
Abstract
There are a broad range of disease-modifying therapies (DMTs) available in relapsing-remitting multiple sclerosis (RRMS), but limited biomarkers exist to personalise DMT choice. All DMTs, including monoclonal antibodies such as rituximab and ocrelizumab, are effective in preventing relapses and preserving neurological function in MS. However, each agent harbours its own risk of therapeutic failure or adverse events. Pharmacogenetics, the study of the effects of genetic variation on therapeutic response or adverse events, could improve the precision of DMT selection. Pharmacogenetic studies of rituximab in MS patients are lacking, but pharmacogenetic markers in other rituximab-treated autoimmune conditions have been identified. This review will outline the wider implications of pharmacogenetics and the mechanisms of anti-CD20 agents in MS. We explore the non-MS rituximab literature to characterise pharmacogenetic variants that could be of prognostic relevance in those receiving rituximab, ocrelizumab or other monoclonal antibodies for MS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
For patients with relapsing-remitting multiple sclerosis (RRMS), available disease-modifying therapies (DMTs) are key, but imperfect, in preventing relapses and disease progression. Delayed effective treatment and prolonged exposure to suboptimal therapy increases the risk of irreversible disability [1,2,3] and adverse events. The sources of inter-individual therapeutic variability, particularly genetic factors, are incompletely understood. Pharmacogenetic markers could optimise long-term disease outcomes by improving treatment selection for a given patient. Existing pharmacogenetic and pharmacogenomic studies of MS DMTs have not yet led to significant changes in prescribing practice, and are largely limited to examinations of interferon-beta and glatiramer acetate [4]. Some pharmacogenetic markers for these two modestly effective agents have been replicated and show promise as predictors of therapeutic success. However, a more complete understanding of the gene-drug interactions that dictate the efficacy of DMTs, particularly higher efficacy therapies including monoclonal antibodies, is required. Rituximab, an anti-CD20 monoclonal antibody with demonstrated efficacy in RRMS and other neurological, rheumatological and haematological conditions, represents a currently unique opportunity to explore and identify these mechanisms.
Pharmacogenetics and Pharmacogenomics
The spectrum of inter-individual pharmacokinetic variability was clarified with assays able to identify those with unusually high or low plasma drug or metabolite levels. The discovery of genetic variation in the cytochrome P-450 (CYP450) family of enzymes, and other metabolic and transport pathways that underpin this unpredictability, was a key achievement [5]. Pharmacogenetics is the study of DNA variations that relate to drug response. Single-nucleotide polymorphisms (SNPs), genetic variations present in at least 1% of a population, are the focus of most genetic studies of complex traits such as drug response. In each human genome, there are approximately 4–5 million SNPs, one for every 1000 nucleotides [6]. Candidate genes, selected for known associations with disease phenotypes, were the initial focus of pharmacogenetics studies. The advent of next-generation sequencing (NGS) revealed the whole exome (protein-coding DNA) and genome (almost all DNA), and has resulted in an exponential increase of known rare and common SNPs in the last two decades [7, 8]. With the availability of electronic health records and associated infrastructure, gene and genome-based therapeutic personalisation has the potential to be diversely and recurrently useful throughout a lifetime [9].
Gene-drug interactions can affect pharmacokinetics (absorption, distribution, metabolism or elimination) or, less commonly, pharmacodynamics (target effects) [10]. Prescribers may therefore benefit from the genetic identification of patients at risk of therapeutic failure or drug toxicity. Although most are not screened for in routine practice, 91 to 99% of patients possess at least one common genetic variant that would prompt more vigilant surveillance, a change in dosing, or avoidance, of certain medications [11, 12]. As of September 2020, the Clinical Pharmacogenetics Implementation Consortium (CPIC) recognises 416 gene interactions with drugs or drug classes, 34 of which have established guidelines (Table 1). Although some increase enzymatic activity (e.g. CYP2C19*17 and CYP2D6 duplications), the majority of these variants effect a partial or complete inhibition of function [5]. Examples include allelic variants of CYP450 enzymes such as CYP2C19 and CYP2D6, which predict outcomes in patients taking antidepressants or antipsychotics [13,14,15,16]. An individual’s metaboliser status can be described on a range between “poor” and “ultrarapid,” with corresponding variations in drug plasma concentrations, efficacy and side effect profile [17]. Codeine and other oral opioid formulations require enzymatic bioactivation for efficacy [18]. Poor CYP2D6 metabolisers are subjected to therapeutic inefficiency or failure, and ultrarapid metabolisers are posed a significantly higher risk of opioid toxicity [19]. Clopidogrel, an antiplatelet that similarly requires activation by CYP2C19, is commonly prescribed for secondary prevention of cardiovascular events. Patients who have poor or intermediate CYP2C19 activity are subjected to significantly worse cardiovascular outcomes, such as in-stent restenosis following percutaneous coronary intervention [20]. Where gene-informed prescribing of clopidogrel is available, multiple studies have shown its superiority, with significantly lower ischaemic and bleeding events compared to usual prescribing practices [21,22,23,24].
Contemporary oncology routinely makes use of tumour sequencing or gene product assays to identify driver mutations, successfully targeting these mutations with molecular therapies [25]. Gene product targets include human epidermal growth factor receptor 2 (HER2) in breast cancer (trastuzumab), epidermal growth factor receptor (EGFR) in colorectal cancer (cetuximab) and CD20 in chronic lymphocytic leukaemia and non-Hodgkin’s lymphoma (rituximab). With selective cytotoxicity against CD20-positive B cells, rituximab became the first clinically used chemotherapeutic monoclonal antibody in 1997; it has since shown efficacy in various autoimmune conditions such as rheumatoid arthritis [26] and multiple sclerosis [27].
B Cells and MS
Although initially thought to be primarily driven by aberrantly activated cytotoxic (CD8+) and helper (CD4+) T cells, evidence now suggests the inflammation in MS stems from more complex and bidirectional interactions between T cells and antigen-presenting cells (APCs) such as B cells and myeloid cells (macrophages, dendritic cells and microglia) [28,29,30,31]. The pro-inflammatory pathogenicity of B cells in MS most likely involves antibody-independent processes such as antigen presentation, cytokine production and formation of ectopic lymphoid organs in the central nervous system (CNS) [32, 33].
Antibody Production
Plasma cells derived from B cells fulfil the function of humoral immunity in the adaptive immune system. B cell receptor (BCR) binding to a specific antigen results in clonal expansion of the B cell, maturation to plasma cells and a coordinated antibody response. Antibodies have been implicated in active MS lesions, where they can be found along myelin sheathes, and within phagocytosing macrophages [34, 35]. A signature of humoral activity is the intrathecal presence of oligoclonal bands: clonally restricted antibodies, produced by plasma cells that undergo clonal expansion in the cerebrospinal fluid (CSF) [36,37,38]. Autoantibodies are an unlikely primary pathogenic mechanism in MS, particularly because anti-CD20 therapies—which deplete B cells, but usually not plasma cells or immunoglobulin levels—are highly effective in the disease [27, 39].
Antigen Presentation
Unlike myeloid APCs such as macrophages and neutrophils, B cells are highly selective for antigens bound to their unique BCR [40]. The antigen-BCR complex is internalised and processed, before being presented externally to CD4+ T cells (Th1 and Th17) with complementary T cell receptors. This process is dependent on major histocompatibility complex class II (MHC-II) and costimulatory molecules (CD80, CD86 and CD40). In MS, priming of T cells is caused by autoreactive B cells, which more densely express MHC-II and costimulatory molecules and demonstrate higher levels of antigen-presenting activity compared to B cells of healthy controls [41, 42].
Cytokine Production
The binding of autoantigen to BCR also causes aberrant B cells to produce effector cytokines that perpetuate the inflammatory milieu. In MS, the effector cytokine profile is abnormally inflammatory compared to controls, with elevated interleukin 6 (IL-6) and depressed IL-10 levels [43, 44]. Anti-inflammatory effects are mediated by regulatory B (B-reg) cells and cytokines such as IL-10 and IL-35; in patients with MS, these B-reg cells respond and differentiate aberrantly compared to controls [45]. IL-6 supports differentiation of CD4+ T cells into pro-inflammatory Th17 cells and diverts the pool away from regulatory T cells [46, 47], an effect that is mitigated by B cell depletion [48]. Other B cell–secreted cytokines upregulated in MS include granulocyte macrophage–colony-stimulating factor (GM-CSF), which increases mobility and activity of myeloid populations [49], and B cell activation factor (BAFF), which supports B cell self-propagation [50].
Ectopic Lymphoid Follicle Formation
In this landscape of pro-inflammatory cytokines, chemokines and lymphotoxin signalling, B cells support the development of ectopic lymphoid follicles (ELFs), also known as tertiary lymphoid organs [51]. The presence of ELFs in the CNS of MS patients is associated with progressive phenotypes and an earlier onset of the disease [52]. Over time, the primary influence of B cells shifts from infiltrating primed cells that cause focal relapses, to the support and seeding of autonomous populations sequestered in ELFs that drive grey and white matter atrophy [53]. ELFs are similarly described in autoimmune diseases such as systemic lupus erythematosus, as well as cancers and organ transplant rejection, where they perpetuate chronic inflammation in target tissues [54]. B cells residing in transplant-associated ELFs appear relatively protected from anti-B cell therapy: this may in part be due to paracrine secretion of BAFF [55]. In MS, this is further compounded by a relative lack of drug access to the CNS [56, 57].
Anti-CD20 Therapies and Mechanisms in MS
Therapies for RRMS such as alemtuzumab, cladribine and mitoxantrone cause cytolysis of B cells. Fingolimod sequesters lymphocytes including B cells, teriflunomide is cytostatic, while natalizumab prevents their trafficking across the blood-brain barrier. However, it was the efficacy of the anti-CD20 agents in preventing relapse and progression that led to the appreciation of B cells as key to aberrant inflammation in MS [40, 58]. Anti-CD20 therapies deplete cells displaying CD20 (pre-B cells, mature and memory B cells and some plasmablasts), but not B cell progenitors (pro-B cells) and terminally differentiated plasma cells [40, 59]. Administration therefore causes selective loss of B cell lineage cells responsible for antigen presentation and cytokine production, without affecting B cell reconstitution or pre-existing humoral immunity [39]. T cells expressing CD20 are also affected [60, 61]: this could be relevant as patients with MS have an increased proportion of CD20-expressing anti-myelin T cells compared to controls [62, 63]. A negligible peripheral B cell count can be seen as early as 4 days following infusion, with radiological benefit demonstrable at 4 weeks and clinical benefit apparent at an average of 8 weeks [64]. The duration of effect is variable but thought to be typically 6 to 9 months, and is followed by repopulation with B cells that are more naïve and less autoreactive than the mature and memory B cells they replace [65].
Upon binding to B cells, anti-CD20 agents induce both complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC). In CDC, C1q binding to the Fc region of the anti-CD20 antibody results in activation of the complement cascade, complement deposition onto the B cell and apoptosis due to membrane attack complex (MAC) insertion [66]. In ADCC, immune effector cells recognise the Fc portion of the bound anti-CD20 antibody, leading to cytotoxicity or phagocytosis of the B cell [67, 68]. Although CDC is believed to be the major effector mechanism for rituximab, the anti-CD20 family now includes several agents of different constitution, each with a distinct ADCC and CDC profile (Table 2). ADCC is dependent on the binding of the Fc domain to the Ig receptor (FcγR) of effector cells such as natural killer (NK) cells, monocytes, macrophages and dendritic cells [69]. The classes of these receptors include stimulatory (high affinity FcγRI, and low affinity FcγRIIA and FcγRIIIA) and inhibitory FcγRs (FcγRIIB) [70]. FcγR binding also facilitates elimination of immune complexes by myeloid cells, but this mechanism appears to be less significant for agents with cellular targets such as CD20 [71]. Allelic polymorphisms of genes responsible for such receptors are associated with variability in the binding strength of monoclonal antibodies, as discussed next.
All monoclonal antibodies are subclasses of IgG, with metabolism mostly reliant on intracellular lysosomal degradation [72]. Cellular uptake is required for this process and is achieved by either (1) receptor-mediated endocytosis by the target cell following binding of the antibody to its target antigen, or (2) non-specific pinocytosis by phagocytes or endothelial cells of lymphatic vessels [73]. Binding of the monoclonal antibody to its target (for example, the Fab portion of rituximab to CD20 expressed on B cells) triggers endocytosis of the antibody-receptor complex. These target-mediated interactions are dependent on binding affinity and receptor expression, leading to complex non-linear kinetics that vary with repeat administration [74]. At higher doses, receptor-mediated endocytosis becomes a saturated pathway for IgG metabolism, and non-specific pinocytosis by phagocytes and endothelial cells becomes more relevant. The neonatal Fc receptor (FcRn) is a key modulator of IgG degradation by this pathway [75]. Following pinocytosis, FcRn-bound IgG molecules are protected from intracellular catabolism, whereas unbound IgG molecules are degraded by endosomal proteases [76]. Two-thirds of IgG molecules are protected in this way after pinocytosis, and are instead recycled into circulation to contribute to the therapeutic pool and half-life [77]. The expression and binding capacity of FcRn is associated with polymorphisms in the promotor region of its gene, FCGRT.
In the 2008 placebo-controlled HERMES trial, rituximab, a chimeric mouse-human monoclonal antibody first trialled in lymphoid cancers a decade prior, was shown to reduce clinical and radiological disease activity in people with RRMS [27]. In primary progressive multiple sclerosis (PPMS), rituximab was shown to reduce disease progression in younger patients and patients with contrast-enhancing MRI lesions. [78]. For commercial reasons, rituximab never progressed to phase 3 studies in RRMS. Contemporary use of rituximab in MS is off-label with variable uptake worldwide; it has largely been supplanted by ocrelizumab, a humanised anti-CD20 agent [79]. Rituximab has also been used off-label for secondary progressive MS (SPMS), with reductions in disease progression shown in retrospective analyses [80]. Value has also been seen in other autoimmune neurological conditions such as neuromyelitis optica spectrum disorder (NMOSD) [81], myasthenia gravis [82] and autoimmune encephalitis [83]. First-dose infusion reactions, likely due to cytokine release accompanying B cell lysis [84], were reported in 78.3% of patients (compared to 40.3% in the placebo group) in the HERMES trial, ...although incidence dropped off significantly with repeat infusion. Hypogammaglobulinaemia, not explicitly defined in the HERMES study but commonly defined as a serum IgG level of less than 6 g/L [85, 86], was seen more frequently in the treated compared to placebo group (7.9% vs 3.0%). Urinary tract infections (UTIs) and sinusitis were more common with rituximab compared to placebo, but serious or opportunistic infective complications are only rarely reported with rituximab in MS [87] and other autoimmune conditions [88]. Hepatitis B (HBV) reactivation has been reported in rituximab-treated patients with systemic autoimmune or lymphoproliferative disorders [89]. Progressive multifocal leukoencephalopathy (PML), due to the opportunistic reactivation of John Cunningham virus in myelin-producing oligodendrocytes, has also been reported in these populations [90]. PML carries a significant risk of mortality or irreversible disability, of particular concern in this setting as the treatment effects of rituximab cannot be reversed.
The efficacy of ocrelizumab, a humanised anti-CD20 agent, was established in the OPERA trials, with treatment resulting in a 46 to 47% lower annualised relapse rate (ARR) compared to subcutaneous interferon-beta 1a in patients with RRMS [91]. Treatment benefit was also seen for disease progression and MRI metrics, and benefit for all endpoints was sustained over most demographic and clinical stratification subgroups [92]. The ORATORIO trial demonstrated the efficacy of ocrelizumab over placebo in PPMS. The reduction in clinical and radiological progression in this trial supports the significance of B cells in the pathogenesis of progressive MS phenotypes [93]. In the OPERA trials, infusion reactions were seen in 34% of patients and lessened with subsequent infusions. Again, these are thought to be mostly due to cytokine release by lysed B cells and are usually manageable with slowed infusion rates, and administration of antihistamines and corticosteroids. In these trials, treatment with ocrelizumab was associated with a higher incidence of upper respiratory tract and oral herpes infections compared to placebo. Cellulitis and UTIs were also more common in a study of rheumatoid arthritis patients [94]. HBV reactivation [95] and hypogammaglobulinaemia [96] have been described. Given its mechanism of action, PML is expected to rarely occur.
Other anti-CD20 monoclonal agents have been the subject of MS trials. Ofatumumab is the first subcutaneously administered anti-CD20 agent approved for relapsing forms of MS. It demonstrates more effective CDC than rituximab, and patients have reduced B cell depletion and faster repletion kinetics compared to intravenous alternatives [97, 98]. Its superiority for the prevention of relapses and disability progression was recently demonstrated over teriflunomide, an oral DMT [99]. Obinutuzumab is humanised while ublituximab is chimeric; both are used in B cell lymphoid malignancies, with stronger ADCC activation compared to rituximab [100]. Phase 2 placebo-controlled data demonstrated the safety and efficacy of ublituximab in RRMS [101], and phase 3 trials are ongoing (ULTIMATE I and II).
Pharmacogenetic Prediction of Rituximab Outcomes
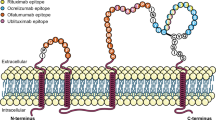
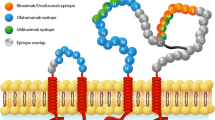
Genotypic differences relating to monoclonal antibody recognition, metabolism and disease-related signalling are likely to explain a significant amount of variability seen in their efficacy and toxicity [69]. To date, no pharmacogenetic studies of rituximab or ocrelizumab efficacy in MS have been performed. However, genotype-phenotype studies of rituximab response in various diseases including lymphoid malignancies and rheumatoid arthritis have yielded several positive results. The identified polymorphisms affect the FcγR family (FCγRIIIA and FCγRIIA), FcRn, BAFF, C1qA, CD20 and IL-6 (Fig. 1 and Table 3).

Selected polymorphisms relevant to rituximab efficacy
The FcγR Family
Genetic polymorphisms in FcγR affect the cytotoxic function of macrophages and NK cells, which could alter monoclonal antibody efficacy (Table 4). The most studied of these is a dimorphism of the FCGR3A gene that modulates the strength of interaction with the lower hinge region of IgG1, characterised by either phenylalanine (F) or valine (V) at residue 158 [102]. Although monocytes and macrophages possess a combination of stimulatory and inhibitory FcγRs, NK cells only express the stimulatory FCγRIIIA. The NK cells of V/V homozygotes participate in more effective ADCC compared to those of with V/F or F/F allotypes, thought to be due to stronger binding affinity for Fc seen in the FCGR3A-158V genotype [103, 104]. There is also evidence of increased NK cell expression of FcγRIIIA in V/V homozygotes [105, 106].
The relationship between FCGR3A variants and rituximab efficacy has been mostly examined in B cell malignancies [107, 108] and rheumatoid arthritis [109]. An analysis of 212 patients with rheumatoid arthritis found a significantly higher rate of clinical response to rituximab in V/V homozygotes (89.5%), compared to the V/F and F/F groups (66% and 66.2%, respectively). Loss of response to rituximab was seen only in V/F and F/F patients (10.8% and 16.4%, compared to none in the V/V group) [110]. In patients given rituximab as part of chemotherapy for follicular lymphoma, the V/V and F/F genotypes have been associated with 12-month response rates of 75% and 30%, respectively [111]. This relationship has been shown to be specific for rituximab-containing regimens [112, 113]. One study of 85 patients with RRMS has reported a lack of relationship between FCGR3A-158 genotype and outcomes with alemtuzumab, an anti-CD52 monoclonal antibody [114]. Although there have been no other analyses of FcγR variant relationship to monoclonal antibody treatment response in MS patients, a key study of rituximab-treated patients with NMOSD showed that V allele carriage at FCGR3A-158 was associated with lower relapse risk (OR 0.35, 95% CI 0.12–0.91) and longer time to retreatment [115]. The V/V genotype is associated with enhanced B cell depletion [116], whereas poorer outcomes seen with F/F homozygosity are associated with incomplete memory B cell depletion. In a study of NK cells from patients with B cell malignancies, the rituximab concentration required to lyse 50% of target cells was 4.2 times lower in for FCGR3A-158V, compared to F/F homozygotes [117]. It has therefore been suggested that F/F homozygotes may require more frequent dosing.
A similar relationship exists between rituximab and polymorphisms affecting FcγRIIA, another activating Fc receptor found on monocytes and macrophages. The strength of IgG binding at this receptor varies depending on a polymorphism at residue 131, with improved response to monoclonal antibodies predicted with homozygosity for histidine (H) compared to arginine (R) [118]. For example, the response rates to rituximab in follicular lymphoma for FCGR2A-131 H/H homozygotes (55% at 12 months) is superior to patients with H/R or R/R genotypes (26%) [111]. This stratification is enhanced by combined FCGR2A-131 and FCGR3A-158 genotyping: 100% of patients with both -131H/H and -158V/V maintained clinical response at 12 months, compared to 54% of those with either -131H/H or -158V/V, and 18% of those with neither.
The distinctions in rituximab response between FCGR3A-158 and FCGR2A-131 genotypes appear generalisable to monoclonal antibodies with non-CD20 targets. Trastuzumab utilises FcγR for ADCC in its effect against breast cancer, and significantly poorer clinical response rates are seen with FCGR3A-158 V/F and F/F genotypes (42% and 35%, respectively), compared to V/V homozygotes (82%) [119]. Clinical response rates and in vitro antibody-mediated cytotoxicity are also improved in those with the FCGR2A-131 H/H genotype [119]. Genetic variants affecting FcγR also appear relevant to metastatic colorectal cancer patients treated with cetuximab, an anti-EGFR monoclonal antibody: one study found progression-free survival to be significantly longer for FCGR3A-158 V/V compared to F/F homozygotes (5.5 vs 3.0 months) [120].
FcγR family polymorphisms could be similarly useful in predicting adverse events due to rituximab (Table 5). Hypogammaglobulinaemia following rituximab is predicted by FCGR3A-158 status in patients with non-Hodgkin’s lymphoma [121]. In a study with comparable baseline immunoglobulin levels between genotype groups, post-rituximab IgG levels were significantly lower in FCGRA-158 F/F homozygotes compared to V allele carriers. The effect was not seen in ten controls treated with transplantation using conditioning regimens without rituximab. This relationship is contrary to what was initially hypothesised by the authors. A possible explanation is increased rituximab uptake by lymphomatous B cells in carriers of the higher-affinity V allele could lead to reduced anti-CD20 effects on non-malignant cells. Carriage of the F allele at FCGRA-158 was associated with a higher risk of bloodstream infections after rituximab therapy in a cohort of ABO blood group incompatible liver transplant recipients, although a significant difference in IgG levels was not seen [122]. FCGR3A-158 status also stratifies the risk of rituximab-induced late-onset neutropaenia. In a cohort of patients treated for rheumatological conditions, each V allele carried conferred a fourfold increase in odds ratio for developing neutropaenia after 4 weeks [123]. Another study of diffuse large B cell lymphoma found 50% of homozygotes of the higher-affinity V allele experienced late-onset neutropaenia, compared to 7% of heterozygotes and 2% of F/F homozygotes [124]. Hypotheses for this effect include bystander neutrophil loss due to increased lysosomal enzyme release, higher risk of anti-neutrophil autoantibody development, and greater disturbance of bone marrow homeostasis [125, 126]. Similarly, the higher-affinity H allele at FCGR2A-131 predicts a greater risk of post-treatment anaemia compared to R/R homozygotes (24.2% vs 7.9%) [113]. The H allele is also associated with lower immunoglobulin levels and increased risk of bloodstream infections in liver transplant recipient [122]. There could be a shared mechanism for rituximab efficacy and the genesis of these adverse events; for example, late-onset neutropaenia has been identified as a positive prognostic factor following rituximab for rheumatological and lymphoproliferative disorders, and can predict remission without need for re-treatment [123, 127]. Late-onset neutropaenia has been described in MS patients receiving rituximab, but it is unclear whether its occurrence has similar prognostic implications [128].
Interestingly, the effects of these FcγR polymorphisms are reversed for TNF-α inhibitors, a monoclonal antibody class that does not rely on ADCC for efficacy. This reversal is thought to be due to the relationship between the binding strength of these agents and their endocytic and pinocytic metabolism. The expression of higher-affinity FcγR genotypes (FCGR3A-158V and FCGR2A-131H) on monocytes, macrophages and dendritic cells results in accelerated drug clearance and reduced efficacy [69, 71]. In a meta-analysis of TNF-alpha inhibitors in rheumatoid arthritis, H allele carriage at FCGR2A-131 predicted a reduced response to adalimumab therapy [129]. Infliximab non-response in rheumatoid arthritis has been associated with V allele carriage at FCGR3A-158 (2.2%, compared to 24.1% for F/F homozygotes), and H allele carriage at FCGR2A-131 (33.3% vs. 60% for R/R homozygotes) [130]. Similar findings were seen in rheumatoid and psoriatic arthritis patients [131]. The relationship has also been described in V/V homozygous patients with Crohn’s disease, with increased rates of infliximab elimination, biochemical inflammation, clinical relapse and treatment discontinuation [132].
Neonatal Fc Receptor
The neonatal Fc receptor (FcRn) is a critical mediator of antibody metabolism that requires consideration, although no rituximab-specific pharmacogenetic data exists. Higher FcRn levels protect IgG from intracellular catabolism; FcRn-knockout mice eliminate antibodies more efficiently [133] and FcRn-blocking monoclonal antibodies are associated with a dose-dependent reduction in circulating IgG levels [134]. FcRn expression is modulated by a variable number of tandem repeats (VNTR) polymorphism within the promoter region of its gene FCGRT. VNTR3 (three repetitions of a 37 base-pair VNTR) homozygosity is associated with increased FcRn expression and a lower clearance rate of administered immunoglobulins compared to VNTR2/VNTR3 heterozygotes [135]. Greater infliximab and adalimumab blood levels are seen in VNTR3 homozygotes treated for inflammatory bowel disease, compared to VNTR2/VNTR3 heterozygotes [136]. For cetuximab, VNTR3 homozygosity is associated with lower distribution clearance due to increased retention in cells and interstitial tissues [137]. As FcRn status is relevant to the survival of all circulating IgG molecules, the potential impact of VNTR status (or other FcRn variants) on the risk of hypogammaglobulinaemia associated with anti-CD20 agents also warrants further investigation.
B Cell Activation Factor
As a B cell survival cytokine, B cell activation factor (BAFF) promotes survival of autoreactive B cells both before and after anti-CD20 therapy. A C/T polymorphism at locus 871 of the BAFF gene modulates rituximab efficacy and corresponds to the binding site of myeloid zinc finger 1 (MZF1), which modulates BAFF mRNA expression. Rheumatoid arthritis patients who are C/C homozygous at BAFF-871 had a higher rate of 6-month rituximab response compared to T/T homozygotes (92% vs. 64%) [138]. At a locus in the same promoter region (BAFF-2704), C/C homozygous patients with ANCA vasculitis experienced a higher risk of rituximab failure, lower immunoglobulin levels and greater B cell reconstitution at 6 months [139]. The common TTTT promoter haplotype, characterised by four SNPs in the BAFF gene regulatory region (871 C>T, 2704 T>C, 2841 T>C and 2701 T>A), significantly predicted a positive rituximab response in patients with seropositive rheumatoid arthritis [140]. Although not yet examined for associations with rituximab outcomes, an insertion-deletion variant (BAFF-var) is associated with over-expression of BAFF, higher B cell and immunoglobulin levels and an increased risk of MS [141]. It has been suggested that the B cell contribution to disease may be greater in patients with BAFF-var, which could translate to a greater response to anti-CD20 therapies [142].
Complement C1q A Chain
The G to A polymorphism at residue 276 of the C1q A chain gene (C1QA) does not alter the C1q protein structure, and appears to affect rituximab outcomes by modulating C1q expression [143]. C1q is the complement cascade trigger and crucial for CDC, but also has a complex relationship with autoimmune disorders. C1q deficiency is associated with autoimmunity, suggesting a role for modulating tolerance to self-antigens [144]. Higher C1q levels and more efficient CDC are seen with G allele of C1QA-276, whereas the A allele is associated with lower C1q levels and a higher incidence of cutaneous lupus [145]. However, C1QA-276 A/A homozygosity is linked to improved rates and duration of rituximab response in B cell malignancies, compared to G allele carriers [143, 146]. It has been hypothesised that, although A/A homozygosity could lead to less effective opsonisation and CDC, this results in more B cell fragments for APC recognition and a more developed and coordinated immune response [146].
CD20
One pharmacogenetic study of rituximab-treated patients with B cell lymphoma demonstrated the clinical significance of a C/T polymorphism at locus 216 in Exon2 of the CD20 gene. Significantly improved rates of remission were reported with the C/C genotype (67%), compared to carriers of the T allele (47%). Exon2-216C/T is a synonymous SNP, with effects likely related to gene expression, splicing efficiency or mRNA stability [147]. The association did not extend to predicting progression-free survival, disease-specific survival or overall prognosis: this may be due to inadequate power, or the possibility that CD20 polymorphisms are important determinants of initial response, but not long-term outcomes. It should be noted that de novo mutations of CD20 in target tissues can also be a source of outcome variability in patients treated with rituximab for lymphoma [148].
Interleukin-6
In rituximab-treated patients with rheumatoid arthritis, G allele carriage at IL6-174, a locus within the promoter region for interleukin-6 (IL-6), was associated with a higher clinical response rate (81.5%) compared to C/C homozygotes (61%) [149]. Although there was no definite link between genotype and baseline serum IL-6 levels in this study, post-rituximab assays were not performed. Persistently elevated IL-6 levels in the sera and synovia have since been described in ungenotyped rheumatoid arthritis patients who failed to respond to rituximab [150]. Interestingly, homozygous carriage of the C allele at IL6-174 is associated with increased risk for the development of MS in Asian populations [151]. IL-6 has various pro-inflammatory functions and is known to promote survival and proliferation of B cells [152]. Levels in MS patients are elevated and result in increased T cell differentiation towards the pro-inflammatory Th17 phenotype [46, 47].
Conclusion
Given the expanding range of DMTs available for use in RRMS, pharmacogenetic studies are a promising avenue towards personalised therapy. Studies of rituximab in rheumatological and haematological diseases suggest anti-CD20 therapy outcomes could be predicted by SNPs that affect ADCC, CDC and the signalling and survival of B cells. That these polymorphisms predicted efficacy but not baseline clinical or demographic differences suggests treatment response and phenotype expression are not generally linked. The effects of some of these variants appear generalisable to monoclonal agents with non-CD20 targets. Based on experiences seen with other disease examples highlighted in this review, pharmacogenetics and pharmacogenomics to predict MS outcomes in monoclonal antibody therapy have significant potential, and could be rapidly translated to clinical care.
References
Jokubaitis VG, Spelman T, Kalincik T, et al. Predictors of disability worsening in clinically isolated syndrome. Annals of Clinical and Translational Neurology. 2015;2:479–491.
Jokubaitis VG, Spelman T, Kalincik T, et al. Predictors of long-term disability accrual in relapse-onset multiple sclerosis: Predictors of MS Outcomes. Annals of Neurology. 2016;80:89–100.
Kalincik T, Manouchehrinia A, Sobisek L, et al. Towards personalized therapy for multiple sclerosis: prediction of individual treatment response. Brain. 2017;140:2426–2443.
Hočevar K, Ristić S, Peterlin B. Pharmacogenomics of Multiple Sclerosis: A Systematic Review. Front Neurol. 2019;10:134.
Roden DM, McLeod HL, Relling MV, et al. Pharmacogenomics. The Lancet. 2019;394:521–532.
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74.
Peterson JF, Roden DM, Orlando LA, et al. Building evidence and measuring clinical outcomes for genomic medicine. The Lancet. 2019;394:604–610.
Telenti A, Pierce LCT, Biggs WH, et al. Deep sequencing of 10,000 human genomes. Proc Natl Acad Sci USA. 2016;113:11901–11906.
Scott RH, Fowler TA, Caulfield M. Genomic medicine: time for health-care transformation. The Lancet. 2019;394:454–456.
Weinshilboum RM, Wang L. Pharmacogenomics: Precision Medicine and Drug Response. Mayo Clinic Proceedings. 2017;92:1711–1722.
Van Driest S, Shi Y, Bowton E, et al. Clinically Actionable Genotypes Among 10,000 Patients With Preemptive Pharmacogenomic Testing. Clin Pharmacol Ther. 2014;95:423–431.
Ji Y, Skierka JM, Blommel JH, et al. Preemptive Pharmacogenomic Testing for Precision Medicine. The Journal of Molecular Diagnostics. 2016;18:438–445.
Dinama O, Warren AM, Kulkarni J. The role of pharmacogenomic testing in psychiatry: Real world examples. Australian & New Zealand Journal of Psychiatry. 2014;48:778–778.
Hicks J, Bishop J, Sangkuhl K, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 Genotypes and Dosing of Selective Serotonin Reuptake Inhibitors. Clin Pharmacol Ther. 2015;98:127–134.
Hicks JK, Stowe D, Willner MA, et al. Implementation of Clinical Pharmacogenomics within a Large Health System: From Electronic Health Record Decision Support to Consultation Services. Pharmacotherapy. 2016;36:940–948.
Crews KR, Gaedigk A, Dunnenberger HM, et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for Cytochrome P450 2D6 Genotype and Codeine Therapy: 2014 Update. Clin Pharmacol Ther. 2014;95:376–382.
Bertilsson L, Dahl M-L, Dalén P, et al. Molecular genetics of CYP2D6: clinical relevance with focus on psychotropic drugs. Br J Clin Pharmacol. 2002;53:111–122.
Cavallari LH, Johnson JA. A case for genotype-guided pain management. Pharmacogenomics. 2019;20:705–708.
Gasche Y, Daali Y, Fathi M, et al. Codeine intoxication associated with ultrarapid CYP2D6 metabolism. N Engl J Med. 2004;351:2827–2831.
Mega JL, Simon T, Collet J-P, et al. Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: a meta-analysis. JAMA. 2010;304:1821–1830.
Notarangelo FM, Maglietta G, Bevilacqua P, et al. Pharmacogenomic Approach to Selecting Antiplatelet Therapy in Patients With Acute Coronary Syndromes: The PHARMCLO Trial. J Am Coll Cardiol. 2018;71:1869–1877.
Cavallari LH, Lee CR, Beitelshees AL, et al. Multisite Investigation of Outcomes With Implementation of CYP2C19 Genotype-Guided Antiplatelet Therapy After Percutaneous Coronary Intervention. JACC: Cardiovascular Interventions. 2018;11:181–191.
Ozawa T, Suda M, Ikegami R, et al. Dual Antiplatelet Therapy Guided by CYP2C19 Polymorphisms after Implantation of Second-Generation Drug-Eluting Stents for Management of Acute Coronary Syndrome. Int Heart J. 2018;59:21–26.
Joo HJ, Ahn SG, Park JH, et al. Effects of genetic variants on platelet reactivity and one-year clinical outcomes after percutaneous coronary intervention: A prospective multicentre registry study. Sci Rep. 2018;8:1229.
Nakagawa H, Fujita M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018;109:513–522.
Edwards JCW, Szczepanski L, Szechinski J, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581.
Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688.
Filippi M, Bar-Or A, Piehl F, et al. Multiple sclerosis. Nat Rev Dis Primers. 2018;4:43.
Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron. 2018;97:742–768.
Gresle MM, Jordan MA, Stankovich J, et al. Multiple sclerosis risk variants regulate gene expression in innate and adaptive immune cells. Life Science Alliance. 2020;3:e202000650.
Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15:545–558.
Sabatino JJ, Pröbstel A-K, Zamvil SS. B cells in autoimmune and neurodegenerative central nervous system diseases. Nature Reviews Neuroscience. 2019;20:728–745.
Wanleenuwat P, Iwanowski P. Role of B cells and antibodies in multiple sclerosis. Multiple Sclerosis and Related Disorders. 2019;36:101416.
Breij ECW, Brink BP, Veerhuis R, et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol. 2008;63:16–25.
Prineas JW, Wright RG. Macrophages, lymphocytes, and plasma cells in the perivascular compartment in chronic multiple sclerosis. Lab Invest. 1978;38:409–421.
Qin Y, Duquette P, Zhang Y, et al. Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J Clin Invest. 1998;102:1045–1050.
Qin Y, Duquette P, Zhang Y, et al. Intrathecal B-Cell Clonal Expansion, an Early Sign of Humoral Immunity, in the Cerebrospinal Fluid of Patients with Clinically Isolated Syndrome Suggestive of Multiple Sclerosis. Lab Invest. 2003;83:1081–1088.
Obermeier B, Mentele R, Malotka J, et al. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat Med. 2008;14:688–693.
DiLillo DJ, Hamaguchi Y, Ueda Y, et al. Maintenance of long-lived plasma cells and serological memory despite mature and memory B cell depletion during CD20 immunotherapy in mice. J Immunol. 2008;180:361–371.
Greenfield AL, Hauser SL. B-cell Therapy for Multiple Sclerosis: Entering an era: MS: Entering the Era of B-Cell Therapy. Ann Neurol. 2018;83:13–26.
Mathias A, Perriard G, Canales M, et al. Increased ex vivo antigen presentation profile of B cells in multiple sclerosis. Mult Scler. 2017;23:802–809.
Jelcic I, Al Nimer F, Wang J, et al. Memory B Cells Activate Brain-Homing, Autoreactive CD4+ T Cells in Multiple Sclerosis. Cell. 2018;175:85-100.e23.
Lund FE. Cytokine-producing B lymphocytes-key regulators of immunity. Curr Opin Immunol. 2008;20:332–338.
Duddy M, Niino M, Adatia F, et al. Distinct Effector Cytokine Profiles of Memory and Naive Human B Cell Subsets and Implication in Multiple Sclerosis. J Immunol. 2007;178:6092–6099.
Bar-Or A, Fawaz L, Fan B, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol. 2010;67:452–461.
Korn T, Mitsdoerffer M, Croxford AL, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proceedings of the National Academy of Sciences. 2008;105:18460–18465.
Molnarfi N, Schulze-Topphoff U, Weber MS, et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med. 2013;210:2921–2937.
Barr TA, Shen P, Brown S, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6–producing B cells. The Journal of Experimental Medicine. 2012;209:1001–1010.
Li R, Rezk A, Miyazaki Y, et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med. 2015;7:310ra166.
Kannel K, Alnek K, Vahter L, et al. Changes in Blood B Cell-Activating Factor (BAFF) Levels in Multiple Sclerosis: A Sign of Treatment Outcome. PLoS ONE. 2015;10:e0143393.
Serafini B, Rosicarelli B, Magliozzi R, et al. Detection of Ectopic B-cell Follicles with Germinal Centers in the Meninges of Patients with Secondary Progressive Multiple Sclerosis. Brain Pathology. 2004;14:164–174.
Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2006;130:1089–1104.
Lassmann H. New concepts on progressive multiple sclerosis. Curr Neurol Neurosci Rep. 2007;7:239–244.
Corsiero E, Nerviani A, Bombardieri M, et al. Ectopic Lymphoid Structures: Powerhouse of Autoimmunity. Front Immunol. 2016;7:430.
Thaunat O, Patey N, Gautreau C, et al. B cell survival in intragraft tertiary lymphoid organs after rituximab therapy. Transplantation. 2008;85:1648–1653.
Monson NL, Cravens PD, Frohman EM, et al. Effect of Rituximab on the Peripheral Blood and Cerebrospinal Fluid B Cells in Patients With Primary Progressive Multiple Sclerosis. Arch Neurol. 2005;62:258–264.
Larouche J-F, Bergeron M, Hampson G, et al. Rituximab Cerebrospinal Fluid Levels in Patients with Primary Central Nervous System Lymphoma Treated with Intravenous High Dose Rituximab. Blood. 2011;118:1644–1644.
Krumbholz M, Derfuss T, Hohlfeld R, et al. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nature Reviews Neurology. 2012;8:613–623.
Leandro MJ. B-cell subpopulations in humans and their differential susceptibility to depletion with anti-CD20 monoclonal antibodies. Arthritis Res Ther. 2013;15 Suppl 1:S3.
Gingele S, Jacobus TL, Konen FF, et al. Ocrelizumab Depletes CD20+ T Cells in Multiple Sclerosis Patients. Cells. 2018;8:12.
Palanichamy A, Jahn S, Nickles D, et al. Rituximab Efficiently Depletes Increased CD20-Expressing T Cells in Multiple Sclerosis Patients. JI. 2014;193:580–586.
Sabatino JJ, Wilson MR, Calabresi PA, et al. Anti-CD20 therapy depletes activated myelin-specific CD8 + T cells in multiple sclerosis. Proc Natl Acad Sci USA. 2019;116:25800–25807.
Schuh E, Berer K, Mulazzani M, et al. Features of Human CD3+CD20+ T Cells. J Immunol. 2016;197:1111–7. Available from: https://www.jimmunol.org/content/early/2016/07/13/jimmunol.1600089. Accessed 20 Jul 2020
Barkhof F, Kappos L, Wolinsky JS, et al. Onset of clinical and MRI efficacy of ocrelizumab in relapsing multiple sclerosis. Neurology. 2019;93:e1778–e1786.
Roll P, Palanichamy A, Kneitz C, et al. Regeneration of B cell subsets after transient B cell depletion using anti-CD20 antibodies in rheumatoid arthritis. Arthritis Rheum. 2006;54:2377–2386.
Zhou X, Hu W, Qin X. The Role of Complement in the Mechanism of Action of Rituximab for B-Cell Lymphoma: Implications for Therapy. The Oncologist. 2008;13:954–966.
Maloney DG. Anti-CD20 Antibody Therapy for B-Cell Lymphomas. N Engl J Med. 2012;366:2008–2016.
Klein C, Lammens A, Schäfer W, et al. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. mAbs. 2013;5:22–33.
Shek D, Read SA, Ahlenstiel G, et al. Pharmacogenetics of anticancer monoclonal antibodies. Cancer Drug Resist 2019;2:69–81. Available from: https://cdrjournal.com/article/view/3004. Accessed 8 Mar 2020
Yan L, Beckman RA. Pharmacogenetics and pharmacogenomics in oncology therapeutic antibody development. BioTechniques. 2005;39:S565–S568.
Abuqayyas L, Balthasar JP. Application of knockout mouse models to investigate the influence of FcγR on the tissue distribution and elimination of 8C2, a murine IgG1 monoclonal antibody. Int J Pharm. 2012;439:8–16.
Leipold D, Prabhu S. Pharmacokinetic and Pharmacodynamic Considerations in the Design of Therapeutic Antibodies. Clinical and Translational Science. 2019;12:130–139.
Ryman JT, Meibohm B. Pharmacokinetics of Monoclonal Antibodies: Pharmacokinetics of Monoclonal Antibodies. CPT: Pharmacometrics & Systems Pharmacology. 2017;6:576–588.
Glassman PM, Balthasar JP. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol Med. 2014;11:20–33.
Dirks NL, Meibohm B. Population Pharmacokinetics of Therapeutic Monoclonal Antibodies. Clin Pharmacokinet. 2010;49:633–659.
Kuo TT, Baker K, Yoshida M, et al. Neonatal Fc receptor: from immunity to therapeutics. J Clin Immunol. 2010;30:777–789.
Kim J, Hayton WL, Robinson JM, et al. Kinetics of FcRn-mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism-based model. Clin Immunol. 2007;122:146–155.
Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66:460–471.
Berntsson SG, Kristoffersson A, Boström I, et al. Rapidly increasing off-label use of rituximab in multiple sclerosis in Sweden — Outlier or predecessor? Acta Neurologica Scandinavica. 2018;138:327–331.
Naegelin Y, Naegelin P, von Felten S, et al. Association of Rituximab Treatment With Disability Progression Among Patients With Secondary Progressive Multiple Sclerosis. JAMA Neurol. 2019;76:274.
Collongues N, de Seze J. An update on the evidence for the efficacy and safety of rituximab in the management of neuromyelitis optica. Ther Adv Neurol Disord. 2016;9:180–188.
Iorio R, Damato V, Alboini PE, et al. Efficacy and safety of rituximab for myasthenia gravis: a systematic review and meta-analysis. J Neurol. 2015;262:1115–1119.
Lancaster E. The Diagnosis and Treatment of Autoimmune Encephalitis. J Clin Neurol. 2016;12:1–13.
Winkler U, Jensen M, Manzke O, et al. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood. 1999;94:2217–2224.
Barmettler S, Ong M-S, Farmer JR, et al. Association of Immunoglobulin Levels, Infectious Risk, and Mortality With Rituximab and Hypogammaglobulinemia. JAMA Network Open. 2018;1:e184169.
Roberts DM, Jones RB, Smith RM, et al. Rituximab-associated hypogammaglobulinemia: incidence, predictors and outcomes in patients with multi-system autoimmune disease. J Autoimmun. 2015;57:60–65.
Hu Y, Nie H, Yu H-H, et al. Efficacy and safety of rituximab for relapsing-remitting multiple sclerosis: A systematic review and meta-analysis. Autoimmunity Reviews. 2019;18:542–548.
van Vollenhoven RF, Fleischmann RM, Furst DE, et al. Longterm Safety of Rituximab: Final Report of the Rheumatoid Arthritis Global Clinical Trial Program over 11 Years. J Rheumatol. 2015;42:1761–1766.
Evens AM, Jovanovic BD, Su Y-C, et al. Rituximab-associated hepatitis B virus (HBV) reactivation in lymphoproliferative diseases: meta-analysis and examination of FDA safety reports. Ann Oncol. 2011;22:1170–1180.
Berger JR, Malik V, Lacey S, et al. Progressive multifocal leukoencephalopathy in rituximab-treated rheumatic diseases: a rare event. J Neurovirol. 2018;24:323–331.
Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. New England Journal of Medicine. 2017;376:221–234.
Turner B, Cree BAC, Kappos L, et al. Ocrelizumab efficacy in subgroups of patients with relapsing multiple sclerosis. J Neurol. 2019;266:1182–1193.
Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. New England Journal of Medicine. 2017;376:209–220.
Emery P, Rigby W, Tak PP, et al. Safety with Ocrelizumab in Rheumatoid Arthritis: Results from the Ocrelizumab Phase III Program. Schooling CM, editor. PLoS ONE. 2014;9:e87379.
Ciardi MR, Iannetta M, Zingaropoli MA, et al. Reactivation of Hepatitis B Virus With Immune-Escape Mutations After Ocrelizumab Treatment for Multiple Sclerosis. Open Forum Infect Dis. 2019;6:ofy356.
ECTRIMS 2019 – Oral Presentations. Mult Scler. 2019;25:3–130.
Bar-Or A, Grove RA, Austin DJ, et al. Subcutaneous ofatumumab in patients with relapsing-remitting multiple sclerosis: The MIRROR study. Neurology. 2018;90:e1805–e1814.
Teeling JL, French RR, Cragg MS, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood. 2004;104:1793–1800.
Hauser SL, Bar-Or A, Cohen JA, et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. New England Journal of Medicine. 2020;383:546–557.
Payandeh Z, Bahrami AA, Hoseinpoor R, et al. The applications of anti-CD20 antibodies to treat various B cells disorders. Biomedicine & Pharmacotherapy. 2019;109:2415–2426.
Fox E, Lovett-Racke AE, Gormley M, et al. A phase 2 multicenter study of ublituximab, a novel glycoengineered anti-CD20 monoclonal antibody, in patients with relapsing forms of multiple sclerosis. Mult Scler. 2020;1352458520918375.
Sondermann P, Huber R, Oosthuizen V, et al. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature. 2000;406:267–273.
Koene HR, Kleijer M, Algra J, et al. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood. 1997;90:1109–1114.
Wu J, Edberg JC, Redecha PB, et al. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest. 1997;100:1059–1070.
Vance BA, Huizinga TW, Wardwell K, et al. Binding of monomeric human IgG defines an expression polymorphism of Fc gamma RIII on large granular lymphocyte/natural killer cells. J Immunol. 1993;151:6429–6439.
Hatjiharissi E, Xu L, Santos DD, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood. 2007;110:2561–2564.
Cartron G, Dacheux L, Salles G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcγRIIIa gene. Blood. 2002;99:754–758.
Treon SP, Hansen M, Branagan AR, et al. Polymorphisms in FcγRIIIA (CD16) Receptor Expression Are Associated With Clinical Response to Rituximab in Waldenström’s Macroglobulinemia. JCO. 2005;23:474–481.
Ruyssen-Witrand A, Rouanet S, Combe B, et al. Fcγ receptor type IIIA polymorphism influences treatment outcomes in patients with rheumatoid arthritis treated with rituximab. Ann Rheum Dis. 2012;71:875–877.
Quartuccio L, Fabris M, Pontarini E, et al. The 158VV Fcgamma receptor 3A genotype is associated with response to rituximab in rheumatoid arthritis: results of an Italian multicentre study. Ann Rheum Dis. 2014;73:716–721.
Weng W-K, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940–3947.
Persky DO, Dornan D, Goldman BH, et al. Fc gamma receptor 3a genotype predicts overall survival in follicular lymphoma patients treated on SWOG trials with combined monoclonal antibody plus chemotherapy but not chemotherapy alone. Haematologica. 2012;97:937–942.
Ahlgrimm M, Pfreundschuh M, Kreuz M, et al. The impact of Fc-γ receptor polymorphisms in elderly patients with diffuse large B-cell lymphoma treated with CHOP with or without rituximab. Blood. 2011;118:4657–4662.
Keller CW, Ruck T, McHugh D, et al. Impact of FcγR variants on the response to alemtuzumab in multiple sclerosis. Ann Clin Transl Neurol. 2019;6:2586–2594.
Kim S-H, Jeong IH, Hyun J-W, et al. Treatment Outcomes With Rituximab in 100 Patients With Neuromyelitis Optica: Influence of FCGR3A Polymorphisms on the Therapeutic Response to Rituximab. JAMA Neurol. 2015;72:989–995.
Anolik JH, Campbell D, Felgar RE, et al. The relationship of FcgammaRIIIa genotype to degree of B cell depletion by rituximab in the treatment of systemic lupus erythematosus. Arthritis Rheum. 2003;48:455–459.
Dall’Ozzo S, Tartas S, Paintaud G, et al. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 2004;64:4664–4669.
Warmerdam PA, van de Winkel JG, Vlug A, et al. A single amino acid in the second Ig-like domain of the human Fc gamma receptor II is critical for human IgG2 binding. J Immunol. 1991;147:1338–1343.
Musolino A, Naldi N, Bortesi B, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol. 2008;26:1789–1796.
Bibeau F, Lopez-Crapez E, Di Fiore F, et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27:1122–1129.
Nishio M, Endo T, Fujimoto K, et al. FCGR3A-158V/F polymorphism may correlate with the levels of immunoglobulin in patients with non-Hodgkin’s lymphoma after rituximab treatment as an adjuvant to autologous stem cell transplantation. European Journal of Haematology. 2009;82:143–147.
Sakai H, Tanaka Y, Tazawa H, et al. Effect of Fc-γ Receptor Polymorphism on Rituximab-Mediated B Cell Depletion in ABO-Incompatible Adult Living Donor Liver Transplantation. Transplant Direct. 2017;3:e164.
Ajeganova S, Tesfa D, Hägglund H, et al. Effect of FCGR polymorphism on the occurrence of late-onset neutropenia and flare-free survival in rheumatic patients treated with rituximab. Arthritis Research & Therapy. 2017;19:44.
Keane C, Nourse JP, Crooks P, et al. Homozygous FCGR3A-158V alleles predispose to late onset neutropenia after CHOP-R for diffuse large B-cell lymphoma: Impact of LON and FCGR3A-V158F in DLBCL. Internal Medicine Journal. 2012;42:1113–1119.
Weng W-K, Negrin RS, Lavori P, et al. Immunoglobulin G Fc Receptor FcγRIIIa 158 V/F Polymorphism Correlates With Rituximab-Induced Neutropenia After Autologous Transplantation in Patients With Non-Hodgkin’s Lymphoma. Journal of Clinical Oncology. 2010;28:279–284.
Li S-C, Chen Y-C, Evens AM, et al. Rituximab-induced late onset neutropenia in newly-diagnosed B-cell lymphoma correlates with Fc receptor FcγRIIIa 158(V/F) polymorphism. American Journal of Hematology. 2010;85:810–812.
Hincks I, Woodcock BE, Thachil J. Is Rituximab-induced Late-onset Neutropenia a good prognostic indicator in lymphoproliferatve disorders?: Correspondence. British Journal of Haematology. 2011;153:411–413.
Rigal J, Ciron J, Lépine Z, et al. Late-onset neutropenia after RITUXIMAB therapy for multiple sclerosis, neuromyelitis optica spectrum disorders and MOG-antibody-associated diseases. Mult Scler Relat Disord. 2020;41:102019.
Lee YH, Bae S-C. Associations between PTPRC rs10919563 A/G and FCGR2A R131H polymorphisms and responsiveness to TNF blockers in rheumatoid arthritis: a meta-analysis. Rheumatol Int. 2016;36:837–844.
Cañete JD, Suárez B, Hernández MV, et al. Influence of variants of Fc gamma receptors IIA and IIIA on the American College of Rheumatology and European League Against Rheumatism responses to anti-tumour necrosis factor alpha therapy in rheumatoid arthritis. Ann Rheum Dis. 2009;68:1547–1552.
Tutuncu Z, Kavanaugh A, Zvaifler N, et al. Fcγ receptor type IIIA polymorphisms influence treatment outcomes in patients with inflammatory arthritis treated with tumor necrosis factor α-blocking agents. Arthritis Rheum. 2005;52:2693–2696.
Ternant D, Berkane Z, Picon L, et al. Assessment of the Influence of Inflammation and FCGR3A Genotype on Infliximab Pharmacokinetics and Time to Relapse in Patients with Crohn’s Disease. Clin Pharmacokinet. 2015;54:551–562.
Borvak J, Richardson J, Medesan C, et al. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int Immunol. 1998;10:1289–1298.
Baldwin WM, Valujskikh A, Fairchild RL. The neonatal Fc receptor: Key to homeostasic control of IgG and IgG-related biopharmaceuticals. American Journal of Transplantation. 2019;19:1881–1887.
Gouilleux-Gruart V, Chapel H, Chevret S, et al. Efficiency of immunoglobulin G replacement therapy in common variable immunodeficiency: correlations with clinical phenotype and polymorphism of the neonatal Fc receptor: IgG efficiency and FcRn in CVID. Clinical & Experimental Immunology. 2013;171:186–194.
Billiet T, Dreesen E, Cleynen I, et al. A Genetic Variation in the Neonatal Fc-Receptor Affects Anti-TNF Drug Concentrations in Inflammatory Bowel Disease: American Journal of Gastroenterology. 2016;111:1438–1445.
Passot C, Azzopardi N, Renault S, et al. Influence of FCGRT gene polymorphisms on pharmacokinetics of therapeutic antibodies. mAbs. 2013;5:614–619.
Ruyssen-Witrand A, Rouanet S, Combe B, et al. Association between -871C>T promoter polymorphism in the B-cell activating factor gene and the response to rituximab in rheumatoid arthritis patients. Rheumatology. 2013;52:636–641.
Alberici F, Smith RM, Fonseca M, et al. Association of a TNFSF13B (BAFF) regulatory region single nucleotide polymorphism with response to rituximab in antineutrophil cytoplasmic antibody–associated vasculitis. Journal of Allergy and Clinical Immunology. 2017;139:1684-1687.e10.
Fabris M, Quartuccio L, Vital E, et al. The TTTT B lymphocyte stimulator promoter haplotype is associated with good response to rituximab therapy in seropositive rheumatoid arthritis resistant to tumor necrosis factor blockers. Arthritis & Rheumatism. 2013;65:88–97.
Steri M, Orrù V, Idda ML, et al. Overexpression of the Cytokine BAFF and Autoimmunity Risk. New England Journal of Medicine. 2017;376:1615–1626.
Comabella M. Neuroimmunology: B cells and variant BAFF in autoimmune disease. Nat Rev Neurol. 2017;13:453–454.
Jin X, Ding H, Ding N, et al. Homozygous A polymorphism of the complement C1qA276 correlates with prolonged overall survival in patients with diffuse large B cell lymphoma treated with R-CHOP. J Hematol Oncol. 2012;5:51.
Stegert M, Bock M, Trendelenburg M. Clinical presentation of human C1q deficiency: How much of a lupus? Mol Immunol. 2015;67:3–11.
Racila DM, Sontheimer CJ, Sheffield A, et al. Homozygous single nucleotide polymorphism of the complement C1QA gene is associated with decreased levels of C1q in patients with subacute cutaneous lupus erythematosus. Lupus. 2003;12:124–132.
Racila E, Link BK, Weng W-K, et al. A Polymorphism in the Complement Component C1qA Correlates with Prolonged Response Following Rituximab Therapy of Follicular Lymphoma. Clinical Cancer Research. 2008;14:6697–6703.
Ding H, Jin X, Ding N, et al. Single nucleotide polymorphisms of CD20 gene and their relationship with clinical efficacy of R-CHOP in patients with diffuse large B cell lymphoma. Cancer Cell Int. 2013;13:58.
Terui Y, Mishima Y, Sugimura N, et al. Identification of CD20 C-terminal deletion mutations associated with loss of CD20 expression in non-Hodgkin’s lymphoma. Clin Cancer Res. 2009;15:2523–2530.
Fabris M, Quartuccio L, Lombardi S, et al. The CC homozygosis of the -174G>C IL-6 polymorphism predicts a lower efficacy of rituximab therapy in rheumatoid arthritis. Autoimmun Rev. 2012;11:315–320.
Das S, Vital EM, Horton S, et al. Abatacept or tocilizumab after rituximab in rheumatoid arthritis? An exploratory study suggests non-response to rituximab is associated with persistently high IL-6 and better clinical response to IL-6 blocking therapy. Ann Rheum Dis. 2014;73:909–912.
Hu S, Chen Y, Sun X-D, et al. Association between IL-6-174G/C polymorphism and risk of multiple sclerosis: a meta-analysis. Genet Test Mol Biomarkers. 2014;18:127–130.
Fonseca JE, Santos MJ, Canhão H, et al. Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun Rev. 2009;8:538–542.
Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793–2806.
Emery P, Fleischmann R, Filipowicz-Sosnowska A, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54:1390–1400.
Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–232.
Salles G, Barrett M, Foà R, et al. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv Ther. 2017;34:2232–2273.
Coiffier B, Haioun C, Ketterer N, et al. Rituximab (anti-CD20 monoclonal antibody) for the treatment of patients with relapsing or refractory aggressive lymphoma: a multicenter phase II study. Blood. 1998;92:1927–1932.
Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378:1779–1787.
Morschhauser F, Marlton P, Vitolo U, et al. Results of a phase I/II study of ocrelizumab, a fully humanized anti-CD20 mAb, in patients with relapsed/refractory follicular lymphoma. Ann Oncol. 2010;21:1870–1876.
Rigby W, Tony H-P, Oelke K, et al. Safety and efficacy of ocrelizumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: Results of a forty-eight-week randomized, double-blind, placebo-controlled, parallel-group phase III trial. Arthritis & Rheumatism. 2012;64:350–359.
Tak PP, Mease PJ, Genovese MC, et al. Safety and efficacy of ocrelizumab in patients with rheumatoid arthritis and an inadequate response to at least one tumor necrosis factor inhibitor: Results of a forty-eight-week randomized, double-blind, placebo-controlled, parallel-group phase III trial. Arthritis & Rheumatism. 2012;64:360–370.
Coiffier B, Lepretre S, Pedersen LM, et al. Safety and efficacy of ofatumumab, a fully human monoclonal anti-CD20 antibody, in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: a phase 1-2 study. Blood. 2008;111:1094–1100.
Freeman CL, Sehn LH. A tale of two antibodies: obinutuzumab versus rituximab. Br J Haematol. 2018;182:29–45.
Mössner E, Brünker P, Moser S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115:4393–4402.
Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370:1101–1110.
Marcus R, Davies A, Ando K, et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N Engl J Med. 2017;377:1331–1344.
Le Garff-Tavernier M, Herbi L, de Romeuf C, et al. Antibody-dependent cellular cytotoxicity of the optimized anti-CD20 monoclonal antibody ublituximab on chronic lymphocytic leukemia cells with the 17p deletion. Leukemia. 2014;28:230–233.
Babiker HM, Glode AE, Cooke LS, et al. Ublituximab for the treatment of CD20 positive B-cell malignancies. Expert Opinion on Investigational Drugs. 2018;27:407–412.
Kim DH, Jung HD, Kim JG, et al. FCGR3A gene polymorphisms may correlate with response to frontline R-CHOP therapy for diffuse large B-cell lymphoma. Blood. 2006;108:2720–2725.
Kastbom A, Cöster L, Ärlestig L, et al. Influence of FCGR3A genotype on the therapeutic response to rituximab in rheumatoid arthritis: an observational cohort study. BMJ Open. 2012;2:e001524.
Pál I, Szamosi S, Hodosi K, et al. Effect of Fcγ-receptor 3a (FCGR3A) gene polymorphisms on rituximab therapy in Hungarian patients with rheumatoid arthritis. RMD Open. 2017;3:e000485.
Acknowledgment
Required Author Forms Disclosure forms provided by the authors are available with the online version of this article. Figure 1 was created with BioRender.com.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 734 kb)
Rights and permissions
About this article

Cite this article
Zhong, M., van der Walt, A., Campagna, M.P. et al. The Pharmacogenetics of Rituximab: Potential Implications for Anti-CD20 Therapies in Multiple Sclerosis. Neurotherapeutics 17, 1768–1784 (2020). https://doi.org/10.1007/s13311-020-00950-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00950-2