disorder patients [165]. In the majority of studies conducted, there have been no strong correlations with time to positivity, maximum fluorescence or time to 50% fluorescence with clinical aspects of Parkinson’s disease such as motor burden [166, 167]. There was one study where mild to moderate correlations of time to threshold and maximum fluorescence were observed with disease duration and motor burden on the unified Parkinson’s disease rating scale but this has not been replicated [167]. While not routinely performed in clinical assays, serial dilution of biological samples can be used to calculate an SD50 value, the seeding dose at which 50% of wells will turn positive, using a Spearman-Karber method [171] (Fig. 1). In two studies, aSyn-SAA SD50 values correlated with disease duration and higher SD50 values were noted with higher degrees of pathological aSyn deposits in postmortem autopsy analyses [160, 167, 172]; however, it has not been consistently related to other disease features and such methods are time consuming and unlikely to be scaled for routine use. Regarding pure autonomic failure, subjects with this condition may phenoconvert to PD, DLB, or multiple systems atrophy (MSA) and there is some evidence that the kinetics of the aSyn-SAA curve (i.e., maximum fluorescence and time to positivity) and additional information from neurofilament light chain testing in CSF may offer prognostic information about which synucleinopathy a subject is likely to phenoconvert to [163, 165]. Samples from patients with MSA in some studies have a faster time to positive but lower maximum fluorescence and this may reflect properties of different aSyn strains in these associated diseases as numerous biochemical and structural differences between the aSyn species in MSA and Lewy body disorders have been described [163, 165, 168].
In the last few years, several attempts have been made to adapt aSyn-SAA assays from CSF above to peripheral tissue and fluid samples, which could potentially offer a less invasive manner of diagnosing the presence of aSyn seeds (Fig. 2). Much of this initial work was pursued because of the well documented observations of abnormally phosphorylated aSyn deposits in skin, colon, submandibular gland, and other tissues in PD patients both at autopsy and in vivo from biopsy studies discussed above [116, 134]. aSyn-SAA from skin biopsies appear to offer similarly high sensitivity and specificity comparable to CSF in several studies in PD and RBD patients [21, 172,173,174,175,176]. Olfactory mucosa samples may be useful as well, but sampling requires accessing very deep structures, often using a rigid scope and with an otolaryngologist operator, which may limit feasibility [177]. Seeding from olfactory mucosa samples in PD, MSA, DLB, and REM sleep disorder patients has been demonstrated though [174, 177,178,179]. aSyn seeding activity has also been demonstrated from not only submandibular gland biopsies but also from saliva itself [180,181,182]. Lastly, seeding activity can be demonstrated from colonic biopsies, where phosphorylated aSyn has been known to deposit [116, 134, 174, 183]. See Table 4 for selected aSyn-SAA studies.

aSyn assays from biofluids and tissue. Summary of static aSyn assessments, peripheral tissue immunohistochemistry, immunofluorescence and aSyn-SAA assays in different tissues and fluids studied currently with spinal fluid aSyn-SAA and skin aSyn-SAA and immunofluorescence assays showing the greatest accuracy to date but with many other assays still in development. Created with Biorender.com
The majority of the above studies have been performed in clinically defined cohorts, and in those studies where neuropathological confirmation has been performed, co-pathologies are not typically assessed in a standardized fashion [17, 160]. While aSyn aggregates in Lewy bodies and Lewy neurites are noted in brainstem, limbic and neocortical areas in PD and DLB, Lewy bodies, and Lewy neurites are present in the amygdala and nearby limbic structures in about 50% of sporadic Alzheimer’s disease patients and around 90% of familial Alzheimer’s disease cases with presenilin mutations [184,185,186,187]. Such cases are unlikely to exhibit PD or DLB like clinical phenotypes [24]. Two studies recently have addressed whether current CSF assays can detect aSyn seeds in these amygdala-predominant cases and both found that CSF assays detected aSyn seeds in these cases at much lower rates than in cases with limbic or neocortical stage Lewy pathology [188, 189]. Both studies also show that positivity of these assays is dependent on aSyn stage and not masked or significantly influenced by the co-occurrence of Alzheimer’s pathology [188, 189]. Direct seeding assays from frozen amygdala samples from amygdala-predominant cases also showed a mix of positive and negative reactions in one of these studies [189]. Further studies are needed to understand whether this variability in seeding activity is due to a lower overall dose of aSyn seeds or if there are differences in the aSyn species in amygdala-predominant cases that result in lower seeding activity. Such studies are important to understand the interpretation of aSyn-SAA results when applied to a larger population where subjects may harbor incidental Lewy bodies or amygdala predominant Lewy bodies. Furthermore, several population-based cohorts would suggest that the baseline prevalence of aSyn pathology is around 20–30%, and in some cases, this pathology can be widespread without causing clinical symptoms [23, 35, 190,191,192,193]. While it appears that these assays may be somewhat less sensitive to detect these cases of incidental Lewy body disease and amygdala-predominant disease, further studies will be needed [168, 188]. Additionally, some patients with (LRRK2 mutations and most, if not all, patients parkin PBR E3 ubiquitin protein ligase (PRKN) will not have pathological aSyn accumulations at autopsy [156, 194] and therefore will be less likely to exhibit seeding activity or aSyn deposits [153, 195,196,197]. Therefore, there is likely a role of integrating genetic testing information into the application of these assays in PD.
At this time, there is also a commercially available CSF aSyn-SAA assay SynTAP (Amprion Laboratories) that is not FDA approved but did receive FDA breakthrough designation in 2019. In the SynTAP assay, which is a slightly modified version of Amprion’s research assay (formerly referred to as PMCA), samples are run in triplicate using glass beads with fluorescence measured less frequently than the research assay to allow for higher throughput [189, 198]. The SynTap assay has shown similarly high accuracy to Amprion’s research assay [189].
AD Fluid Biomarkers in PD and DLB
As noted previously, autopsy studies of PD patients typically reveal 35–50% of PD patients with dementia and more than 70% of DLB patients have moderate to high levels of AD neuropathologic change [29,30,31,32,33,34,35]. AD co-pathology in PD has been associated with older age of onset, shorter disease duration, faster time to dementia, greater likelihood of amnestic memory deficits and greater likelihood of an akinetic rigid motor phenotype in several studies [31, 32, 36,37,38,39,40,41,42]. These findings are not universal however, and in cluster analyses of PD, no major changes in rates of AD co-pathology of CSF AD biomarkers in studies comparing so called diffuse-malignant subtypes of PD with mild motor-predominant forms [199, 200]. Still, understanding the interplay of aSyn, Aβ, and tau pathology in PD and DLB is of interest as it will inform the interpretation of AD biomarkers in these populations as these assays become more widely available and stratifying clinical trials by the presence or absence of AD co-pathology may be of interest [201].
PD and DLB patients tend to have lower levels of CSF Aβ42 and tau species than normal controls in groupwise comparisons early in the disease [45, 46, 49, 73, 202,203,204]. In PD, lower levels of CSF Aβ42 is related to worse cognition cross-sectionally, longitudinally, and is related to higher likelihood of AD co-pathology at death [44, 46, 47, 64, 73, 202, 204, 205]. Interestingly, one study showed an increase in CSF Aβ42 in PD patients with freezing of gait compared to PD patients who did not [206]; thus, clinical heterogeneity of PD may influence biomarker interpretation as well. While total and p-tau 181 is on average lower than controls in early PD, levels may increase later in the disease in some patients which is also associated with a greater likelihood of dementia [207,208,209,210]. While optimal cut-offs for these Aβ42, t-tau, and p-tau 181 and their ratios have been well established in Alzheimer’s disease, it is not clear if the same cutoffs apply in PD and other Lewy body disorders [211, 212]. Indeed, in rare autopsy-confirmed work, there is data to suggest CSF Aβ42 may be associated with increasing aSyn pathology independent of plaque burden in LBD [47].
More recently, plasma assays (Aβ1-42, t-tau, p-tau 181, p-tau 217, and p-tau 231) are being developed for use in AD but are already being studied in PD as well [213,214,215,216]. Plasma Aβ42 may be related to more severe gait impairment and severity of akinetic rigid symptoms [217, 218]. Plasma p-tau 181 and p-tau 217 levels correlate with degree of tau PET and Aβ PET status [219]. In studies of DLB, where tau co-pathology is more likely, plasma p-tau 181 and 231 have been associated with faster cognitive declines [219, 220]. Higher levels of plasma p-tau 181 are reported in PD patients when compared to healthy controls and these levels correlate with plasma aSyn markers [221]. However, in some studies plasma p-tau 181 has not clearly been linked to cognitive decline in PD and plasma t-tau and neurofilament light chain measurements have had stronger correlations with cognitive dysfunction [95, 105, 222]. In DLB, in particular, where rates of AD co-pathology are often quite high, stratification by the presence of these AD biomarkers may prove especially important for clinical trial enrollment of more biologically homogenous patients or those who may benefit from combination therapies [201].
Conclusion
aSyn-specific biomarkers have long been an unmet need in the field of neurodegenerative medicine. While the search for biomarkers with strong associations with disease pathology continues, several new fluid and tissue based biomarkers are being developed which offer the ability to detect aSyn species in patients with PD, DLB, and also in prodromal states, which is critical for therapeutic trials targeting aSyn mechanisms. CSF aSyn and plasma aSyn species detected by current assays may be limited but further development with newer second-generation immunoassays or other methods of detection may provide additional opportunities for biomarker development. Please see Table 3 for a summary of CSF (Table 1), plasma (Table 2), and aSyn-SAA and immunofluorescence (Table 4) biomarker data findings in PD. aSyn immunofluorescence from skin samples and aSyn-SAA assays both from CSF and peripheral tissues appear promising and will likely be of imminent use in clinic and research settings which will likely provide accurate methods of categorically assessing for the presence of aSyn deposits and aSyn seeds [138]. More work will be needed to determine of more labor-intensive methods like calculating SD50 will provide quantitative readouts of aSyn seeding that have relevance for disease activity, but initial studies suggest some significant correlations with disease duration and pathological burden. Most studies of aSyn-SAA to date have been done in clinically defined cohorts of PD and other synucleinopathies, some with autopsy validation [164, 167, 168]. However, given the sensitivity of some of these assays in detecting aSyn seeds or clinicians may have to grapple shortly interpretation of a positive result in patients without a defined synucleinopathy syndrome, and it is not entirely clear if these patients are universally destined to phenoconvert. The integration of other biomarkers like hyposmia, polysomnograms for RBD, and DAT scans will likely further be of use to stratify those aSyn positive cases who are more likely to develop a parkinsonian syndrome. When combined with CSF or plasma biomarkers for AD, a more comprehensive picture of both primary and commonly occurring AD co-pathologies can be constructed for PD patients. These assays will likely prove useful in augmenting enrollment of homogenous populations into clinical trials. Focuses for future work to bring these skin immunofluorescence and aSyn-SAAs to clinical use include assay standardization and research in autopsy-confirmed cohorts to clarify the complex relationships between pathology in the brain and those detected from peripheral tissues and biofluids. aSyn assays that have quantitative value for disease activity remain a major unmet need, but the exciting development of these assays will allow for clinical assessments to be augmented by aSyn-specific biomarkers in a manner which has not been previously available for living patients.
References
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–40.
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211.
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord [Internet]. 2015/10/17. 2015;30(12):1591–601. Available from: https://movementdisorders.onlinelibrary.wiley.com/doi/10.1002/mds.26424
Adler CH, Beach TG, Hentz JG, Shill HA, Caviness JN, Driver-Dunckley E, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology [Internet]. 2014/07/01. 2014;83(5):406–12. Available from: http://www.neurology.org/content/83/5/406.
Hughes AJ, Ben-Shlomo Y, Daniel SE, Lees AJ. What features improve the accuracy of clinical diagnosis in Parkinson’s disease: a clinicopathologic study. Neurology. 1992;42(6):1142–6.
Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125(Pt 4):861–70.
Rajput AH, Pahwa R, Pahwa P, Rajput A. Prognostic significance of the onset mode in parkinsonism. Neurology. 1993;43(4):829–30.
Litvan I, MacIntyre A, Goetz CG, Wenning GK, Jellinger K, Verny M, et al. Accuracy of the clinical diagnoses of Lewy body disease, Parkinson disease, and dementia with Lewy bodies: a clinicopathologic study. Arch Neurol. 1998;55(7):969–78.
Postuma RB, Poewe W, Litvan I, Lewis S, Lang AE, Halliday G, et al. Validation of the MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord [Internet]. 2018 [cited 2022 Oct 12];33(10):1601–8. Available from: https://pubmed.ncbi.nlm.nih.gov/30145797/.
Mollenhauer B, El-Agnaf OMA, Marcus K, Trenkwalder C, Schlossmacher MG. Quantification of α-synuclein in cerebrospinal fluid as a biomarker candidate: review of the literature and considerations for future studies. Biomark Med [Internet]. 2010 [cited 2022 Nov 27];4(5):683–9. Available from: https://www.futuremedicine.com/doi/10.2217/bmm.10.90.
Mollenhauer B. Quantification of α-synuclein in cerebrospinal fluid: How ideal is this biomarker for Parkinson’s disease? Parkinsonism Relat Disord. 2014;20(SUPPL.1):S76–9.
Wang Y, Shi M, Chung KA, Zabetian CP, Leverenz JB, Berg D, et al. Phosphorylated α-synuclein in Parkinson’s disease. Sci Transl Med [Internet]. 2012 [cited 2022 Nov 27];4(121). Available from: https://www.science.org/doi/10.1126/scitranslmed.3002566.
Ng ASL, Tan YJ, Lu Z, Ng EYL, Ng SYE, Chia NSY, et al. Plasma alpha‐synuclein detected by single molecule array is increased in PD. Ann Clin Transl Neurol [Internet]. 2019 [cited 2022 Nov 23];6(3):615. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6414476/.
Li QX, Mok SS, Laughton KM, McLean CA, Cappai R, Masters CL, et al. Plasma α-synuclein is decreased in subjects with Parkinson’s disease. Exp Neurol. 2007;204(2):583–8.
Gibbons CH, Garcia J, Wang N, Shih LC, Freeman R. The diagnostic discrimination of cutaneous α-synuclein deposition in Parkinson disease. Neurology [Internet]. 2016 [cited 2022 Nov 5];87(5):505–12. Available from: https://pubmed.ncbi.nlm.nih.gov/27385742/.
Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White III CL, et al. Multi-organ distribution of phosphorylated α-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol [Internet]. 2010 [cited 2022 Nov 4];119(6):689–702. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2866090/.
Fairfoul G, McGuire LI, Pal S, Ironside JW, Neumann J, Christie S, et al. Alpha-synuclein RT-Qu IC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol. 2016;3(10):812–8.
Concha-Marambio L, Shahnawaz M, Soto C. Detection of misfolded α-synuclein aggregates in cerebrospinal fluid by the protein misfolding cyclic amplification platform. Methods Mol Biol [Internet]. 2019 [cited 2022 Apr 15];1948:35–44. Available from: https://link.springer.com/protocol/10.1007/978-1-4939-9124-2_4.
Shahnawaz M, Tokuda T, Waragai M, Mendez N, Ishii R, Trenkwalder C, et al. Development of a biochemical diagnosis of Parkinson disease by detection of α-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017;74(2):163–72.
Groveman BR, Orrù CD, Hughson AG, Raymond LD, Zanusso G, Ghetti B, et al. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol Commun. 2018;6(1):7.
Manne S, Kondru N, Jin H, Serrano GE, Anantharam V, Kanthasamy A, et al. Blinded RT-QuIC analysis of α-synuclein biomarker in skin tissue from Parkinson’s disease patients. Mov Disord [Internet]. 2020 [cited 2021 Jul 26];35(12):2230–9. Available from: https://movementdisorders.onlinelibrary.wiley.com/doi/full/10.1002/mds.28242.
Hurtig HI, Trojanowski JQ, Galvin J, Ewbank D, Schmidt ML, Lee V-Y, et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology. 2000;54(10):1916–21.
Coughlin DG, Petrovitch H, White LR, Noorigian J, Masaki KH, Ross GW, et al. Most cases with lewy pathology in a population-based cohort adhere to the braak progression pattern but ‘failure to fit’is highly dependent on staging system applied. Parkinsonism Relat Disord. 2019.
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J-P, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies Fourth consensus report of the DLB Consortium. Neurology. 2017. https://doi.org/10.1212/WNL.0000000000004058.
Beach TG, White CL, Hamilton RL, Duda JE, Iwatsubo T, Dickson DW, et al. Evaluation of alpha-synuclein immunohistochemical methods used by invited experts. Acta Neuropathol. 2008;116(3):277–88.
Adler CH, Beach TG, Zhang N, Shill HA, Driver-Dunckley E, Caviness JN, et al. Unified staging system for Lewy body disorders: clinicopathologic correlations and comparison to Braak staging. J Neuropathol Exp Neurol [Internet]. 2019 [cited 2022 Feb 3];78(10):891–9. Available from: https://academic.oup.com/jnen/article/78/10/891/5561420.
Bayram E, Coughlin DG, Banks SJ, Litvan I. Sex differences for phenotype in pathologically defined dementia with Lewy bodies. J Neurol Neurosurg Psychiatry [Internet]. 2021 [cited 2021 Jul 27];92(7):745–50. Available from: https://jnnp.bmj.com/content/92/7/745.
Bayram E, Coughlin DG, Litvan I. Sex differences for clinical correlates of Alzheimer’s pathology in people with Lewy body pathology. Mov Disord [Internet]. 2022 [cited 2022 Nov 27];37(7):1505–15. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.29044.
Jellinger KA, Korczyn AD. Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med. 2018;16(1):34.
Jellinger KA. Neuropathological aspects of Alzheimer disease, Parkinson disease and frontotemporal dementia. Neurodegener Dis. 2008;5(3–4):118–21.
Jellinger KA, Seppi K, Wenning GK, Poewe W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson’s disease. J Neural Transm. 2002;109(3):329–39.
Irwin DJ, White MT, Toledo JB, Xie SX, Robinson JL, Van Deerlin V, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. 2012;72(4):587–98.
Smith C, Malek N, Grosset K, Cullen B, Gentleman S, Grosset DG. Neuropathology of dementia in patients with Parkinson’s disease: a systematic review of autopsy studies. J Neurol Neurosurg Psychiatry [Internet]. 2019 [cited 2021 Jul 29];90(11):1234–43. Available from: https://jnnp.bmj.com/content/90/11/1234.
Marui W, Iseki E, Kato M, Akatsu H, Kosaka K. Pathological entity of dementia with Lewy bodies and its differentiation from Alzheimer’s disease. Acta Neuropathol. 2004;108(2):121–8.
Wakisaka Y, Furuta A, Tanizaki Y, Kiyohara Y, Iida M, Iwaki T. Age-associated prevalence and risk factors of Lewy body pathology in a general population: the Hisayama study. Acta Neuropathol [Internet]. 2003;106(4):374–82. Available from: https://doi.org/10.1007/s00401-003-0750-x.
Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol. 2017;16(1):55–65.
Compta Y, Parkkinen L, O’sullivan SS, Vandrovcova J, Holton JL, Collins C, et al. Lewy-and Alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain. 2011;134(5):1493–505.
Halliday G, Hely M, Reid W, Morris J. The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol. 2008;115(4):409–15.
Howlett DR, Whitfield D, Johnson M, Attems J, O’brien JT, Aarsland D, et al. Regional multiple pathology scores are associated with cognitive decline in Lewy body dementias. Brain Pathol. 2015;25(4):401–8.
Sabbagh MN, Adler CH, Lahti TJ, Connor DJ, Vedders L, Peterson LK, et al. Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Disord. 2009;23(3):295–7.
Kotzbauer PT, Cairns NJ, Campbell MC, Willis AW, Racette BA, Tabbal SD, et al. Pathologic accumulation of alpha-synuclein and Abeta in Parkinson disease patients with dementia. Arch Neurol. 2012;69(10):1326–31.
Coughlin D, Xie SX, Liang M, Williams A, Peterson C, Weintraub D, et al. Cognitive and pathological influences of tau pathology in Lewy body disorders. Ann Neurol. 2019;85(2):259–71.
Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement [Internet]. 2018 [cited 2021 Aug 3];14(4):535–62. Available from: https://alz-journals.onlinelibrary.wiley.com/doi/full/10.1016/j.jalz.2018.02.018.
Siderowf A, Xie SX, Hurtig H, Weintraub D, Duda J, Chen-Plotkin A, et al. CSF amyloid beta 1–42 predicts cognitive decline in Parkinson disease. Neurology. 2010;75(12):1055–61.
Kang JH, Mollenhauer B, Coffey CS, Toledo JB, Weintraub D, Galasko DR, et al. CSF biomarkers associated with disease heterogeneity in early Parkinson’s disease: the Parkinson’s Progression Markers Initiative study. Acta Neuropathol [Internet]. 2016/03/30. 2016;131(6):935–49. Available from: https://link.springer.com/content/pdf/10.1007%2Fs00401-016-1552-2.pdf.
Irwin DJ, Fedler J, Coffey CS, Caspell‐Garcia C, Kang JH, Simuni T, et al. Evolution of Alzheimer’s disease cerebrospinal fluid biomarkers in early Parkinson’s disease. Ann Neurol [Internet]. 2020 [cited 2021 Aug 3];88(3):574–87. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/ana.25811.
Irwin DJ, Coughlin D, Nevler N, Akhtar RS, McMillan CT, Lee EB, et al. Antemortem CSF tau and A beta biomarkers are predictive of postmortem Alzheimer’s disease pathology in autopsy-confirmed Lewy body disease. In: Annals of Neurology. WILEY 111 RIVER ST, HOBOKEN 07030–5774, NJ USA; 2017. p. S56–S56.
Lemstra AW, de Beer MH, Teunissen CE, Schreuder C, Scheltens P, van der Flier WM, et al. Concomitant AD pathology affects clinical manifestation and survival in dementia with Lewy bodies. J Neurol Neurosurg Psychiatry [Internet]. 2017 [cited 2021 Jan 25];88(2):113–8. Available from: https://pubmed.ncbi.nlm.nih.gov/27794030/.
van Steenoven I, Aarsland D, Weintraub D, Londos E, Blanc F, van der Flier WM, et al. Cerebrospinal fluid Alzheimer’s disease biomarkers across the spectrum of Lewy body diseases: results from a large multicenter cohort. J Alzheimer’s Dis. 2016;54(1):287–95.
Beek M van de, Ooms FAH, Ebenau JL, Barkhof F, Scheltens P, Teunissen CE, et al. ATN classification in dementia with Lewy bodies: association with clinical profile, cognitive decline and survival. Alzheimer’s Dement [Internet]. 2021 [cited 2022 Dec 6];17(S4):e052056. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/alz.052056.
Ferreira D, Przybelski SA, Lesnick TG, Lemstra AW, Londos E, Blanc F, et al. β-Amyloid and tau biomarkers and clinical phenotype in dementia with Lewy bodies. Neurology [Internet]. 2020 [cited 2021 Apr 30];95(24):e3257–68. Available from: https://pubmed.ncbi.nlm.nih.gov/32989106/.
Cousins KAQ, Arezoumandan S, Shellikeri S, Ohm D, Shaw LM, Grossman M, et al. CSF biomarkers of Alzheimer disease in patients with concomitant α-synuclein pathology. Neurology [Internet]. 2022 [cited 2022 Nov 27];99(20):e2303–12. Available from: https://n.neurology.org/content/99/20/e2303.
Gaig C, Valldeoriola F, Gelpi E, Ezquerra M, Llufriu S, Buongiorno M, et al. Rapidly progressive diffuse Lewy body disease. Mov Disord [Internet]. 2011 [cited 2022 Dec 6];26(7):1316–23. Available from: https://pubmed.ncbi.nlm.nih.gov/21484863/.
Josephs KA, Ahlskog JE, Parisi JE, Boeve BF, Crum BA, Giannini C, et al. Rapidly progressive neurodegenerative dementias. Arch Neurol [Internet]. 2009 [cited 2022 Dec 6];66(2):201–7. Available from: https://jamanetwork.com/journals/jamaneurology/fullarticle/796577.
Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain. 2018;141(7).
Robinson JL, Richardson H, Xie SX, Suh E, Van Deerlin VM, Alfaro B, et al. The development and convergence of co-pathologies in Alzheimer’s disease. Brain [Internet]. 2021 [cited 2022 Nov 27];144(3):953–62. Available from: https://academic.oup.com/brain/article/144/3/953/6101614.
Coulthard EJ, Love S. A broader view of dementia: multiple co-pathologies are the norm. Brain [Internet]. 2018 [cited 2022 Nov 6];141(7):1894–7. Available from: https://academic.oup.com/brain/article/141/7/1894/5045300.
Mollenhauer B, Zimmermann J, Sixel-Doring F, Focke NK, Wicke T, Ebentheuer J, et al. Monitoring of 30 marker candidates in early Parkinson disease as progression markers. Neurology [Internet]. 2016/05/11. 2016;87(2):168–77. Available from: http://www.neurology.org/content/87/2/168.
Barkovits K, Kruse N, Linden A, Tönges L, Pfeiffer K, Mollenhauer B, et al. Blood contamination in CSF and its impact on quantitative analysis of alpha-synuclein. Cells. 2020;9(2).
Goldman JG, Andrews H, Amara A, Naito A, Alcalay RN, Shaw LM, et al. Cerebrospinal fluid, plasma, and saliva in the BioFIND study: relationships among biomarkers and Parkinson’s disease features. Mov Disord [Internet]. 2018 [cited 2022 Nov 22];33(2):282–8. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.27232.
Abdi IY, Majbour NK, Willemse EAJ, van de Berg WDJ, Mollenhauer B, Teunissen CE, et al. Preanalytical stability of CSF total and oligomeric alpha-synuclein. Front Aging Neurosci. 2021;3(13):85.
Youssef P, Kim WS, Halliday GM, Lewis SJG, Dzamko N. Comparison of different platform immunoassays for the measurement of plasma alpha-synuclein in Parkinson’s disease patients. J Parkinsons Dis [Internet]. 2021 [cited 2023 Mar 21];11(4):1761. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8609717/.
Kruse N, Mollenhauer B. Validation of a commercially available enzyme-linked immunoabsorbent assay for the quantification of human α-Synuclein in cerebrospinal fluid. J Immunol Methods. 2015;1(426):70–5.
Parnetti L, Farotti L, Eusebi P, Chiasserini D, De Carlo C, Giannandrea D, et al. Differential role of CSF alpha-synuclein species, tau, and Abeta42 in Parkinson’s disease. Front Aging Neurosci. 2014;6:53.
Hong Z, Shi M, Chung KA, Quinn JF, Peskind ER, Galasko D, et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain. 2010;133(Pt 3):713–26.
Tokuda T, Salem SA, Allsop D, Mizuno T, Nakagawa M, Qureshi MM, et al. Decreased α-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem Biophys Res Commun. 2006;349(1):162–6.
Wang L, Gao L, Tang H, Nie K, Wang L, Zhao J, et al. Cerebrospinal fluid alpha-synuclein as a biomarker for Parkinson’s disease diagnosis: a systematic review and meta-analysis. Int J Neurosci [Internet]. 2014 [cited 2022 Nov 23];125(9):645–54. Available from: https://www.tandfonline.com/doi/abs/10.3109/00207454.2014.961454.
Eusebi P, Giannandrea D, Biscetti L, Abraha I, Chiasserini D, Orso M, et al. Diagnostic utility of cerebrospinal fluid α-synuclein in Parkinson’s disease: A systematic review and meta-analysis. Mov Disord. 2017;32(10):1389–400.
Mollenhauer B, Caspell-Garcia CJ, Coffey CS, Taylor P, Singleton A, Shaw LM, et al. Longitudinal analyses of cerebrospinal fluid α-synuclein in prodromal and early Parkinson’s disease. Mov Disord [Internet]. 2019 [cited 2023 Mar 23];34(9):1354–64. Available from: https://pubmed.ncbi.nlm.nih.gov/31361367/.
Gao L, Tang H, Nie K, Wang L, Zhao J, Gan R, et al. Cerebrospinal fluid alpha-synuclein as a biomarker for Parkinson’s disease diagnosis: a systematic review and meta-analysis. Int J Neurosci [Internet]. 2015 [cited 2022 Oct 12];125(9):645–54. Available from: https://www.tandfonline.com/doi/abs/10.3109/00207454.2014.961454.
Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, Sixel-Döring F, Trenkwalder C, Schlossmacher MG. α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol [Internet]. 2011 [cited 2022 Oct 13];10(3):230–40. Available from: https://pubmed.ncbi.nlm.nih.gov/21317042/.
Aerts MB, Esselink RAJ, Abdo WF, Bloem BR, Verbeek MM. CSF α-synuclein does not differentiate between parkinsonian disorders. Neurobiol Aging. 2012;33(2):430.e1-430.e3.
Hall S, Surova Y, Ohrfelt A, Zetterberg H, Lindqvist D, Hansson O. CSF biomarkers and clinical progression of Parkinson disease. Neurology. 2015;84(1):57–63.
Jankovic J, Schwartz K, Clemence W, Aswad A, Mordaunt J. A randomized, double-blind, placebo-controlled study to evaluate botulinum toxin type A in essential hand tremor. Mov Disord. 1996;11(3):250–6.
Stewart T, Liu C, Ginghina C, Cain KC, Auinger P, Cholerton B, et al. Cerebrospinal fluid alpha-synuclein predicts cognitive decline in Parkinson disease progression in the DATATOP cohort. Am J Pathol. 2014;184(4):966–75.
Majbour NK, Vaikath NN, Eusebi P, Chiasserini D, Ardah M, Varghese S, et al. Longitudinal changes in CSF alpha-synuclein species reflect Parkinson’s disease progression. Mov Disord [Internet]. 2016 [cited 2022 Oct 12];31(10):1535–42. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.26754.
Hall S, Surova Y, Öhrfelt A, Blennow K, Zetterberg H, Hansson O. Longitudinal measurements of cerebrospinal fluid biomarkers in Parkinson’s disease. Mov Disord [Internet]. 2016 [cited 2023 Mar 18];31(6):898–905. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.26578.
Hall S, Öhrfelt A, Constantinescu R, Andreasson U, Surova Y, Bostrom F, et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch Neurol. 2012;69(11):1445–52.
Slaets S, Vanmechelen E, Le Bastard N, Decraemer H, Vandijck M, Martin JJ, et al. Increased CSF α-synuclein levels in Alzheimer’s disease: correlation with tau levels. Alzheimer’s Dement [Internet]. 2014 [cited 2022 Oct 12];10(5):S290–8. Available from: https://onlinelibrary.wiley.com/doi/full/10.1016/j.jalz.2013.10.004.
Toledo JB, Korff A, Shaw LM, Trojanowski JQ, Zhang J. CSF α-synuclein improves diagnostic and prognostic performance of CSF tau and Aβ in Alzheimer’s disease. Acta Neuropathol [Internet]. 2013 [cited 2022 Oct 12];126(5):683–97. Available from: https://link.springer.com/article/10.1007/s00401-013-1148-z.
Wang ZY, Han ZM, Liu QF, Tang W, Ye K, Yao YY. Use of CSF α-synuclein in the differential diagnosis between Alzheimer’s disease and other neurodegenerative disorders. Int Psychogeriatrics [Internet]. 2015 [cited 2022 Oct 12];27(9):1429–38. Available from: https://www.cambridge.org/core/journals/international-psychogeriatrics/article/use-of-csf-synuclein-in-the-differential-diagnosis-between-alzheimers-disease-and-other-neurodegenerative-disorders/1CFFECD7F5A1E883B4B48ACB5D894DBC.
Kong Y, Chen Z, Wang X, Wang W, Zhang J. Diagnostic utility of cerebrospinal fluid α-synuclein in Creutzfeldt-Jakob disease: a systematic review and meta-analysis. J Alzheimers Dis [Internet]. 2022 [cited 2023 Mar 19];89(2):493–503. Available from: https://pubmed.ncbi.nlm.nih.gov/35912746/.
Hu Y, Yu SY, Zuo LJ, Cao CJ, Wang F, Chen ZJ, et al. Parkinson disease with REM sleep behavior disorder: features, α-synuclein, and inflammation. Neurology [Internet]. 2015 [cited 2023 Mar 23];84(9):888–94. Available from: https://pubmed.ncbi.nlm.nih.gov/25663225/.
Compta Y, Valente T, Saura J, Segura B, Iranzo A, Serradell M, et al. Correlates of cerebrospinal fluid levels of oligomeric-and total-α-synuclein in premotor, motor and dementia stages of Parkinson’s disease. J Neurol. 2015;262(2):294–306.
Schmid AW, Fauvet B, Moniatte M, Lashuel HA. Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies. Mol Cell Proteomics [Internet]. 2013 [cited 2022 Oct 13];12(12):3543–58. Available from: https://pubmed.ncbi.nlm.nih.gov/23966418/.
Stewart T, Sossi V, Aasly JO, Wszolek ZK, Uitti RJ, Hasegawa K, et al. Phosphorylated α-synuclein in Parkinson’s disease: correlation depends on disease severity. Acta Neuropathol Commun [Internet]. 2015 [cited 2022 Oct 13];3(1):7. Available from: https://actaneurocomms.biomedcentral.com/articles/10.1186/s40478-015-0185-3.
Schulz I, Kruse N, Gera RG, Kremer T, Cedarbaum J, Barbour R, et al. Systematic assessment of 10 biomarker candidates focusing on α-synuclein-related disorders. Mov Disord. 2021;36(12):2874–87.
Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med 2017 232 [Internet]. 2017 [cited 2022 Nov 29];23(2):1–13. Available from: https://www.nature.com/articles/nm.4269.
Hansson O, Hall S, Öhrfelt A, Zetterberg H, Blennow K, Minthon L, et al. Levels of cerebrospinal fluid α-synuclein oligomers are increased in Parkinson’s disease with dementia and dementia with Lewy bodies compared to Alzheimer’s disease. Alzheimers Res Ther. 2014;6(3):25.
Majbour NK, Chiasserini D, Vaikath NN, Eusebi P, Tokuda T, Van De Berg W, et al. Increased levels of CSF total but not oligomeric or phosphorylated forms of alpha-synuclein in patients diagnosed with probable Alzheimer’s disease. Sci Rep. 2017;7:40263.
Majbour NK, Vaikath NN, Van Dijk KD, Ardah MT, Varghese S, Vesterager LB, et al. Oligomeric and phosphorylated alpha-synuclein as potential CSF biomarkers for Parkinson’s disease. Mol Neurodegener [Internet]. 2016 [cited 2022 Oct 13];11(1):1–15. Available from: https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-016-0072-9.
Majbour NK, Abdi IY, Dakna M, Wicke T, Lang E, Ali Moussa HY, et al. Cerebrospinal α-Synuclein Oligomers Reflect Disease Motor Severity in DeNoPa Longitudinal Cohort. Mov Disord [Internet]. 2021 [cited 2022 Oct 13];36(9):2048–56. Available from: https://pubmed.ncbi.nlm.nih.gov/33978256/.
Parnetti L, Gaetani L, Eusebi P, Paciotti S, Hansson O, El-Agnaf O, et al. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol [Internet]. 2019 [cited 2022 Oct 13];18(6):573–86. Available from: https://pubmed.ncbi.nlm.nih.gov/30981640/.
Cariulo C, Martufi P, Verani M, Azzollini L, Bruni G, Weiss A, et al. Phospho-S129 alpha-synuclein is present in human plasma but not in cerebrospinal fluid as determined by an ultrasensitive immunoassay. Front Neurosci [Internet]. 2019 [cited 2022 Oct 13];13. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6714598/.
Chen NC, Chen HL, Li SH, Chang YH, Chen MH, Tsai NW, et al. Plasma levels of α-synuclein, Aβ-40 and T-tau as biomarkers to predict cognitive impairment in Parkinson’s disease. Front Aging Neurosci. 2020;28(12):112.
Chang CW, Yang SY, Yang CC, Chang CW, Wu YR. Plasma and serum alpha-synuclein as a biomarker of diagnosis in patients with Parkinson’s disease. Front Neurol [Internet]. 2020 [cited 2023 Mar 22];10. Available from: https://pubmed.ncbi.nlm.nih.gov/32038461/.
Youssef P, Kim WS, Halliday GM, Lewis SJG, Dzamko N. Comparison of different platform immunoassays for the measurement of plasma alpha-synuclein in Parkinson’s disease patients. J Parkinsons Dis [Internet]. 2021 [cited 2022 Nov 23];11(4):1761. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8609717/.
Duran R, Barrero FJ, Morales B, Luna JD, Ramirez M, Vives F. Plasma α-synuclein in patients with Parkinson’s disease with and without treatment. Mov Disord [Internet]. 2010 [cited 2022 Nov 22];25(4):489–93. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.22928.
Lee PH, Lee G, Park HJ, Bang OY, Joo IS, Huh K. The plasma alpha-synuclein levels in patients with Parkinson’s disease and multiple system atrophy. J Neural Transm 2006 11310 [Internet]. 2006 [cited 2022 Nov 22];113(10):1435–9. Available from: https://link.springer.com/article/10.1007/s00702-005-0427-9.
Fan Z, Pan YT, Zhang ZY, Yang H, Yu SY, Zheng Y, et al. Systemic activation of NLRP3 inflammasome and plasma α-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J Neuroinflammation [Internet]. 2020 [cited 2023 Mar 22];17(1). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6950934/.
Lin CH, Yang SY, Horng HE, Yang CC, Chieh JJ, Chen HH, et al. Plasma α-synuclein predicts cognitive decline in Parkinson’s disease. J Neurol Neurosurg Psychiatry [Internet]. 2017 [cited 2023 Mar 22];88(10):818–24. Available from: https://jnnp.bmj.com/content/88/10/818.
Caranci G, Piscopo P, Rivabene R, Traficante A, Riozzi B, Castellano AE, et al. Gender differences in Parkinson’s disease: Focus on plasma alpha-synuclein. J Neural Transm [Internet]. 2013 [cited 2022 Nov 22];120(8):1209–15. Available from: https://link.springer.com/article/10.1007/s00702-013-0972-6.
Shim KH, Kim SC, Youn YC, Sung YH, An SSA. Decreased plasma α-synuclein in idiopathic Parkinson’s disease patients after adjusting hemolysis factor. Mol Cell Toxicol [Internet]. 2020 [cited 2023 Mar 22];16(4):477–84. Available from: https://link.springer.com/article/10.1007/s13273-020-00104-7.
Gorostidi A, Bergareche A, Ruiz-Martínez J, Martí-Massó JF, Cruz M, Varghese S, et al. α-Synuclein levels in blood plasma from LRRK2 mutation carriers. PLoS One [Internet]. 2012 [cited 2022 Nov 22];7(12):e52312. Available from: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0052312.
Lin WT, Shaw JS, Cheng FY, Chen PH. Plasma total tau predicts executive dysfunction in Parkinson’s disease. Acta Neurol Scand [Internet]. 2022 [cited 2022 Nov 22];145(1):30–7. Available from: https://onlinelibrary.wiley.com/doi/full/10.1111/ane.13517.
Foulds PG, Diggle P, Mitchell JD, Parker A, Hasegawa M, Masuda-Suzukake M, et al. A longitudinal study on α-synuclein in blood plasma as a biomarker for Parkinson’s disease. Sci Reports [Internet]. 2013 [cited 2022 Nov 22];3(1):1–6. Available from: https://www.nature.com/articles/srep02540.
Ikemura M, Saito Y, Sengoku R, Sakiyama Y, Hatsuta H, Kanemaru K, et al. Lewy body pathology involves cutaneous nerves. J Neuropathol Exp Neurol [Internet]. 2008 [cited 2022 Nov 4];67(10):945–53. Available from: https://pubmed.ncbi.nlm.nih.gov/18800013/.
Gelpi E, Navarro-Otano J, Tolosa E, Gaig C, Compta Y, Rey MJ, et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov Disord [Internet]. 2014 [cited 2022 Nov 4];29(8):1010–8. Available from: https://pubmed.ncbi.nlm.nih.gov/24395122/.
Gibbons CH, Wang N, Freeman R. Cutaneous alpha-synuclein from paraffin embedded autopsy specimens in Parkinson’s disease. J Parkinsons Dis [Internet]. 2017 [cited 2021 Jul 26];7(3):503–9. Available from: https://pubmed.ncbi.nlm.nih.gov/28582870/.
Beach TG, Adler CH, Serrano G, Sue LI, Walker DG, Dugger BN, et al. Prevalence of submandibular gland synucleinopathy in Parkinson’s disease, dementia with Lewy bodies and other Lewy body disorders. J Parkinsons Dis [Internet]. 2016 [cited 2022 Nov 4];6(1):153. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5498170/.
Braak H, De Vos RAI, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett [Internet]. 2006 [cited 2022 Nov 4];396(1):67–72. Available from: https://pubmed.ncbi.nlm.nih.gov/16330147/.
Braak H, Rüb U, Gai WP, Del Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm [Internet]. 2003 [cited 2022 Nov 4];110(5):517–36. Available from: https://pubmed.ncbi.nlm.nih.gov/12721813/.
Kim S, Kwon S-H, Kam T-I, Panicker N, Karuppagounder SS, Lee S, et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron [Internet]. 2019 [cited 2021 Aug 3];103(4):627–641.e7. Available from: https://pubmed.ncbi.nlm.nih.gov/31255487/.
Beach TG, Adler CH, Sue LI, Shill HA, Driver-Dunckley E, Mehta SH, et al. Vagus nerve and stomach synucleinopathy in Parkinson’s disease, Incidental Lewy body disease, and normal elderly subjects: evidence against the “body-first” hypothesis. J Parkinsons Dis. 2021;11(4):1833–43.
Pouclet H, Lebouvier T, Coron E, Bruley des Varannes S, Rouaud T, Roy M, et al. A comparison between rectal and colonic biopsies to detect Lewy pathology in Parkinson’s disease. Neurobiol Dis [Internet]. 2012 [cited 2022 Nov 4];45(1):305–9. Available from: https://pubmed.ncbi.nlm.nih.gov/21878391/.
Chahine LM, Beach TG, Brumm MC, Adler CH, Coffey CS, Mosovsky S, et al. In vivo distribution of α-synuclein in multiple tissues and biofluids in Parkinson disease. Neurology [Internet]. 2020 [cited 2022 Nov 4];95(9):e1267–84. Available from: https://n.neurology.org/content/95/9/e1267.
Pouclet H, Lebouvier T, Coron E, Des Varannes SB, Neunlist M, Derkinderen P. A comparison between colonic submucosa and mucosa to detect Lewy pathology in Parkinson’s disease. Neurogastroenterol Motil [Internet]. 2012 [cited 2022 Nov 4];24(4). Available from: https://pubmed.ncbi.nlm.nih.gov/22292943/.
Corbillé AG, Letournel F, Kordower JH, Lee J, Shanes E, Neunlist M, et al. Evaluation of alpha-synuclein immunohistochemical methods for the detection of Lewy-type synucleinopathy in gastrointestinal biopsies. Acta Neuropathol Commun [Internet]. 2016 [cited 2022 Nov 4];4:35. Available from: https://pubmed.ncbi.nlm.nih.gov/27044604/.
Beach TG, Corbillé A-G, Letournel F, Kordower JH, Kremer T, Munoz DG, et al. Multicenter assessment of immunohistochemical methods for pathological alpha-synuclein in sigmoid colon of autopsied Parkinson’s disease and control subjects. J Parkinsons Dis [Internet]. 2016 [cited 2022 Nov 4];6(4):761–70. Available from: https://www.medra.org/servlet/aliasResolver?alias=iospress&doi=10.3233/JPD-160888.
Adler CH, Dugger BN, Hinni ML, Lott DG, Driver-Dunckley E, Hidalgo J, et al. Submandibular gland needle biopsy for the diagnosis of Parkinson disease. Neurology [Internet]. 2014 [cited 2022 Nov 4];82(10):858. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3959757/.
Adler CH, Dugger BN, Hentz JG, Hinni ML, Lott DG, Driver-Dunckley E, et al. Peripheral synucleinopathy in early Parkinson’s disease: submandibular gland needle biopsy findings. Mov Disord [Internet]. 2016 [cited 2022 Nov 4];31(2):250–6. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.26476.
Tredici K Del, Hawkes CH, Ghebremedhin E, Braak H. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathol [Internet]. 2010 [cited 2022 Nov 4];119(6):703–13. Available from: https://pubmed.ncbi.nlm.nih.gov/20229352/.
Donadio V, Doppler K, Incensi A, Kuzkina A, Janzen A, Mayer G, et al. Abnormal α-synuclein deposits in skin nerves: intra- and inter-laboratory reproducibility. Eur J Neurol [Internet]. 2019 [cited 2021 Jul 26];26(10):1245–51. Available from: https://pubmed.ncbi.nlm.nih.gov/30770596/.
Donadio V, Incensi A, Rizzo G, Scaglione C, Capellari S, Fileccia E, et al. Spine topographical distribution of skin α-synuclein deposits in idiopathic Parkinson disease. J Neuropathol Exp Neurol [Internet]. 2017 [cited 2022 Nov 5];76(5):384–9. Available from: https://academic.oup.com/jnen/article/76/5/384/3573469.
Donadio V, Incensi A, Rizzo G, Capellari S, Pantieri R, Stanzani Maserati M, et al. A new potential biomarker for dementia with Lewy bodies. Neurology [Internet]. 2017 [cited 2022 Dec 6];89(4):318–26. Available from: https://n.neurology.org/content/89/4/318.
Doppler K, Jentschke HM, Schulmeyer L, Vadasz D, Janzen A, Luster M, et al. Dermal phospho-alpha-synuclein deposits confirm REM sleep behaviour disorder as prodromal Parkinson’s disease. Acta Neuropathol [Internet]. 2017 [cited 2022 Nov 5];133(4):535–45. Available from: https://pubmed.ncbi.nlm.nih.gov/28180961/.
Doppler K, Weis J, Karl K, Ebert S, Ebentheuer J, Trenkwalder C, et al. Distinctive distribution of phospho-alpha-synuclein in dermal nerves in multiple system atrophy. Mov Disord [Internet]. 2015 [cited 2022 Nov 5];30(12):1688–92. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.26293.
Doppler K, Ebert S, Üçeyler N, Trenkwalder C, Ebentheuer J, Volkmann J, et al. Cutaneous neuropathy in Parkinson’s disease: a window into brain pathology. Acta Neuropathol [Internet]. 2014 [cited 2021 Jul 26];128(1):99–109. Available from: https://pubmed.ncbi.nlm.nih.gov/24788821/.
Navarro-Otano J, Casanova-Mollà J, Morales M, Valls-Solé J, Tolosa E. Cutaneous autonomic denervation in Parkinson’s disease. J Neural Transm [Internet]. 2015 [cited 2022 Nov 5];122(8):1149–55. Available from: https://pubmed.ncbi.nlm.nih.gov/25536890/.
Miki Y, Tomiyama M, Ueno T, Haga R, Nishijima H, Suzuki C, et al. Clinical availability of skin biopsy in the diagnosis of Parkinson’s disease. Neurosci Lett. 2010;469(3):357–9.
Donadio V, Incensi A, Piccinini C, Cortelli P, Giannoccaro MP, Baruzzi A, et al. Skin nerve misfolded α-synuclein in pure autonomic failure and Parkinson disease. Ann Neurol [Internet]. 2016 [cited 2022 Nov 5];79(2):306–16. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/ana.24567.
Donadio V, Incensi A, Leta V, Giannoccaro MP, Scaglione C, Martinelli P, et al. Skin nerve α-synuclein deposits: a biomarker for idiopathic Parkinson disease. Neurology [Internet]. 2014 [cited 2022 Nov 5];82(15):1362–9. Available from: https://pubmed.ncbi.nlm.nih.gov/24634456/.
Wang N, Garcia J, Freeman R, Gibbons CH. Phosphorylated alpha-synuclein within cutaneous autonomic nerves of patients with Parkinson’s disease: the implications of sample thickness on results. J Histochem Cytochem [Internet]. 2020 [cited 2023 Mar 29];68(10):669. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7534099/.
Beach TG, Serrano GE, Kremer T, Canamero M, Dziadek S, Sade H, et al. Immunohistochemical method and histopathology judging for the systemic synuclein sampling study (S4). J Neuropathol Exp Neurol [Internet]. 2018 [cited 2021 Jul 26];77(9):793–802. Available from: https://academic.oup.com/jnen/article/77/9/793/5063596.
Zange L, Noack C, Hahn K, Stenzel W, Lipp A. Phosphorylated α-synuclein in skin nerve fibres differentiates Parkinson’s disease from multiple system atrophy. Brain [Internet]. 2015 [cited 2022 Nov 5];138(Pt 8):2310–21. Available from: https://pubmed.ncbi.nlm.nih.gov/26017579/.
Doppler K, Ebert S, Uçeyler N, Trenkwalder C, Ebentheuer J, Volkmann J, et al. Cutaneous neuropathy in Parkinson’s disease: a window into brain pathology. Acta Neuropathol [Internet]. 2014 [cited 2021 Jul 26];128(1):99–109. Available from: https://pubmed.ncbi.nlm.nih.gov/24788821/.
Al-Qassabi A, Tsao TS, Racolta A, Kremer T, Cañamero M, Belousov A, et al. Immunohistochemical detection of synuclein pathology in skin in idiopathic rapid eye movement sleep behavior disorder and parkinsonism. Mov Disord [Internet]. 2021 [cited 2022 Nov 6];36(4):895–904. Available from: https://pubmed.ncbi.nlm.nih.gov/33232556/.
Donadio V, Wang Z, Incensi A, Rizzo G, Fileccia E, Vacchiano V, et al. In Vivo Diagnosis of synucleinopathies: a comparative study of skin biopsy and RT-QuIC. Neurology [Internet]. 2021 [cited 2022 Nov 6];96(20):e2513–24. Available from: https://pubmed.ncbi.nlm.nih.gov/33837116/.
Wang Z, Becker K, Donadio V, Siedlak S, Yuan J, Rezaee M, et al. Skin α-synuclein aggregation seeding activity as a novel biomarker for Parkinson disease. JAMA Neurol. 2020.
Scott GD, Arnold MR, Beach TG, Gibbons CH, Kanthasamy AG, Lebovitz RM, et al. Fluid and tissue biomarkers of Lewy body dementia: report of an LBDA symposium. Front Neurol [Internet]. 2022 [cited 2022 Nov 29];12. Available from: https://pubmed.ncbi.nlm.nih.gov/35173668/.
Donadio V. Skin nerve α-synuclein deposits in Parkinson’s disease and other synucleinopathies: a review. Clin Auton Res [Internet]. 2019 [cited 2022 Nov 4];29(6):577–85. Available from: https://link.springer.com/article/10.1007/s10286-018-0581-4.
Iranzo A, Fernández-Arcos A, Tolosa E, Serradell M, Molinuevo JL, Valldeoriola F, et al. Neurodegenerative disorder risk in idiopathic REM sleep behavior disorder: study in 174 patients. PLoS One [Internet]. 2014 [cited 2022 Nov 6];9(2):e89741. Available from: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0089741.
Coon EA, Mandrekar JN, Berini SE, Benarroch EE, Sandroni P, Low PA, et al. Predicting phenoconversion in pure autonomic failure. Neurology [Internet]. 2020 [cited 2022 Nov 6];95(7):E889–97. Available from: https://pubmed.ncbi.nlm.nih.gov/32546656/.
Miglis MG, Zitser J, Schneider L, During E, Jaradeh S, Freeman R, et al. Cutaneous α-synuclein is correlated with autonomic impairment in isolated rapid eye movement sleep behavior disorder. Sleep [Internet]. 2021 [cited 2022 Nov 6];44(12). Available from: https://pubmed.ncbi.nlm.nih.gov/34244806/.
Puligheddu M, Figorilli M, Antelmi E, Arnaldi D, Casaglia E, d’Aloja E, et al. Predictive risk factors of phenoconversion in idiopathic REM sleep behavior disorder: the Italian study “FARPRESTO.” Neurol Sci [Internet]. 2022 [cited 2022 Nov 6];1:1–10. Available from: https://link.springer.com/article/10.1007/s10072-022-06374-4.
Donadio V, Incensi A, Rizzo G, De Micco R, Tessitore A, Devigili G, et al. Skin biopsy may help to distinguish multiple system atrophy-parkinsonism from Parkinson’s disease with orthostatic hypotension. Mov Disord [Internet]. 2020 [cited 2022 Nov 6];35(9):1649–57. Available from: https://pubmed.ncbi.nlm.nih.gov/32557839/.
Gibbons C, Wang N, Rajan S, Kern D, Palma J-A, Kaufmann H, et al. Cutaneous α-synuclein signatures in patients with multiple system atrophy and Parkinson disease. Neurology [Internet]. 2023 [cited 2023 Mar 23]; https://doi.org/10.1212/WNL.0000000000206772. Available from: https://pubmed.ncbi.nlm.nih.gov/36657992/.
Senard JM, Raï S, Lapeyre-Mestre M, Brefel C, Rascol O, Rascol A, et al. Prevalence of orthostatic hypotension in Parkinson’s disease. J Neurol Neurosurg Psychiatry [Internet]. 1997 [cited 2022 Nov 6];63(5):584–9. Available from: https://jnnp.bmj.com/content/63/5/584.
Velseboer DC, de Haan RJ, Wieling W, Goldstein DS, de Bie RMA. Prevalence of orthostatic hypotension in Parkinson’s disease: a systematic review and meta-analysis. Parkinsonism Relat Disord. 2011;17(10):724–9.
Donadio V, Incensi A, Del Sorbo F, Rizzo G, Infante R, Scaglione C, et al. Skin nerve phosphorylated α-synuclein deposits in Parkinson disease with orthostatic hypotension. J Neuropathol Exp Neurol [Internet]. 2018 [cited 2022 Nov 6];77(10):942–9. Available from: https://pubmed.ncbi.nlm.nih.gov/30137426/.
Infante R, Scaglione C, Incensi A, Rizzo G, Liguori R, Donadio V. A longitudinal skin biopsy study of phosphorylated alpha-synuclein in a patient with Parkinson disease and orthostatic hypotension. J Neuropathol Exp Neurol [Internet]. 2020 [cited 2022 Nov 6];79(7):813–6. Available from: https://academic.oup.com/jnen/article/79/7/813/5856234.
Yang J, Wang H, Yuan Y, Fan S, Li L, Jiang C, et al. Peripheral synucleinopathy in Parkinson disease with LRRK2 G2385R variants. Ann Clin Transl Neurol [Internet]. 2021 [cited 2022 Nov 6];8(3):592–602. Available from: https://pubmed.ncbi.nlm.nih.gov/33527742/.
Isonaka R, Goldstein DS, Zhu W, Yoon E, Ehrlich D, Schindler AB, et al. α‐synuclein deposition in sympathetic nerve fibers in genetic forms of Parkinson’s disease. Mov Disord [Internet]. 2021 [cited 2022 Nov 6];36(10):2346–57. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.28667.
Doppler K, Brockmann K, Sedghi A, Wurster I, Volkmann J, Oertel WH, et al. Dermal phospho-alpha-synuclein deposition in patients with Parkinson’s disease and mutation of the glucocerebrosidase gene. Front Neurol [Internet]. 2018 [cited 2022 Nov 6];9. Available from: https://pubmed.ncbi.nlm.nih.gov/30619053/.
Carmona-Abellan M, Gabilondo I, Murueta-Goyena A, Khurana V, Tijero B, Luquin MR, et al. Small fiber neuropathy and phosphorylated alpha-synuclein in the skin of E46K-SNCA mutation carriers. Parkinsonism Relat Disord [Internet]. 2019 [cited 2022 Nov 6];65:139–45. Available from: https://pubmed.ncbi.nlm.nih.gov/31178336/.
Poulopoulos M, Levy OA, Alcalay RN. The neuropathology of genetic Parkinson’s disease. Mov Disord [Internet]. 2012 [cited 2022 Nov 6];27(7):831–42. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.24962.
Poulopoulos M, Cortes E, Vonsattel JPG, Fahn S, Waters C, Cote LJ, et al. Clinical and pathological characteristics of LRRK2 G2019S patients with PD. J Mol Neurosci [Internet]. 2012 [cited 2022 Nov 6];47(1):139. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3335886/.
Gibbons CH, Freeman R, Bellaire B, Adler CH, Moore D, Levine T. Synuclein-One study: Skin biopsy detection of phosphorylated α-synuclein for diagnosis of synucleinopathies. Biomark Med [Internet]. 2022 [cited 2023 Mar 21];16(7):499–509. Available from: https://www.futuremedicine.com/doi/10.2217/bmm-2021-0646.
Scott GD, Arnold MR, Beach TG, Gibbons CH, Kanthasamy AG, Lebovitz RM, et al. Fluid and tissue biomarkers of Lewy body dementia: report of an LBDA symposium. Front Neurol [Internet]. 2021 [cited 2023 Mar 21];12:805135. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8841880/.
Sano K, Atarashi R, Satoh K, Ishibashi D, Nakagaki T, Iwasaki Y, et al. Prion-like seeding of misfolded alpha-synuclein in the brains of dementia with Lewy body patients in RT-QUIC. Mol Neurobiol. 2018;55(5):3916–30.
Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, et al. Prion-like spreading of pathological α-synuclein in brain. Brain. 2013;136(4):1128–38.
Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. 2010;11(3):155.
Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nat 2020 5787794 [Internet]. 2020 [cited 2021 Aug 7];578(7794):273–7. Available from: https://www.nature.com/articles/s41586-020-1984-7.
Saijo E, Groveman BR, Kraus A, Metrick M, Orru CD, Hughson AG, et al. Ultrasensitive RT-QuIC seed amplification assays for disease-associated tau, alpha-synuclein, and prion aggregates. Methods Mol Biol. 2019;1873:19–37.
Singer W, Schmeichel AM, Shahnawaz M, Schmelzer JD, Boeve BF, Sletten DM, et al. Alpha‐synuclein oligomers and neurofilament light chain in spinal fluid differentiate multiple system atrophy from Lewy body synucleinopathies. Ann Neurol [Internet]. 2020 [cited 2022 Sep 25];88(3):503–12. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/ana.25824.
Kang UJ, Boehme AK, Fairfoul G, Shahnawaz M, Ma TC, Hutten SJ, et al. Comparative study of cerebrospinal fluid α-synuclein seeding aggregation assays for diagnosis of Parkinson’s disease. Mov Disord. 2019;34(4):536–44.
Russo MJ, Orru CD, Concha-Marambio L, Giaisi S, Groveman BR, Farris CM, et al. High diagnostic performance of independent alpha-synuclein seed amplification assays for detection of early Parkinson’s disease. Acta Neuropathol Commun [Internet]. 2021 [cited 2022 Sep 25];9(1). Available from: https://pubmed.ncbi.nlm.nih.gov/34742348/.
Rossi M, Candelise N, Baiardi S, Capellari S, Giannini G, Orrù CD, et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol. 2020.
Iranzo A, Fairfoul G, Ayudhaya ACN, Serradell M, Gelpi E, Vilaseca I, et al. Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: a longitudinal observational study. Lancet Neurol. 2021;20(3):203–12.
Poggiolini I, Gupta V, Lawton M, Lee S, El-Turabi A, Querejeta-Coma A, et al. Diagnostic value of cerebrospinal fluid alpha-synuclein seed quantification in synucleinopathies. Brain [Internet]. 2022 [cited 2022 Nov 2];145(2):584–95. Available from: https://pubmed.ncbi.nlm.nih.gov/34894214/.
Dougherty R. Animal virus titration techniques. Harris R, editor. Vol. 178, Techniques in experimental virology. New York: Academic Press; 1964. 183–6 p.
Kuzkina A, Bargar C, Schmitt D, Rößle J, Wang W, Schubert AL, et al. Diagnostic value of skin RT-QuIC in Parkinson’s disease: a two-laboratory study. NPJ Park Dis [Internet]. 2021 [cited 2022 Nov 2];7(1). Available from: https://pubmed.ncbi.nlm.nih.gov/34782640/.
Mammana A, Baiardi S, Quadalti C, Rossi M, Donadio V, Capellari S, et al. RT-QuIC detection of pathological α-synuclein in skin punches of patients with Lewy body disease. Mov Disord [Internet]. 2021 [cited 2022 Nov 2];36(9):2173–7. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.28651.
Bargar C, Wang W, Gunzler SA, LeFevre A, Wang Z, Lerner AJ, et al. Streamlined alpha-synuclein RT-QuIC assay for various biospecimens in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol Commun [Internet]. 2021 [cited 2022 Nov 2];9(1). Available from: https://pubmed.ncbi.nlm.nih.gov/33827706/.
Iranzo A, Mammana A, Muñoz-Lopetegi A, Dellavalle S, Mayà ; Gerard, Rossi M, et al. Misfolded α-synuclein assessment in skin and CSF by RT-QuIC in isolated REM sleep behavior disorder. 2023.
Wang Z, Becker K, Donadio V, Siedlak S, Yuan J, Rezaee M, et al. Skin α-synuclein aggregation seeding activity as a novel biomarker for Parkinson disease. JAMA Neurol [Internet]. 2021 [cited 2023 Mar 29];78(1):1. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7522783/.
De Luca CMG, Elia AE, Portaleone SM, Cazzaniga FA, Rossi M, Bistaffa E, et al. Efficient RT-QuIC seeding activity for α-synuclein in olfactory mucosa samples of patients with Parkinson’s disease and multiple system atrophy. Transl Neurodegener [Internet]. 2019 [cited 2021 Jul 26];8(1):24. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31406572.
Perra D, Bongianni M, Novi G, Janes F, Bessi V, Capaldi S, et al. Alpha-synuclein seeds in olfactory mucosa and cerebrospinal fluid of patients with dementia with Lewy bodies. Brain Commun [Internet]. 2021 [cited 2022 Nov 2];3(2). Available from: https://pubmed.ncbi.nlm.nih.gov/33870192/.
Stefani A, Iranzo A, Holzknecht E, Perra D, Bongianni M, Gaig C, et al. Alpha-synuclein seeds in olfactory mucosa of patients with isolated REM sleep behaviour disorder. Brain [Internet]. 2021 [cited 2022 Nov 2];144(4):1118–26. Available from: https://pubmed.ncbi.nlm.nih.gov/33855335/.
Manne S, Kondru N, Jin H, Anantharam V, Huang X, Kanthasamy A, et al. α-Synuclein real-time quaking-induced conversion in the submandibular glands of Parkinson’s disease patients. Mov Disord. 2020;35(2):268–78.
Luan M, Sun Y, Chen J, Jiang Y, Li F, Wei L, et al. Diagnostic value of salivary real-time quaking-induced conversion in Parkinson’s disease and multiple system atrophy. Mov Disord [Internet]. 2022 [cited 2022 Nov 2];37(5):1059–63. Available from: https://pubmed.ncbi.nlm.nih.gov/35278004/.
Vivacqua G, Mason M, De Bartolo MI, Węgrzynowicz M, Calò L, Belvisi D, et al. Salivary α-synuclein RT-QuIC correlates with disease severity in de novo Parkinson’s disease. Mov Disord [Internet]. 2022 [cited 2022 Nov 11]; Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.29246.
Shannon KM, Keshavarzian A, Mutlu E, Dodiya HB, Daian D, Jaglin JA, et al. Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov Disord. 2012;27(6):709–15.
Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using α-synuclein immunohistochemistry. Brain Pathol. 2000;10(3):378–84.
Leverenz JB, Fishel MA, Peskind ER, Montine TJ, Nochlin D, Steinbart E, et al. Lewy body pathology in familial Alzheimer disease: evidence for disease-and mutation-specific pathologic phenotype. Arch Neurol. 2006;63(3):370–6.
Kotzbauer PT, Trojanowski JQ, Lee VMY. Lewy body pathology in Alzheimer’s disease. J Mol Neurosci [Internet]. 2001 [cited 2022 Nov 3];17(2):225–32. Available from: https://pubmed.ncbi.nlm.nih.gov/11816795/.
Roudil J, Deramecourt V, Dufournet B, Dubois B, Ceccaldi M, Duyckaerts C, et al. Influence of Lewy pathology on Alzheimer’s disease phenotype: a retrospective clinico-pathological study. J Alzheimers Dis [Internet]. 2018 [cited 2022 Nov 3];63(4):1317–23. Available from: https://pubmed.ncbi.nlm.nih.gov/29758938/.
Hall S, Orrù CD, Serrano GE, Galasko D, Hughson AG, Groveman BR, et al. Performance of αSynuclein RT-QuIC in relation to neuropathological staging of Lewy body disease. Acta Neuropathol Commun [Internet]. 2022 [cited 2022 Nov 3];10(1). Available from: https://pubmed.ncbi.nlm.nih.gov/35733234/.
Arnold MR, Coughlin DG, Brumbach BH, Smirnov DS, Concha-Marambio L, Farris CM, et al. α-Synuclein seed amplification in CSF and brain from patients with different brain distributions of pathological α-synuclein in the context of co-pathology and non-LBD diagnoses. Ann Neurol [Internet]. 2022 [cited 2022 Nov 3];92(4):650–62. Available from: https://pubmed.ncbi.nlm.nih.gov/35808984/.
Zaccai J, Brayne C, Matthews FE, Ince PG. Alpha-synucleinopathy and neuropsychological symptoms in a population-based cohort of the elderly. Alzheimers Res Ther. 2015;7(1):19.
Parkkinen L, Pirttila T, Tervahauta M, Alafuzoff I. Widespread and abundant alpha-synuclein pathology in a neurologically unimpaired subject. Neuropathology. 2005;25(4):304–14.
Milber JM, Noorigian JV, Morley JF, Petrovitch H, White L, Ross GW, et al. Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology. 2012;79(24):2307–14.
Zaccai J, Brayne C, McKeith I, Matthews F, Ince PG. Patterns and stages of alpha-synucleinopathy: Relevance in a population-based cohort. Neurology. 2008;70(13):1042–8.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44(4):601–7.
Brockmann K, Quadalti C, Lerche S, Rossi M, Wurster I, Baiardi S, et al. Association between CSF alpha-synuclein seeding activity and genetic status in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol Commun [Internet]. 2021 [cited 2022 Nov 29];9(1):1–11. Available from: https://actaneurocomms.biomedcentral.com/articles/10.1186/s40478-021-01276-6.
Garrido A, Fairfoul G, Tolosa E, Marti MJ, Ezquerra M, Green AJE. Brain and cerebrospinal fluid α-synuclein real-time quaking-induced conversion identifies Lewy body pathology in LRRK2-PD. Mov Disord [Internet]. 2023 [cited 2023 Mar 23];38(2):333–8. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.29284.
Siderowf A, Concha-Marambio L, Lafontant DE, Farris CM, Ma Y, Urenia PA, Nguyen H, Alcalay RN, Chahine LM, Foroud T, Galasko D. Assessment of heterogeneity among participants in the Parkinson's Progression Markers Initiative cohort using α-synuclein seed amplification: a cross-sectional study. The Lancet Neurology. 2023 May 1;22(5):407-17
Concha-Marambio L, Pritzkow S, Shahnawaz M, Farris CM, Soto C. Seed amplification assay for the detection of pathologic alpha-synuclein aggregates in cerebrospinal fluid. Nat Protoc 2023 [Internet]. 2023 [cited 2023 Mar 21];1–18. Available from: https://www.nature.com/articles/s41596-022-00787-3.
Fereshtehnejad SM, Romenets SR, Anang JB, Latreille V, Gagnon JF, Postuma RB. New clinical subtypes of Parkinson disease and their longitudinal progression: a prospective cohort comparison with other phenotypes. JAMA Neurol [Internet]. 2015/06/16. 2015;72(8):863–73. Available from: http://archneur.jamanetwork.com/pdfaccess.ashx?url=/data/journals/neur/934259/noi150022.pdf.
Fereshtehnejad SM, Zeighami Y, Dagher A, Postuma RB. Clinical criteria for subtyping Parkinson’s disease: biomarkers and longitudinal progression. Brain [Internet]. 2017/05/27. 2017;140(7):1959–76. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28549077.
Toledo JB, Abdelnour C, Weil RS, Ferreira D, Rodriguez-Porcel F, Pilotto A, et al. Dementia with Lewy bodies: impact of co-pathologies and implications for clinical trial design. Alzheimer’s Dement [Internet]. 2022 [cited 2022 Nov 6]; Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/alz.12814.
Alves G, Bronnick K, Aarsland D, Blennow K, Zetterberg H, Ballard C, et al. CSF amyloid-beta and tau proteins, and cognitive performance, in early and untreated Parkinson’s disease: the Norwegian ParkWest study. J Neurol Neurosurg Psychiatry. 2010;81(10):1080–6.
Kang JH, Irwin DJ, Chen-Plotkin AS, Siderowf A, Caspell C, Coffey CS, et al. Association of cerebrospinal fluid beta-amyloid 1–42, T-tau, P-tau181, and alpha-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA Neurol. 2013;70(10):1277–87.
Montine TJ, Shi M, Quinn JF, Peskind ER, Craft S, Ginghina C, et al. CSF Aβ42 and tau in Parkinson’s disease with cognitive impairment. Mov Disord. 2010;25(15):2682–5.
Compta Y, Pereira JB, Rios J, Ibarretxe-Bilbao N, Junque C, Bargallo N, et al. Combined dementia-risk biomarkers in Parkinson’s disease: a prospective longitudinal study. Parkinsonism Relat Disord. 2013;19(8):717–24.
Hatcher-Martin JM, McKay JL, Pybus AF, Sommerfeld B, Howell JC, Goldstein FC, et al. Cerebrospinal fluid biomarkers in Parkinson’s disease with freezing of gait: an exploratory analysis. NPJ Park Dis [Internet]. 2021 [cited 2023 Mar 14];7(1):1–7. Available from: https://www.nature.com/articles/s41531-021-00247-x.
Compta Y, Martí MJ, Ibarretxe-Bilbao N, Junqué C, Valldeoriola F, Munoz E, et al. Cerebrospinal tau, phospho-tau, and beta-amyloid and neuropsychological functions in Parkinson’s disease. Mov Disord. 2009;24(15):2203–10.
Liu C, Cholerton B, Shi M, Ginghina C, Cain KC, Auinger P, et al. CSF tau and tau/Abeta42 predict cognitive decline in Parkinson’s disease. Parkinsonism Relat Disord. 2015;21(3):271–6.
Mollenhauer B, Trenkwalder C, von Ahsen N, Bibl M, Steinacker P, Brechlin P, et al. Beta-amlyoid 1–42 and tau-protein in cerebrospinal fluid of patients with Parkinson’s disease dementia. Dement Geriatr Cogn Disord. 2006;22(3):200–8.
Baek MS, Lee MJ, Kim HK, Lyoo CH. Temporal trajectory of biofluid markers in Parkinson’s disease. Sci Reports [Internet]. 2021 [cited 2022 Nov 22];11(1):1–12. Available from: https://www.nature.com/articles/s41598-021-94345-8.
Irwin DJ, Xie SX, Coughlin D, Nevler N, Akhtar RS, McMillan CT, et al. CSF tau and amyloid-beta predict cerebral synucleinopathy in autopsied Lewy body disorders. Neurology. 2018. https://doi.org/10.1212/WNL.0000000000005166.
Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, et al. Cerebrospinal fluid beta-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66(3):382–9.
Mattsson N, Zetterberg H, Janelidze S, Insel PS, Andreasson U, Stomrud E, et al. Plasma tau in Alzheimer disease. Neurology [Internet]. 2016 [cited 2022 Nov 22];87(17):1827–35. Available from: https://n.neurology.org/content/87/17/1827.
Barthélemy NR, Horie K, Sato C, Bateman RJ. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J Exp Med [Internet]. 2020 [cited 2022 Nov 22];217(11). Available from: https://doi.org/10.1084/jem.20200861.
Chen SD, Huang YY, Shen XN, Guo Y, Tan L, Dong Q, et al. Longitudinal plasma phosphorylated tau 181 tracks disease progression in Alzheimer’s disease. Transl Psychiatry [Internet]. 2021 [cited 2022 Nov 22];11(1):1–10. Available from: https://www.nature.com/articles/s41398-021-01476-7.
Ashton NJ, Pascoal TA, Karikari TK, Benedet AL, Lantero-Rodriguez J, Brinkmalm G, et al. Plasma p-tau231: a new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol [Internet]. 2021 [cited 2022 Nov 22];141(5):709–24. Available from: https://pubmed.ncbi.nlm.nih.gov/33585983/.
Ding J, Zhang J, Wang X, Zhang L, Jiang S, Yuan Y, et al. Relationship between the plasma levels of neurodegenerative proteins and motor subtypes of Parkinson’s disease. J Neural Transm [Internet]. 2017 Mar 1 [cited 2022 Nov 22];124(3):353–60. Available from: https://link.springer.com/article/10.1007/s00702-016-1650-2.
Walker IM, Cousins KA, Siderowf A, Duda JE, Morley JF, Dahodwala N, et al. Non-tremor motor dysfunction in Lewy body dementias is associated with AD biomarkers. Parkinsonism Relat Disord [Internet]. 2022 [cited 2022 Nov 27];100:33–6. Available from: https://pubmed.ncbi.nlm.nih.gov/35700626/.
Hall S, Janelidze S, Londos E, Leuzy A, Stomrud E, Dage JL, et al. Plasma phospho-tau identifies Alzheimer’s co-pathology in patients with Lewy body disease. Mov Disord [Internet]. 2021 [cited 2022 Nov 22];36(3):767–71. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/mds.28370.
Gonzalez MC, Ashton NJ, Gomes BF, Tovar-Rios DA, Blanc F, Karikari TK, et al. Association of plasma p-tau181 and p-tau231 concentrations with cognitive decline in patients with probable dementia with Lewy bodies. JAMA Neurol [Internet]. 2022 [cited 2022 Nov 22];79(1):32–7. Available from: https://pubmed.ncbi.nlm.nih.gov/34807233/.
Ren J, Pan C, Wang Y, Xue C, Lin H, Xu J, et al. Plasma α-synuclein and phosphorylated tau 181 as a diagnostic biomarker panel for de novo Parkinson’s disease. J Neurochem [Internet]. 2022 [cited 2022 Nov 22];161(6):506–15. Available from: https://onlinelibrary.wiley.com/doi/full/10.1111/jnc.15601.
Batzu L, Rota S, Hye A, Heslegrave A, Trivedi D, Gibson LL, et al. Plasma p-tau181, neurofilament light chain and association with cognition in Parkinson’s disease. npj Park Dis 2022 81 [Internet]. 2022 [cited 2022 Nov 22];8(1):1–7. Available from: https://www.nature.com/articles/s41531-022-00384-x.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Coughlin, D.G., Irwin, D.J. Fluid and Biopsy Based Biomarkers in Parkinson’s Disease. Neurotherapeutics 20, 932–954 (2023). https://doi.org/10.1007/s13311-023-01379-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-023-01379-z