
Abstract
In humans, different B-cell subpopulations can be distinguished in peripheral bloodand other tissues on the basis of differential expression of various surface markers.These different subsets correspond to different stages of maturation, activation anddifferentiation. B-cell depletion therapy based on rituximab, an anti-CD20 mAb, iswidely used in the treatment of various malignant and autoimmune diseases. Rituximabinduces a very significant depletion of B-cell subpopulations in the peripheral bloodusually for a period of 6 to 9 months after one cycle of therapy. Cells detectedcirculating during depletion are mainly CD20 negative plasmablasts. Data on depletionof CD20-expressing B cells in solid tissues are limited but show that depletion issignificant but not complete, with bone marrow and spleen being more easily depletedthan lymph nodes. Factors influencing depletion are thought to include not only thetotal drug dose administered and distribution into various tissues, but also B-cellintrinsic and microenvironment factors influencing recruitment of effector mechanismsand antigen and effector modulation. Available studies show that the degree ofdepletion varies between individuals, even if treated with the same dose, but that ittends to be consistent in the same individual. This suggests that individual factorsare important in determining the final extent of depletion.
Similar content being viewed by others
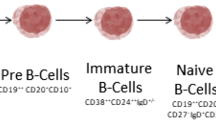

Introduction to B-cell subpopulations
In humans from birth all new B cells originate from common precursors in the bonemarrow. In the bone marrow, peripheral blood and secondary lymphoid tissues, differentB-cell subpopulations can be distinguished corresponding to different stages ofmaturation, activation and differentiation. B-cell subpopulations are characterisedmainly by the differential expression of different cell surface markers that includevarious cluster of differentiation (CD) molecules and different surface immunoglobulinisotypes (B-cell antigen receptor). B-cell development can be separated into an earlierantigen-independent phase, which takes place in the bone marrow, and a laterantigen-dependent phase that takes place mainly in secondary lymphoid tissues. In asimplified way, the different B-cell lineage subsets include pro-B cells, pre-B cells,immature and transitional B cells, mature naïve B cells, memory B cells,plasmablasts and plasma cells (Figure 1). Plasmablasts arerecently differentiated antibody-producing cells that are usually short-lived but canrecirculate and home to tissues such as the mucosa or the bone marrow, where they candifferentiate into fully mature plasma cells. In addition, centroblasts and centrocytesare B cells participating in germinal centre reactions.

Simplified scheme of B-cell subpopulations in humans and CD20expression.
B-cell precursor subpopulations are found in the bone marrow. In the peripheral blood,transitional, naïve mature and memory B cells and plasmablasts, and more rarelyplasma cells, can be identified. Plasma cells are more frequently seen in the bonemarrow and peripheral lymphoid tissues. Centrocytes and centroblasts are found insecondary lymphoid tissues where germinal centre reactions take place, and are not foundcirculating in peripheral blood. Marginal zone B cells can be found in the marginal zoneof the spleen and similar populations are described in particular locations in othersecondary lymphoid tissues [1]. Marginal zone B cells in human adults are mainly memory B cells. There isstill controversy on what drives formation of human marginal zone B cells, to whatextent they are similar to mice marginal zone B cells and what is their relationshipwith circulating IgM+ memory B-cell subsets [1, 2].
Immunophenotyping of B cells with multiparameter flow cytometry has allowedidentification of an increasing number of different subpopulations, increasing ourknowledge of normal B-cell biology and, in particular, changes associated with differentdisease states. For example, different memory B-cell subsets have now been described inperipheral blood including subsets that do not express CD27, a marker previously thoughtto be present on all memory B cells [3, 4]. Memory B-cell subpopulations include pre-switchIgD+IgM+CD27+ memory B cells,IgD-IgM+CD27+ memory B cells (IgMonly memory Bcells), post-switch IgA+CD27+ and IgG+CD27+ memory B cells and also IgA+CD27- andIgG+CD27- memory B cells [5]. These memory subpopulations show different frequencies of somatic mutationand different replication histories that are thought to reflect their formation onprimary or secondary germinal centres or outside germinal centre reactions [5]. A potential new marker for human memory B-cell subpopulations has beenidentified recently [6]. A proposal has been made that immunophenotyping of peripheral blood B cellsshould include the markers CD19, CD20, CD24, CD27, CD38 and IgD to be able todistinguish the major subpopulations [7]. More detailed information including separation into further subsets andsubtle differences in activation status that may be important when looking at diseasestates may require use of other markers such as different immunoglobulin isotopes,activation markers or chemokine receptors [6, 8–14].
Anti-CD20 monoclonal antibodies-rituximab
Anti-CD20 mAbs were developed in the late 1980s and in the 1990s for the treatment ofnon-Hodgkin's lymphoma of B-cell origin. Rituximab (MabThera®,Rituxan®; Roche, Basel, Switzerland) was licensed for the treatmentof follicular lymphoma in 1997/98 and later for diffuse large non-Hodgkin's lymphoma andchronic lymphocytic leukaemia. In 2006 rituximab was licensed for the treatment ofrheumatoid arthritis (RA). Rituximab is also used off-license for the treatment of otherB-cell malignant diseases, in transplantation and for the treatment of a variety ofother autoimmune diseases, predominantly diseases associated with the presence ofautoantibodies. Various other therapeutic anti-CD20 mAbs are either available on themarket (Ofatumumab-Arzerra®; GlaxoSmithKlein, UK-licensed for thetreatment of chronic lymphocytic leukaemia), undergoing clinical trials or underdevelopment [15].
The CD20 antigen is expressed by the majority of cells in the B-lymphocyte lineage, butnot by haematopoietic stem cells, the earliest B-cell precursors (pro-B cells) orterminally differentiated plasmablasts and plasma cells (Figure 1). The CD20 molecule is a transmembrane protein thought to function as acalcium channel and to be involved in B-cell activation and proliferation. A recent casereport of a patient with CD20 deficiency suggested a role in T-cell-independent antibody responses[16].
Because haematopoietic stem cells are not directly depleted by anti-CD20 antibodies, onecourse of treatment with rituximab is followed by B-cell repopulation of the peripheralblood starting usually within 6 to 9 months-but it can take several months or even yearsfor total B-cell numbers in the peripheral blood to recover to pretreatment levels.Repopulation occurs mainly with naïve B cells, with increased frequency and numbersof transitional B cells similar to that seen after bone marrow transplantation [14, 17]. The time at which B-cell repopulation of the peripheral blood starts isprobably determined by the extent of earlier depletion, drug clearance and the capacityof the bone marrow to regenerate. Variability in time to repopulation in primate animalmodels did not seem to be dose dependent [18]. Factors influencing B-cell precursor formation in humans are poorlyunderstood, as are factors that determine to what extent a fully functional B-cellrepertoire is regenerated and how long it takes. Whether age or other individualcharacteristics influence repopulation is not known [19, 20].
The fact that plasma cells are also not directly depleted by anti-CD20 antibodiesexplains why, in the majority of patients, serum total immunoglobulin levels remainwithin the normal range after treatment with one course of rituximab. Several studieshave shown that serum levels of several autoantibodies decrease after treatment withrituximab (although they do not usually become undetectable) and do so proportionallymore than total immunoglobulin levels or anti-microbial antibodies [21–23]. This observation suggests that these auto-antibodies are produced byproportionally more short-lived plasma cells and therefore are more dependent on theformation of new plasma cells, which is interrupted by B-cell depletion [23].
Treatment with rituximab is associated with major depletion of normal B cells invivo. Depletion in the peripheral blood is frequently higher than 99% butdepletion in other tissues has been less well studied, with several studies documentingthat depletion in solid tissues with rituximab is frequently not complete and can showconsiderable variation between individuals. In vitro, rituximab depletesmalignant B cells by antibody-dependent cellular cytotoxicity, complement-mediatedcytotoxicity and induction of apoptosis. In vivo, rituximab is thought to actmainly by inducing antibody-dependent cellular cytotoxicity with activation ofcomplement also contributing [24]. One of the consistent findings in several of the animal and earlier humanstudies is the variability of depletion seen with anti-CD20 mAbs in differentindividuals even when treated with the same dose [18, 25, 26]. Interestingly, depletion in the same individual tends to be consistent indifferent tissues, suggesting that individual characteristics are important.
Resistance to depletion with anti-CD20 monoclonal antibodies
Because depletion is achieved by binding of the mAbs to the cell surface CD20 molecules,the final extent of depletion will necessarily depend on the relationship between totalnumber of B cells and total dose of rituximab administered, on accessibility of the drugand effector immune cells to the tissues where B cells are located, on intrinsic orextrinsic factors that may influence B-cell survival and on the efficacy of recruitedhost immune mechanisms responsible for depletion.
Former small dose-ranging studies in lymphoma and in animal models have shown that Bcells in the peripheral blood are readily killed by anti-CD20 antibodies but that higherdoses and higher serum levels are needed for depletion in extravascular sites [18, 24, 25].
Factors influencing antigen and effector modulation are thought to be important indetermining the final extent of depletion achieved (Table 1) [18, 27, 28]. Antigen modulation refers to antigen endocytosis/modulation after binding tothe antibody. Contrary to what was originally thought, this can be seen with the CD20molecule after binding with certain anti-CD20 antibodies including rituximab [29]. This can lead to less recruitment of Fcγ receptors on effector immunecells and to decreased serum drug levels. Effector modulation refers to genetic andacquired mechanisms that can enhance or diminish effector immune cell function andtherefore influence the extent of depletion. For example, a Fcγ receptor IIIapolymorphism that can influence affinity for IgG has been associated with clinicalresponse in lymphoma [28]. Profound complement depletion as seen during treatment of chroniclymphocytic leukaemia with rituximab can be a limiting factor for further depletion [28].
Intrinsic B-cell factors that may influence depletion include high expression ofcomplement regulatory proteins as seen in chronic lymphocytic leukaemia [28]. In cynomolgus monkeys, different sensitivities to rituximab were associatedwith, but not fully explained by, different levels of expression of CD20 [30]. Binding of rituximab to CD20 leads to translocation of the CD20 molecule tolipid rafts. Alterations in lipid raft composition and treatment with statins have beenassociated with less good responses to rituximab [28]. To what extent external B-cell survival factors, in particular the cytokineB-cell activating factor (BAFF), influence depletion is not known, although it has beensuggested that local high levels of BAFF may contribute to resistance to depletion byrituximab [31].
In animal models, certain subpopulations have been shown to be more resistant todepletion with anti-CD20 antibodies but this varies with the mice strain used andwhether they were studies using human CD20 transgenic mice treated with anti-human CD20mAbs or non-transgenic mice treated with anti-mouse CD20 mAbs [32, 33]. Populations that were found to be more resistant to depletion wereperitoneal B1-type B cells, germinal centre B cells and marginal zone B cells [32, 33]. Insufficient depletion of peritoneal B1 cells is thought to be due to thelack of effector cells in the peritoneal space [33]. Differential sensitivity of germinal centre and marginal zone B cells toanti-CD20 antibodies has also been described in cynomologous monkeys, with differencesappearing more prominent in the lymph nodes than in the spleen [30]. The relative resistance of some populations is thought to be related toB-cell and micro-environment differences responsible for antigen or effector modulationor related to direct resistance of the B cells involved. In an autoimmune mouse model oflupus, B cells were more resistant to depletion when compared with nonautoimmune miceand more frequent administration of larger doses increased efficacy of depletion [34]. Less good depletion has also been associated with acquired defects inantibody-dependent cellular cytotoxicity in the same autoimmune mouse model of lupus [35].
To what extent the differential susceptibility of various B-cell subsets demonstrated insome of the animal models reflects what happens in humans in vivo is not known.Different B-cell malignancies deriving from B cells at different stages ofdifferentiation and different tumour locations are also associated with differentialresponses to treatment with anti-CD20 mAbs but susceptibility of the correspondentnormal human B-cell sub-populations is expected to be substantially different. Whetherthere are any differences in susceptibility to depletion of autoreactive human B-cellclones when compared with nonautoreactive ones, as suggested by mouse models [34], and whether there are any significant differences in susceptibility todepletion of disease-associated B-cell clones between different autoimmune diseases arealso not known.
In addition, administration of chimaeric anti-CD20 mAbs such as rituximab can beassociated with formation of human anti-chimaeric antibodies that can influence drugaction and clearance. Although most large studies show no association between thepresence of human anti-chimaeric antibodies and clinical response or depletion, thisassociation has been described, for example, in small studies in systemic lupuserythematosus patients [36, 37].
With evidence showing that not all B cells that bind rituximab are depleted there is aninterest in knowing what exactly happens to these cells in vivo during theperiod of depletion. Are they eventually depleted later on, particularly if theyrecirculate in peripheral blood? Are they functionally impaired? Are they able to expandin an environment with less competition and raised BAFF levels? Kamburova and colleaguestried to address some of these issues by studying the in vitro effects ofincubation with rituximab on proliferation, activation and differentiation ofnondepleted human normal peripheral blood B cells [38]. They reported that incubation with rituximab (for 30 minutes at 5μl/ml) inhibited the proliferation of stimulated CD27- naïve Bcells but not of CD27+ memory B cells and this was associated with a relativeincrease of B cells with an activated naïve phenotype. B cells stimulated in thepresence of rituximab induced stronger T-cell proliferation and the T-cell populationshowed a more Th2-like phenotype. These results suggest that B cells which are exposedto rituximab but are not depleted may have altered function and that naïve andmemory B cell populations may be differentially affected. Whether any of these phenomenaoccur in vivo and what their implications would be are unclear. Interestingly,and similar to what happens after bone marrow transplantation, the residual B cells arenot able to expand and repopulate the peripheral blood, even in the presence of abundantBAFF.
B-cell depletion in peripheral blood
Administration of rituximab is usually associated with a rapid and profound depletionof circulating B cells in the peripheral blood [18]. Major depletion effector cells are probably macrophages from thereticulo-endothelial system [24]. Studies in autoimmune diseases-in particular, RA and systemic lupuserythematosus-have documented variable degrees and durations of B-cell depletion inperipheral blood in different individuals following treatment with rituximab withstandard doses [17, 36, 37, 39–41]. Incomplete B-cell depletion in the peripheral blood, as defined by B-cellcounts >5 cells/μl after treatment with rituximab, has been well documentedin cases of patients with autoimmune diseases, more frequently in systemic lupuserythematosus than in RA [17, 36, 37]. Persistent presence of circulating B cells has also been documented withhigh-sensitivity flow cytometry and has been associated with no or less good responseto treatment [39, 40]. Insufficient depletion can be seen on retreatment with documented veryrapid clearance of rituximab in association with a marked human anti-chimaericantibody response [42]. Other mechanisms underlying incomplete depletion in the peripheral bloodhave not been well studied but are probably a consequence of more rapid clearance ofthe drug and/or antigen and effector modulation phenomena [17, 24, 36, 37].
The very small numbers of circulating B cells that can be detected during periods ofdepletion usually show a phenotype of plasmablasts but cells with memory or evennaïve B cells have also been reported [17, 40, 41, 43]. The CD20 antigen cannot usually be detected in these memory B cells,suggesting that it is masked by binding to rituximab because the drug can be detectedin the circulation for several months [26]. Mei and colleagues described that, similarly to their controls, themajority of circulating plasmablasts/plasma cells detected during depletion werepositive for IgA and a reasonable proportion expressed markers suggesting they hadbeen formed in mucosal tissue and were circulating back to mucosal areas [44]. These results suggest that depletion in mucosal-associated lymphoidtissue may be particularly less pronounced.
Repopulation of the peripheral blood after treatment with a standard dose ofrituximab usually starts 6 to 9 months after treatment with predominantlytransitional and naïve B cells as previously mentioned. Frequently, repopulationwith larger numbers of memory B cells and/or plasmablasts has been associated withearlier relapse [17, 40, 45]. At repopulation, the decrease from baseline in the frequency ofpre-switch memory B cells (CD27+IgD+)was larger than thedecrease in the switched memory B-cell population (CD27+IgD-) [46]. However, to what extent circulating memory B cells at repopulation areold memory B cells that have not been depleted by rituximab or recentlydifferentiated memory B cells is not known. We therefore do not know whether relativefrequencies of the different B-cell subpopulations at repopulation can tell usanything about the subpopulations of cells that may have resisted depletion.
In RA, nonresponse has been associated with higher numbers of plasmablasts beforetreatment and early relapse has been associated with higher numbers of CD27+ memory B cells before treatment [39, 45]. Again, to what extent this may indicate less susceptibility andinsufficient depletion of memory B-cell subsets in association with no response orwith a shorter response is not known.
B-cell depletion in bone marrow and secondary lymphoid tissues
Unfortunately, there are limited data on the degree of depletion of normal B cells insecondary lymphoid organs and other solid tissues in human individuals treated withrituximab, and hardly any data on differential susceptibility to depletion ofdifferent subpopulations in different tissues except for the expected resistance ofCD20- plasmablasts and plasma cells to depletion [47]. Animal studies in primates showed that increasingly higher doses areneeded to deplete bone marrow, spleen and lymph nodes in this order [18, 48, 49]. These studies also showed that B-cell depletion in solid tissues wasfrequently significant, but not complete, and that it varied from site to site andfrom individual to individual even when the same doses were used. Interestingly,consistency regarding the degree of depletion achieved in different lymph nodes inthe same individual was described [18, 20, 48, 49]. As previously mentioned, mice studies suggested that B cells resident intissues other than peripheral blood may be partly resistant to depletion by anti-CD20antibodies either because of local defective effector mechanisms or because the Bcells have a particular phenotype that renders them resistant to depletion inassociation with their specific state of maturation, activation ordifferentiation.
In bone marrow samples of RA patients treated with rituximab a relatively high numberof B-cell precursors subpopulations can be seen [50–52]. This has been documented at 1 month or 3 to 4 months after treatment, ata time when peripheral blood repopulation had not yet started [50, 51]. Persistence of CD20- plasma cells has been observed asexpected [50, 51]. In the two studies where phenotyping was more detailed, the cells foundwere mainly B-cell precursors and recirculating memory B cells [50, 52]. Once again, variability between individuals was observed [50, 52].
The presence of cells of B-cell lineage that presumably should be expressing CD20 hastherefore been well documented and rituximab is probably still present and binds tothe CD20 molecule, preventing its detection in flow cytometry as discussed above [50, 51]. Alternatively, antigen endocytosis/modulation could occur. Whether thedeveloping B cells are eventually depleted by anti-CD20 recruited mechanisms orwhether their full maturation is prevented by binding of rituximab to CD20 is notknown.
In a study of autopsy samples of lymph node and spleen of patients with lymphomatreated with rituximab monotherapy or with rituximab and chemotherapy, a substantialreduction of B-cell populations was documented-with only three out of eight patientsshowing any reactivity for markers of cells of B-cell lineage in the lymph nodes andonly one out of eight in the spleen by immunohistochemistry [53]. Similarly, a study in patients with idiopathic thrombocytopenic purpurashowed major and prolonged depletion of B cells in the spleen of 10 patients treatedwith rituximab [54]. The number of residual B cells correlated with time from rituximabtreatment but was <5% of spleen lymphocytes in eight out of nine patients studiedup to 10 months after rituximab treatment. Plasma cells were detected at increasedfrequencies when compared with patients with idiopathic thrombocytopenic purpura nottreated with rituximab. In a patient with idiopathic thrombocytopenic purpura,analysis of spleen and bone marrow samples by flow cytometry revealed completedepletion of B cells 3 months after treatment with rituximab [55]. In another patient with idiopathic thrombocytopenic purpura, B cells inthe spleen 3 months after rituximab treatment were only present in very low numbers(around 0.1%) [56]. Interestingly, in this later study persistence of memory B cells againstvaccinia virus in the spleen of patients previously treated with rituximab wasdocumented [56]. In kidney transplant patients that had as plenectomy 3 to 12 days aftertreatment with rituximab, naïve B cells were reduced but not memory B cells orplasma cells [57].
Vaccination studies in patients treated with rituximab can provide indirect data onB-cell subpopulations that may be resistant to depletion with anti-CD20 mAbs.However, published data are difficult to interpret because of the small number ofpatients, effects of concomitant therapy and the background disease itself on thehumoral response to vaccines and, in particular, because studies included patients atvarious stages of B-cell depletion or repopulation at the time of vaccination. Moststudies have looked at responses to influenza vaccines and showed absent or decreasedhumoral responses to vaccination in patients previously treated with rituximab whencompared with normal controls or patients not treated with rituximab [58–64]. Some studies described a positive relationship between the antibodyresponses to vaccination and number of circulating B cells at the time of vaccination [64] or the time from last rituximab treatment [60, 62]. Interestingly, when circulating influenza-specific B cells were studied 6days after vaccination, specific IgM-B cells were decreased in patients treated withrituximab 6 months previously when compared with controls but IgA B cells and IgG Bcells were similar [61]. In a study in lymphoma patients, responses to recall antigens in theinfluenza vaccine were also seen but not to the new antigen [65]. These studies suggest that memory B cells are more resistant to depletionthan naïve B cells and can survive treatment with rituximab and be recruited ina secondary immune response.
B-cell depletion in other solid tissues
In patients with RA, several studies have documented significant but variabledepletion of B cells in samples of synovial tissue of involved joints and persistenceof CD20- plasma cells [66–68]. Variability in depletion between individuals was not explained bydifferences in rituximab serum levels [69]. In a study in patients with Sjogren's syndrome, repeated salivary glandbiopsies 3 months after treatment with rituximab showed incomplete depletion of Bcells [70]. A previous study had shown complete depletion at 4 months [71]. In a study of renal explanted grafts in two patients treated with onedose (4 months earlier) or two doses (10 months earlier) of rituximab, despitedepletion of peripheral blood, tertiary lymphoid structures containing B cells wereseen [72].
Conclusion
In summary, although there are several studies looking at the degree and duration ofB-cell depletion induced by rituximab in the peripheral blood, there is very littleinformation on the exact degree of depletion in solid tissues - and, in particular, fewdefinite data on whether different subtypes of CD20-expressing B cells are more or lesssusceptible to depletion by anti-CD20 antibodies. The data available suggest that thereis variability between individuals on the extent and duration of depletion induced andthat this may have clinical correlations with response and duration of response inautoimmune diseases. Understanding what underlies this variability - and, in particular,whether drug clearance and antigen and effector modulation phenomena are involved - hasthe potential to lead to more effective B-cell depleting strategies and to increasingour understanding of the role that different B-cell subtypes play in the pathogenesis ofthe different autoimmune diseases.
Declarations
This article has been published as part of Arthritis Research & Therapy Volume 15 Supplement 1, 2013: B cells in autoimmune diseases: Part 2. Thesupplement was proposed by the journal and content was developed in consultation withthe Editors-in-Chief. Articles have been independently prepared by the authors and haveundergone the journal's standard peer review process. Publication of the supplement wassupported by Medimmune.
Abbreviations
- BAFF:
-
B-cell activating factor
- CD:
-
cluster of differentiation
- mAb:
-
monoclonalantibody
- RA:
-
rheumatoid arthritis
- Th:
-
T-helper type.
References
Weill JC, Weller S, Reynaud CA: Human marginal zone B cells. Annu Rev Immunol. 2009, 27: 267-285. 10.1146/annurev.immunol.021908.132607.
Lammers AJJ, de Porto APNA, Bennink RJ, van Leeuwen EM, Biemond BJ, Goslings JC, van Marle J, ten Berge IJ, Speelman P, Hoekstra JB: Hyposplenism: comparison of different methods for determining splenic function. Am J Hematol. 2012, 87: 484-489. 10.1002/ajh.23154.
Klein U, Rajewsky K, Kuppers R: Human immunoglobulin (Ig)M+IgD+ peripheral blood B cellsexpressing the CD27 cell surface antigen carry somatically mutated variable regiongenes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med. 1998, 188: 1679-1689. 10.1084/jem.188.9.1679.
Fecteau JF, Cote G, Neron S: A new memory CD27-IgG+ B cell population in peripheral bloodexpressing VH genes with low frequency of somatic mutation. J Immunol. 2006, 177: 3728-3736.
Berkowska MA, Driessen GJA, Bikos V, Grosserichter-Wagener C, Stamatopoulos K, Cerutti A, He B, Biermann K, Lange JF, van der Burg M, van Dongen JJ, van Zelm MC: Human memory B cells originate from three distinct germinal center-dependent and-independent maturation pathways. Blood. 2011, 118: 2150-2158. 10.1182/blood-2011-04-345579.
Koethe S, Zander L, Koster S, Annan A, Ebenfelt A, Spencer J, Bemark M: Pivotal advance: CD45RB glycosylation is specifically regulated during humanperipheral B cell differentiation. J Leukoc Biol. 2011, 90: 5-19. 10.1189/jlb.0710404.
Maecker HT, McCoy JP, Nussenblatt R: Standardizing immunophenotyping for the Human Immunology Project. Nat Rev Immunol. 2012, 12: 191-200.
Bohnhorst JO, Bjorgan MB, Thoen JE, Natvig JB, Thompson KM: Bm1-Bm5 classification of peripheral blood B cells reveals circulating germinalcentre founder cells in healthy individuals and disturbance in the B cellsubpopulations in patients with primary Sjogren's syndrome. J Immunol. 2001, 167: 3610-3618.
Sims GP, Ettinger R, Shirota Y, Yarboro CH, Illei GG, Lipsky PE: Identification and characterization of circulating human transitional B cells. Blood. 2005, 105: 4390-4398. 10.1182/blood-2004-11-4284.
Palanichamy A, Barnard J, Zheng B, Owen T, Quach T, Wei C, Looney RJ, Sanz I, Anolik JH: Novel human transitional B cell populations revealed by B cell depletiontherapy. J Immunol. 2009, 182: 5982-5993. 10.4049/jimmunol.0801859.
Lee J, Kuchen S, Fischer R, Chang S, Lipsky PE: Identification and characterization of a human CD5+ pre-naïve Bcell population. J Immunol. 2009, 182: 4116-4126. 10.4049/jimmunol.0803391.
Suryani S, Fulcher DA, Santner-Nanan B, Nanan R, Wong M, Shaw PJ, Gibson J, Williams A, Tangye SG: Differential expression of CD21 identifies developmentally and functionallydistinct subsets of human transitional B cells. Blood. 2010, 115: 519-529. 10.1182/blood-2009-07-234799.
Wei C, Jung J, Sanz I: OMIP-003: phenotypic analysis of human memory B cells. Cytometry A. 2011, 79: 894-896.
Bemark M, Holmqvist J, Abrahamsson J, Mellgren K: Translational mini-review series on B cell subsets in disease. Reconstitutionafter haematopoietic stem cell transplantation-revelation of B cell developmentalpathways and lineage phenotypes. Clin Exp Immunol. 2011, 167: 15-25.
Alduaji W, Illidge TM: The future of anti-CD20 monoclonal antibodies: are we making progress?. Blood. 2011, 117: 2993-3001. 10.1182/blood-2010-07-298356.
Kuijpers TW, Bende RJ, Baars PA, Grummels A, Derks IA, Dolman KM, Beaumont T, Tedder TF, van Noesel CJ, Eldering E, van Lier RA: CD20 deficiency in humans results in impaired T cell-independent antibodyresponses. J Clin Investig. 2010, 120: 214-222. 10.1172/JCI40231.
Leandro MJ, Cambridge G, Ehrenstein M, Edwards JC: Reconstitution of peripheral blood B cells after depletion with rituximab inpatients with rheumatoid arthritis. Arthritis Rheum. 2006, 54: 613-620. 10.1002/art.21617.
Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R, Newman RA, Hanna N, Anderson DR: Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody toCD20. Blood. 1994, 83: 435-445.
Bulati M, Buffa S, Candore G, Caruso C, Dunn-Walters DK, Pellicano M, Wu YC, Colonna Romano G: B cells and immunosenescence: a focus on IgG+IgD- CD27- B cells in aged humans. Ageing Res Rev. 2011, 10: 274-284. 10.1016/j.arr.2010.12.002.
Keren Z, Naor S, Nussbaum S, Golan K, Itkin T, Sasaki Y, Scmidt-Supprian M, Lapidot T, Melamed D: B-cell depletion reactivates B lymphopoiesis in the BM and rejuvenates the Blineage in aging. Blood. 2011, 117: 3104-3112. 10.1182/blood-2010-09-307983.
Cambridge G, Leandro MJ, Edwards JC, Ehrenstein MR, Salden M, Bodman-Smith M, Webster AD: Serologic changes following B lymphocyte depletion therapy for rheumatoidarthritis. Arthritis Rheum. 2003, 48: 2146-2154. 10.1002/art.11181.
Cambridge G, Isenberg DA, Edwards JC, Leandro MJ, Migone TS, Teodorescu M, Stohl W: B cell depletion therapy in systemic lupus erythematosus: relationships amongserum B lymphocyte stimulator levels, autoantibody profile and clinicalresponse. Ann Rheum Dis. 2008, 67: 1011-1016.
Teng YKO, Wheater G, Hogan VE, Stocks P, Levarht EWN, Huizinag TWJ, Toes REM, van Laar JM: Induction of long-term B-cell depletion in refractory rheumatoid arthritispatients preferentially afftects autoreactive more than protective humoralimmunity. Arthritis Res Ther. 2012, 14: R57-10.1186/ar3770.
Stevenson FK, Stevenson GT: Follicular lymphoma and the immune system: from pathogenesis to antibodytherapy. Blood. 2012, 119: 3659-3667. 10.1182/blood-2011-11-367730.
Press OW, Appelbaum F, Ledbetter JA, Martin PJ, Zarling J, Kidd P, Thomas ED: Monoclonal antibody 1F5 (anti-CD20) serotherapy of human B cell lymphomas. Blood. 1987, 69: 584-591.
Maloney DG, Liles TM, Czerwinski DK, Waldichuk C, Rosenberg J, Levy R: Phase I clinical trial using escalating single-dose infusion of chimeric anti-CD20monoclonal antibody (IDEC-C2B8) in patients with recurrent B-cell lymphoma. Blood. 1994, 84: 2457-2466.
Beurskens FJ, Lindorfer MA, Farooqui M, Beum PV, Engelberts P, Mackus WJ, Parren PW, Wiestner A, Taylor RP: Exhaustion of cytotoxic effector systems may limit monoclonal antibody-basedimmunotherapy in cancer patients. J Immunol. 2012, 188: 3532-3541. 10.4049/jimmunol.1103693.
Rezvani AR, Maloney DG: Rituximab resistance. Best Pract Res Clin Haematol. 2011, 24: 203-216. 10.1016/j.beha.2011.02.009.
Beers SA, French RR, Chan HTC, Lim SH, Jarrett TC, Vidal RM, Wijayaweera SS, Dixon SV, Kim H, Cox KL, Kerr JP, Johnston DA, Johnson PW, Verbeek JS, Glennie MJ, Cragg MS: Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications forantibody selection. Blood. 2011, 115: 5191-5210.
Vugmeyster Y, Howell K, McKeever K, Combs D, Canova-Davis E: Differential in vivo effects of rituximab on two B-cell subsets in cynomolgusmonkeys. Int Immunopharmacol. 2003, 3: 1477-1481. 10.1016/S1567-5769(03)00147-4.
Quartuccio L, Fabris M, Moretti M, Barone F, Bombardieri M, Rupolo M, Lombardi S, Pitzalis C, Beltrami CA, Curcio F, De Vita S: Resistance to rituximab therapy and local BAFF overexpression in Sjogren'ssyndromerelated myoepithelial sialadenitis and low-grade parotid B-celllymphoma. Open Rheumatol J. 2008, 2: 38-43. 10.2174/1874312900802010038.
Gong Q, Ou Q, Ye S, Lee WP, Cornelius J, Diehl L, Lin WY, Hu Z, Lu Y, Chen Y, Wu Y, Meng YG, Gribling P, Lin Z, Nguyen K, Tran T, Zhang Y, Rosen H, Martin F, Chan AC: Importance of cellular microenvironment and circulatory dynamics in B cellimmunotherapy. J Immunol. 2005, 174: 817-826.
Hamaguchi Y, Uchida J, Cain DW, Venturi GM, Poe JC, Haas KM, Tedder TF: The peritoneal cavity provides a protective niche for B1 and conventional Blymphocytes during anti-CD20 immunotherapy in mice. J Immunol. 2005, 174: 4389-4399.
Ahuja A, Shupe J, Dunn R, Kashgarian M, Kehry MR, Shlomchik MJ: Depletion of B cells in murine lupus: efficacy and resistance. J Immunol. 2007, 179: 3351-3361.
Ahuja A, Teichmann LL, Wang H, Dunn R, Kehry MR, Shlomchik MJ: An acquired defect in IgG-dependent phagocytosis explains the impairment inantibody-mediated cellular depletion in lupus. J Immunol. 2011, 187: 3888-3894. 10.4049/jimmunol.1101629.
Looney RJ, Anolik JH, Campbell D, Felgar RE, Young F, Arend LJ, Sloand JA, Rosenblatt J, Sanz I: B cell depletion as a novel treatment for systemic lupus erythematosus: a phaseI/II dose-escalation trial of rituximab. Arthritis Rheum. 2004, 50: 2580-2589. 10.1002/art.20430.
Albert D, Dunham J, Khan S, Stansberry J, Kolasinski S, Tsai D, Pullman-Mooar S, Barnack F, Striebich C, Looney RJ, Prak ET, Kimberly R, Zhang Y, Eisenberg R: Variability in the biological response to anti-CD20 B cell depletion in systemiclupus erythematosus. Ann Rheum Dis. 2008, 67: 1724-1731. 10.1136/ard.2007.083162.
Kamburova EG, Koenen HJPM, Boon L, Hilbrands LB, Joosten I: In vitro effects of rituximab on the proliferation, activation and differentiationof human B cells. Am J Transplant. 2012, 12: 341-350. 10.1111/j.1600-6143.2011.03833.x.
Vital EM, Dass S, Rawstron AC, Buch MH, Goeb V, Henshaw K, Ponchel F, Emery P: Management of nonresponse to rituximab in rheumatoid arthritis: predictors andoutcome of retreatment. Arthritis Rheum. 2010, 62: 1273-1279. 10.1002/art.27359.
Vital EM, Rawstron AC, Dass S, Henshaw K, Madden J, Emery P, McGonagle D: Reduced-dose rituximab in rheumatoid arthritis: efficacy depends on degree of Bcell depletion. Arthritis Rheum. 2011, 63: 603-608. 10.1002/art.30152.
Vital EM, Dass S, Buch MH, Henshaw K, Pease CT, Martin MF, Ponchel F, Rawstron AC, Emery P: B cell biomarkers of rituximab responses in systemic lupus erythematosus. Arthritis Rheum. 2011, 63: 3038-3047. 10.1002/art.30466.
Tahir H, Rohrer J, Bhatia A, Wegener WA, Isenberg DA: Humanized anti-CD20 monoclonal antibody in the treatment of severe resistantsystemic lupus erythematosus in a patient with antibodies against rituximab. Rheumatology. 2005, 44: 561-562. 10.1093/rheumatology/keh533.
Dass S, Rawstron AC, Vital EM, Henshaw K, McGonagle D, Emery P: Highly sensitive B cell analysis predicts response to rituximab therapy inrheumatoid arthritis. Arthritis Rheum. 2008, 58: 2993-2999. 10.1002/art.23902.
Mei HE, Frolich D, Giesecke C, Loddenkemper C, Reiter K, Schmidt S, Feist E, Daridon C, Tony HP, Radbruch A, Dorner T: Steady state generation of mucosal IgA+ plasmablasts is not abrogatedby B cell depletion therapy with rituximab. Blood. 2010, 116: 5181-5190. 10.1182/blood-2010-01-266536.
Roll P, Dorner T, Tony H-P: Anti-CD20 therapy in patients with rheumatoid arthritis: predictors of responseand B cell subset regeneration after repeated treatment. Arthritis Rheum. 2008, 58: 1566-1575. 10.1002/art.23473.
Palanichamy A, Muhammad K, Roll P, Kleinert S, Dorner T, Tony HP: Rituximab therapy leads to reduced imprints of receptor revision in immunoglobulinκ and λ light chains. J Rheumatol. 2012, 39: 1130-1138. 10.3899/jrheum.111513.
Boumans MJH, Thurlings RM, Gerlag DM, Vos K, Tak PP: Response to rituximab in patients with rheumatoid arthritis in differentcompartments of the immune system. Arthritis Rheum. 2011, 63: 3187-3194. 10.1002/art.30567.
Alwayn IP, Xu Y, Basker M, Wu C, Buhler L, Lambrigts D, Treter S, Harper D, Kitamura H, Vitetta ES, Abraham S, Awwad M, White-Scharf ME, Sachs DH, Thall A, Cooper DK: Effects of specific anti-B and/or anti-plasma cell immunotherapy on antibodyproduction in baboons: depletion of CD20- and CD22-positive B cells does notresult in significantly decreased production of anti-alphaGal antibody. Xenotransplantation. 2001, 8: 157-171. 10.1034/j.1399-3089.2001.008003157.x.
Schroder C, Azimzadeh AM, Wu G, Price JO, Atkinson JB, Pierson RN: Anti- CD20 treatment depletes B-cells in blood and lymphatic tissue of cynomolgusmonkeys. Transpl Immunol. 2003, 12: 19-28. 10.1016/S0966-3274(03)00059-5.
Leandro MJ, Cooper N, Cambridge G, Ehrenstein MR, Edwards JC: Bone marrow B-lineage cells in patients with rheumatoid arthritis followingrituximab therapy. Rheumatology. 2007, 46: 29-36. 10.1093/rheumatology/kel148.
Rehnberg M, Amu S, Tarkowski A, Bokarewa MI, Brisslert M: Short- and long-term effects of anti-CD20 treatment on B cell ontogeny in bonemarrow of patients with rheumatoid arthritis. Arthritis Res Ther. 2009, 11: R123-10.1186/ar2789.
Nakou M, Katsikas G, Sidiropoulos P, Bertsias G, Papadimitraki E, Raptopoulou A, Koutala H, Papadaki HA, Kritikos H, Boumpas DT: Rituximab therapy reduces activated B cells in both the peripheral blood and bonemarrow of patients with rheumatoid arthritis: depletion of memory B cellscorrelates with clinical response. Arthritis Res Ther. 2009, 11: R131-10.1186/ar2798.
Cioc AM, Vanderwerf SM, Peterson BA, Robu VG, Forster CL, Pambuccian SE: Rituximab-induced changes in hematolymphoid tissues found at autopsy. Am J Clin Pathol. 2008, 130: 604-612. 10.1309/UXLE9RHL968TER7B.
Audia S, Samson M, Guy J, Janikashvili N, Fraszczak J, Trad M, Ciudad M, Leguy V, Berthier S, Petrella T, Aho-Glele S, Martin L, Maynadie M, Lorcerie B, Rat P, Cheynel N, Katsanis E, Larmonier N, Bonnotte B: Immunologic effects of rituximab on the human spleen in immunethrombocytopenia. Blood. 2011, 118: 4394-4400. 10.1182/blood-2011-03-344051.
Kneitz C, Wilhelm M, Tony HP: Effective B cell depletion with rituximab in the treatment of autoimmunediseases. Immunobiology. 2002, 206: 519-527. 10.1078/0171-2985-00200.
Mamani-Matsuda M, Cosma A, Weller S, Faili A, Staib C, Garcon L, Hermine O, Beyne-Rauzy O, Fieschi C, Pers JO, Arakelyan N, Varet B, Sauvanet A, Berger A, Paye F, Andrieu JM, Michel M, Godeau B, Buffet P, Reynaud CA, Weill JC: The human spleen is a major reservoir for long-lived vaccinia virus-specificmemory B cells. Blood. 2008, 111: 4653-4659. 10.1182/blood-2007-11-123844.
Ramos EJ, Pollinger HS, Stegall MD, Gloor JM, Dogan A, Grande JP: The effect of desensitization protocols on human splenic B-cell populations invivo. Am J Transplant. 2007, 7: 402-407. 10.1111/j.1600-6143.2006.01632.x.
Gelinck LBS, Teng YKO, Rimmelzwaan GF, van den Bemt BJF, Kroon FP, van Laar JM: Poor serological responses upon influenza vaccination in patients with rheumatoidarthritis treated with rituximab. Ann Rheum Dis. 2007, 66: 1402-1403. 10.1136/ard.2007.071878.
Oren S, Mandelboim M, Braun-Moscovici Y, Paran D, Ablin J, Litinsky I, Comaneshter D, Levartovsky D, Mendelson E, Azar R, Wigler I, Balbir-Gurman A, Caspi D, Elkayam O: Vacciantion against influenza in patients with rheumatoid arthritis: the effect ofrituximab on the humoral response. Ann Rheum Dis. 2008, 67: 937-941.
Van Assen S, Holvast A, Benne CA, Posthumus MD, van Leeuwen MA, Voskuyl AE, Blom M, Risselada AP, ce Haan A, Westra J, Kallenberg CGM, Bijl M: Humoral responses after influenza vaccination are severely reduced in patientswith rheumatoid arthritis treated with rituximab. Arthritis Rheum. 2010, 62: 75-81. 10.1002/art.25033.
Rehnberg M, Brisslert M, Amu S, Zendjanchi K, Hawi G, Bokarewa MI: Vaccination response to protein and carbohydrate antigens in patients withrheumatoid arthritis after rituximab treatment. Arthritis Res Ther. 2010, 12: R111-10.1186/ar3047.
Arad U, Tzadok S, Amir S, Mandelboim M, Mendelson E, Wigler I, Sarbagil-Maman H, Paran D, Caspi D, Elkayam O: The cellular immune response to influenza vaccination is preserved in rheumatoidarthritis patients treated with rituximab. Vaccine. 2011, 29: 1643-1648. 10.1016/j.vaccine.2010.12.072.
Adler S, Krivine A, Weix J, Rozenberg F, Launay O, Huesler J, Guillevin L, Viliger PM: Protective effect of A/H1N1 vaccination in immune-mediated disease-a prospectivelycontrolled vaccination study. Rheumatology. 2012, 51: 695-700. 10.1093/rheumatology/ker389.
Eisenberg R, Jawad AF, Boyer J, Maurer K, McDonald K, Prak ETL, Sullivan KE: Rituximab-treated patients have a poor response to influenza vaccination. J Clin Immunol. 2013, 33: 388-396. 10.1007/s10875-012-9813-x.
Takata T, Suzumiya J, Ishikawa T, Takamatsu Y, Ikematsu H, Tamura K: Attenuated antibody reaction for the primary antigen but not for the recallantigen of influenza vaccination in patients with non-Hodgkin B-cell lymphomaafter the administration of rituximab-CHOP. J Clin Exp Hematopathol. 2009, 49: 9-13. 10.3960/jslrt.49.9.
Vos K, Thurlings RM, Wijbrandts CA, van Schaardenburg D, Gerlag DM, Tak PP: Early effects of rituximab on the synovial cell infiltrate in patients withrheumatoid arthritis. Arthritis Rheum. 2007, 56: 772-778. 10.1002/art.22400.
Kavanaugh A, Rosengren S, Lee SJ, Hammaker D, Firenstein GS, Kalunian K, Wei N, Boyle DL: Assessment of rituximab's immunomodulatory synovial effects (ARISE trial). 1:clinical and synovial biomarker results. Ann Rheum Dis. 2008, 67: 402-408.
Teng YK, Levarht EW, Hashemi M, Bajema IM, Toes RE, Huizinga TW, van Laar JM: Immunohistochemical analysis as a means to predict responsiveness to rituximabtreatment. Arthritis Rheum. 2007, 56: 3909-3918. 10.1002/art.22967.
Thurlings RM, Teng O, Vos K, Gerlag DM, Aarden L, Stapel SO, van Laar JM, Tak PP, Wolbinj GJ: Clinical response, pharmacokinetics, development of human anti-chimaericantibodies, and synovial tissue response to rituximab treatment in patients withrheumatoid arthritis. Ann Rheum Dis. 2010, 69: 409-412. 10.1136/ard.2009.109041.
Pijpe J, Meijer JM, Bootsma H, van der Wal JE, Spijkervet FK, Kallenberg CG, Vissink A, Ihrler S: Clinical and histological evidence of salivary gland restoration supports theefficacy of rituximab treatment in Sjogren's syndrome. Arthritis Rheum. 2009, 60: 3251-3256. 10.1002/art.24903.
Pers JO, Devauchelle V, Daridon C, Bendaoud B, Le Berre R, Bordron A, Hutin P, Renaudineau Y, Dueymes M, Loisel S, Berthou C, Saraux A, Youinou P: BAff-modulated repopulation of B lymphocytes in the blood and salivary glands ofrituximab-treated patients with Sjogren's syndrome. Arthritis Rheum. 2007, 56: 1464-1477. 10.1002/art.22603.
Thaunat O, Patey N, Gautreau C, Lechaton S, Fremeaux-Bacchi V, Dieu-Nosjean MC, Cassuto-Viguier E, Legendre C, Delahousse M, Lang P, Michel JB, Nicoletti A: B cell survival in intragraft tertiary lymphoid organs after rituximab therapy. Transplantation. 2008, 85: 1648-1653. 10.1097/TP.0b013e3181735723.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author has received consultancy fees and funding to attend international medicalmeetings from Roche Pharmaceuticals and consultancy fees and research funding fromGlaxoSmithKlein.
Rights and permissions
About this article
Cite this article
Leandro, M.J. B-cell subpopulations in humans and their differential susceptibility to depletionwith anti-CD20 monoclonal antibodies. Arthritis Res Ther 15 (Suppl 1), S3 (2013). https://doi.org/10.1186/ar3908
Published:
DOI: https://doi.org/10.1186/ar3908