
Abstract
The complex bidirectional communication between the gut and the brain is finely orchestrated by different systems, including the endocrine, immune, autonomic, and enteric nervous systems. Moreover, increasing evidence supports the role of the microbiome and microbiota-derived molecules in regulating such interactions; however, the mechanisms underpinning such effects are only beginning to be resolved. Microbiota–gut peptide interactions are poised to be of great significance in the regulation of gut–brain signaling. Given the emerging role of the gut–brain axis in a variety of brain disorders, such as anxiety and depression, it is important to understand the contribution of bidirectional interactions between peptide hormones released from the gut and intestinal bacteria in the context of this axis. Indeed, the gastrointestinal tract is the largest endocrine organ in mammals, secreting dozens of different signaling molecules, including peptides. Gut peptides in the systemic circulation can bind cognate receptors on immune cells and vagus nerve terminals thereby enabling indirect gut–brain communication. Gut peptide concentrations are not only modulated by enteric microbiota signals, but also vary according to the composition of the intestinal microbiota. In this review, we will discuss the gut microbiota as a regulator of anxiety and depression, and explore the role of gut-derived peptides as signaling molecules in microbiome–gut–brain communication. Here, we summarize the potential interactions of the microbiota with gut hormones and endocrine peptides, including neuropeptide Y, peptide YY, pancreatic polypeptide, cholecystokinin, glucagon-like peptide, corticotropin-releasing factor, oxytocin, and ghrelin in microbiome-to-brain signaling. Together, gut peptides are important regulators of microbiota–gut–brain signaling in health and stress-related psychiatric illnesses.
Similar content being viewed by others


Introduction
In the last decade, the physiological significance of gut peptides has been expanded beyond gastrointestinal (GI) digestion and absorption of nutrients. The more than 20 signaling molecules released from specialized enteroendocrine cells (EECs) in the GI tract have significant endocrine and metabolic functions and are able to communicate with the brain [1]. It is now also becoming clear that the activity of EECs, which represent no more than 1% of the total gut epithelium cell population, is modulated by the gut microbiota [2,3,4]. Enteric bacterial diversity and composition uniquely influence the release of gut peptides, including glucagon-like peptide (GLP-1), peptide YY (PYY), cholecystokinin (CCK), corticotropin-releasing factor (CRF), oxytocin, and ghrelin [1, 5,6,7]. However, the mechanisms underlying EEC–microbiota interactions have yet to be fully resolved.
Relatedly, increased emphasis has been given to the role of the microbiota and its metabolites in health and disease. Alterations in the gut microbiota may influence conditions such as obesity, allergies, autoimmune disorders, irritable bowel syndrome (IBS), inflammatory bowel disease (IBD), and psychiatric disorders [8,9,10,11,12,13]. Although, several animal studies have demonstrated that the gut microbiota modulates behaviors relevant to psychiatric disorders such as anxiety-like [14,15,16,17,18,19] and depressive-like behaviors [20,21,22], the mechanisms underpinning such microbe–host communication has not yet been figured out. Of note, several of the same peptides and their receptors that are released into the gut are also widely expressed in the brain or signal to the brain, where these peptides play well-established roles in the neurobiology of anxiety and depression (for review, see [23, 24]).
Since the expression of these gut-derived peptides is likely to be modulated by changes in the gut microbiota, it has been proposed that these peptide hormones may have an important role in gut–brain communication [21, 25,26,27].
The majority of the gut-derived peptides also play a role in the central regulation of appetite and food intake, in particular via hypothalamus nuclei [28,29,30,31,32]. Interestingly, obesity and psychiatric disorders are commonly associated [33,34,35,36,37], and a Westernized diet appears to exceed beyond nutrition, modulating behavioral reward and the development of psychopathologies [38,39,40,41,42,43].
Thus, an understanding of gut peptide signaling may provide new insights into gut–brain communication to help explain how the gut microbiota may modulate pathophysiological processes relevant to brain disorders such as anxiety and depression. The latter disorders are major causes of disability and contribute considerably to the global health burden [44, 45]. Although antidepressants and anxiolytics are usually effective, these drugs are frequently associated with serious side effects [46,47,48]. Conversely, some patients are resistant to the therapeutic effects of conventional pharmacological therapies. Recent data from human cohorts support preclinical findings suggesting that the modulation of the gut microbiota can influence brain function [49,50,51,52,53]. It is therefore clear that new therapeutic strategies should be developed to prevent and/or alleviate the symptoms of depression and anxiety. Psychobiotics, which target the gut microbiota to affect mental health for the better, may be one such innovative strategy (for review, see [54,55,56]). In this review we will discuss the role of the microbiota–gut–brain axis in anxiety and depression, and how gut-derived signaling peptides are likely important mediators in this process.
The Gut Microbiota
We live in a microbial world where each daily action can shape our microbiome. Mammalian microbial colonization likely begins at birth [57, 58] although a diverse range of microbes have been detected in umbilical cord blood, amniotic fluid, the placenta, and the fetal membranes [59,60,61,62]. Following parturition, the gut microbiota is refined and modified until adult-like communities reach homeostasis in its diversity [63]. A variety of factors can influence the infant microbiota, including birth delivery mode, breast feeding, weaning age, and maternal lifestyle [64,65,66]. In addition, the use of antibiotics, diet, exercise, and disease shape the microbial landscape creating interindividual differences within the gut microbiota [25, 67,68,69]. Typically, within healthy adults, the composition of the intestinal microbiota remains stable over time, and the bacterial phyla Firmicutes (including Lactobacillus, Clostridium, and Enterococcus genus) and Bacteroidetes (e.g., Bacteroides genus) represent > 90% of the intestinal community [70]. These phyla are frequently correlated with the health outcomes in human and animal studies (for review, see [71, 72]).
The diversity and stability of the microbiota are important indices for the overall health of an individual. The gut microbiota may impact host health via the conversion of nondigestible carbohydrates to short-chain fatty acids (SCFAs) [15], transformation of bile acids [73, 74], action against pathogenic bacteria [75], and the modulation of host innate and adaptive immune systems [76]. For example, a reduction in microbial diversity or the complete absence of the gut microbiota alters normal immune system function [77, 78] and decreases the capacity for harvesting energy from the diet [79, 80]. Consumption of complex carbohydrates rich in dietary fibers changes the composition of the gut microbiota often increasing the abundance of Firmicutes and Bacteroides [43, 81, 82]. Prebiotic supplements, here defined as nondigestible fibers selectively metabolized by the enteric microbiome, increase the production of SCFA [15, 83]. SCFA act on G-protein-coupled receptors [free fatty acid (FFA) receptors] located along the GI tract to regulate energy homeostasis via the stimulation of leptin production in adipocytes, and the secretion of gut peptides from colonic EECs [84,85,86,87].
The Microbiota–Gut–Brain Pathways: Focus on Gut Peptides
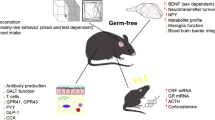
The brain and the gut are engaged in continuous bidirectional communication. Such communication may be important in mediating physiological effects ranging from GI function to the brain and behavior, bringing about the perception of visceral events such as nausea, satiety, and pain. In turn, stressful experiences lead to altered GI secretions and motility. The clinical relevance of such bidirectional communication, along with the modification of the gut microbiota composition, is now being investigated. Bidirectional gut–brain communication may begin as sensory information from the GI tract [88], and is consequently transformed into neural, hormonal, and immunological signals. These signals can independently or cooperatively relay information to the central nervous system (CNS). The neural and immune pathway, when relevant, are discussed throughout the paper. For an in-depth review of the literature regarding the neural and immune pathway, please see [2, 89, 90] and [11, 76, 91], respectively. Figure 1 also provide a schematic view of the gut microbiota–brain axis, including both neural and immune pathways.

Major communication pathways of the microbiota–gut–brain axis. There are numerous mechanisms through which the gut microbiota may signal the brain to control physiological processes. These include the release of gut peptides by enteroendocrine cells (EECs) where they activate cognate receptors of the immune system and on vagus terminals in the gut. Gut peptides such as neuropeptide Y (NPY) can also be released by cytokines under immune stimulation. There have been numerous reports of alterations in the gut microbiota in neuropsychiatric conditions, where gut peptides may play a key signaling role. Only a few examples of gut–brain pathway and gut peptides are represented in this figure
CCK = cholecystokinin; GLP-1 = glucagon-like peptide; CRF = corticotropin-releasing factor; PYY = peptide YY; GABA = γ-aminobutyric acid; SCFA = short-chain fatty acid
The gut endocrine system is comprised of gut peptides and other signaling molecules (i.e., serotonin), which are released by different types of EECs along the GI tract in response to food intake, particularly after ingestion of carbohydrates and fats. EECs represent approximately 1% of epithelial cells in the gut lumen and are divided into subtypes according to location in the gut and class of molecules released, including peptides (Fig. 2). Recently, in an elegant study, Bellono et al. [88] demonstrated that EECs, in particular enterochromaffins, control serotonin release onto 5-HT3 receptor-expressing primary afferent nerve fibers that extend into intestinal villi, enabling them to detect and transduce information from the gut to the nervous system.

Gut peptide distribution in the gastrointestinal tract. Gut peptides are released from different subgroups of enteroendocrine cells (EECs) following appropriate stimulation. The stomach is rich in ghrelin expression from A cells (brown circles), cholecystokinin (CCK) is expressed in the proximal segment of the small intestine by I cells (blue circles). The small and large intestine secretes glucagon-like peptide (GLP-1) and peptide YY (PYY) from L cells (red and green circles, respectively). Only some examples of EEC subgroups and relative hormones secreted are represented in the figure
Peptides are short chains of amino-acid monomers linked by amide bonds (approximately ≤ 50 amino-acid residues) that can be released from specialized neurons in the brain (where they are called neuropeptides) or in EECs as a signaling molecule through binding G protein-coupled receptors [88, 92, 93]. Peptides are able to reach receptors far from the site of their release, and are efficiently metabolized by endogenous enzymes and do not accumulate in tissues [92, 94]. There are > 100 peptides in mammals, with those relevant to gut–brain communication being mostly involved in regulating digestion and satiety. Many of the peptides found in EECs and in CNS regulating the GI tract are also found in the enteric nervous system. For example, tachykinins (i.e., substance P) and calcitonin gene-related peptide are expressed in intrinsic sensory neurons [95, 96]. Gastrin-releasing peptide stimulates the release of gastrin from G cells of the stomach and regulates gastric acid secretion and enteric motor function [97]. Vasoactive intestinal peptide communicates with adjacent postsynaptic targets to regulate circadian rhythm and inhibit gastric acid secretion and absorption from the intestinal lumen [98, 99]. Structurally similar to vasoactive intestinal peptide, the pituitary adenylate cyclase-activating polypeptide can stimulate enterochromaffin-like cells, a type of EEC [100]. Other peptides include the opioid peptides (enkephalin, endorphins), insulin, calcitonin gene-related peptide, oxyntomodulin, glucagon, enteroglucagon, GLP-2, somatostatin, and gastrin inhibitory polypeptide, among others. Owing to their privileged location in the GI tract, it is possible that those peptides may be influenced by changes in the gut microbiota and modulate (directly or indirectly) upstream signaling to the brain. In this review we focus on peptides with known roles in anxiety and depression; for an in-depth review of others, please see [7, 101,102,103,104].
For the peptides discussed in this review, beyond the role in satiety, they can be stimulated by bacterial products that come into contact with the gut epithelium [83, 105, 106]. Distinct subtypes of EECs release different peptides in the gut. For example, the stomach contains X/A-like cells that secrete ghrelin, an orexigenic peptide. Increased ghrelin is associated with the timing of a meal. The proximal small intestine contains I cells and K cells that produce CCK and glucose-dependent insulinotropic polypeptide, respectively [5, 107]. In the distal small and large intestines are L cells, which produce GLP-1/2 and PYY. CRF, which is found in the colon and ileum, is expressed by enterochromaffin cells [108]. Moreover, an additional subset of EECs has the ability to co-express peptides, including CCK, GLP-1, and PYY [109, 110].
Following secretion, gut peptides diffuse throughout the lamina propria, which is occupied by a variety of immune cells, ultimately to reach the bloodstream, to stimulate sensory neurons and potentially to act on the vagus nerve, thereby creating an intersection for gut peptidergic signaling to the brain [111]. In a complementary manner, the blood–brain barrier (BBB) selectively facilitates the transport of some peptides in the blood-to-brain direction, suggesting a direct pathway for some gut peptides that are not quickly degraded after their release [112,113,114,115].
Owing to the wide expression of peptides and their receptors in both brain and gut, in addition to their facilitated transit into the bloodstream, it seems reasonable to assume that these peptides can act beyond their primary signaling function, suggesting a possible role (most likely indirect) of the gut peptides in neuropsychiatric disorders [5]. The dynamic profile of gut peptides, including their direct link with mood disorders, and their broad role in the gut suggests that the microbiota–gut peptide–brain communication axis can be a target for psychobiotics to prevent and treat such disorders (Table 1).
The Gut Microbiota in Anxiety and Depression
The link between gut function and mental health is well appreciated (for review, see [134,135,136]. Depression and anxiety are often accompanied by changes in colonic motility, which, in turn, alters the composition and stability of the gut microbiota, as well as the colonic physiology and morphology [137,138,139,140]. Stress-related disorders can also alter the intestinal barrier, triggering a “leaky gut”, which could allow a microbiota-driven proinflammatory response through the translocation of certain bacterial products from the gut [141,142,143]. For example, Maes et al. [142, 143] found in 2 different studies that lipopolysaccharide (LPS) produced by gut bacteria could increase immune responses in depressed patients. Likewise, in IBS, a disease linked to alterations in the immune system [144], a correlation was found between gut microbiota composition and brain volumes in patients with IBS with early life trauma, suggesting a shift in the Firmicutes/Bacteroidetes ratio, with Firmicutes increased and Bacteriodetes decreased [53]. Interestingly, this study failed to identify correlations between anxiety or depression symptoms scores. However, in depressive patients without IBS symptoms, the Firmicutes/Bacteroidetes ratio was inversely correlated with the microbiota composition profiled in patients with IBS [145,146,147].
Similar observations have been made in animal studies. Yu et al. [148] found that depressive-like rats had the relative abundances of Bacteroidetes significantly increased, whereas that of Firmicutes was markedly decreased. In mice that underwent chronic stress, reductions in the genus Bacteroides were identified [149]. Stressed mice also have been shown to have greater populations of the genus Clostridium, which agrees with the gut microbiota profile in maternally separated rodents [149,150,151]. The genus Clostridium is commonly altered as a result of changes modulated by gut metabolites, such as phenylalanine, tryptophan, and tyrosine [152, 153]. Such metabolites are part of the metabolism of key neurotransmitters in mammals, including serotonin, with implications for ENS and CNS function and thus gut–brain axis signaling [154,155,156,157,158].
Antidepressants are well known to have antimicrobial effects, modulating the pathophysiology of the anxiety and depression by reshaping not only brain biochemistry, but also the gut microbiota [159, 160]. Conversely, certain antibiotics, such as β-lactams and tetracyclines have potential antidepressant properties in rodents and humans [161, 162]. Epidemiologic studies have suggested that some classes of antibiotics such as fluoroquinolones are associated with the development of depression and anxiety [163,164,165,166].
Although the altered gut microbiota and its effects on host metabolic phenotype during psychiatric illness remains unknown, these observations highlight the potential association of the gut microbiota with host depression and anxiety. Thus, the manipulation of the gut microbiota may be a useful tool to decode the physiological role of the gut–brain axis in psychiatric disorders [167]. The use of germ-free rodents, animals with pathogenic bacterial infections, animals exposed to probiotic agents or to antibiotics, prebiotics, and fecal microbiota transfer from another animal or human may lead to important new findings for the development of new therapeutic strategies to treat or prevent anxiety and depression.
The Neuropeptide Y Family
The members of the neuropeptide Y (NPY) family comprise NPY itself, PYY, and pancreatic polypeptide (PP). The functional role of the NPY family in gut–brain signaling is supported by the wide distribution of the 4 main NPY receptors in mammals (Y1,Y2, Y4, and Y5 receptor) (for a review, see [168]). NPY has higher affinity than PYY for the Y1 and Y5 receptors, whereas PYY is the stronger agonist at Y2 receptors [168, 169]. The Y2 receptors might also act as an autoreceptor, regulating the release of NPY, as well as other neurotransmitters, such as γ-amino butyric acid (GABA) [170,171,172]. The Y4 receptors are preferentially bound by PP [169]. The Y1,Y2, and Y4 receptors are found in the colon and small intestine [173]. In addition, the Y1 and Y2 receptors are also widely distributed within the brain, but both Y4 and Y5 receptors are restricted to particular areas in the brain, including the nucleus of the solitary tract (NTS), hypothalamus, and amygdala [174, 175].
The NPY family is expressed at all levels of the gut–brain axis. NPY is the most abundant peptide in the brain and is expressed from the medullary brainstem to the cerebral cortex (for review, see [175]). The presence of NPY in the NTS and the ventral hypothalamus deserves special mention since these areas are the main relays for peripheral signaling, which are particularly important for the gut–brain signaling [24]. Moreover, NPY is also found in enteric nerve plexuses and postganglionic sympathetic neurons [24, 176]. The gut peptides PYY and PP are released mainly by EECs, where PYY is expressed from L cells of the ileum and colon and released in response to feeding (Fig. 2) [5], whereas PP is synthesized from F cells of pancreatic islets and under vagal control [176, 177]. Both PYY and PP are able to cross the BBB by transmembrane diffusion and bind cognate receptors located in the area postrema [115, 178]. Moreover, these gut peptides can activate their cognate receptors located in vagal afferents to signal to the brainstem [179, 180]. In addition, PYY-positive neurons are also found in the NTS, and nerve terminals containing PYY are located in the pons, hypothalamus, and spinal cord [177].
Microbiota and the NPY, PYY, and PP: Relevance to Anxiety and Depression
There is abundant evidence that the central NPY system affects stress-related disorders such as anxiety and depression (for a review, see [181, 182]). Additionally, NPY signaling also has modulatory properties involving neuroprotection, neurogenesis, and neuroinflammation [183, 184]. The Y4 receptor, which is mainly activated by peripheral PP, modulates anxiety and fear-related disorders in rodents [174, 185, 186]. PP is about 100 times more potent than NPY in Y4 activation, suggesting that PP/Y4 receptors can be an important route of gut–brain communication in the pathogenesis of anxiety and depression.
Following a meal, PP and PYY are released and inhibit gastric emptying via the targeting of receptors on the intestinal epithelium, as well as vagus nerve signaling [187,188,189]. By activating Y4 receptors in the gut, PP reduces intestinal motor activity and facilitates nutrient absorption, an effect likewise exerted by PYY [24, 188, 190]. Interestingly, it has been suggested that hunger in rodents could promote fear extinction through Y4 receptor signaling [185, 191]. Although the gut microbiota was not investigated in these studies, the results strongly suggest a potential role for gut–brain communication through Y4 signaling. The expression of NPY in the hypothalamus (where NPY plays a major role in feeding behavior and stress response) is increased in adult, healthy, germ-free mice when compared with conventionally raised mice [116]. In addition, Schéle et al. [116] found that administration of leptin reduced NPY expression in germ-free mice but not in conventionally raised mice, suggesting that the gut microbiota may decrease leptin sensitivity, effects that are dependent on vagal signaling [192].
Despite its elevation by food intake, circulating PYY can also be increased by exercise and stress [193, 194], conditions that also modify the gut microbiota [195, 196]. It is important to note that more studies linking changes in PYY and the gut microbiota are needed. Correlative evidence exists between PYY and the gut microbiota, where elevation in PYY in obese individuals was associated with changes in the gut microbiota [197]. Interestingly, PYY secretion is influenced by activation of FFA receptors by SCFA [85, 198,199,200]. Moreover, the SCFA butyrate can increase the expression of receptors that sense microbial compounds (Toll-like receptors), which, in turn, increases the expression of PYY [201]. Given the importance of microbiota-generated SCFAs such as butyrate, the role of the SCFA in mediating PYY expression through immune cells could be an important link in microbial–host communication and PYY signaling. The use of dietary compounds to modulate the gut microbiota has been a useful approach to investigating the role of intestinal bacteria in PYY expression. For instance, PYY expression is upregulated in the ileum after treatment with prebiotic galacto-oligosaccharides (GOS) [202]. Although plasma PYY is increased after oligofructose treatment in healthy rats [202, 203], oligofructose did not alter plasma PYY levels in obese mice [204]. A separate study in rodents demonstrated that bacterial proteins produced after nutrient-induced Esherichia coli growth may signal the CNS via increased plasma PYY [205]. The same effect was observed after the intake of probiotics. Hong et al. [202] found an upregulation of intestinal PYY with the commensal bacteria Bifidobacterium bifidum in rats. However, a study in humans showed that plasma PYY was unaffected after treatment with barley kernel bread, an indigestible carbohydrate source, and a mixture of the probiotics Bifidobacterium animalis, Lactobacillus reuteri, and Lacobacillus plantarum [206]. Despite the interspecies variability, the contrasting results found in humans and animals could be owing to the selective spectrum of bacteria that can modulate PYY expression. For this, Rajpal et al. [207] investigated selective spectrum antibiotics and found that only an alteration of Gram-negative bacteria (which, in turn, increased the population of Firmicutes) improved metabolic aspects, including PYY, in rodents.
Evidence of direct involvement interaction between the NPY system and the gut microbiota modulating behavior is lacking. Perhaps the most convincing evidence of the gut NPY family signaling brain function is found within the context of depression and anxiety associated with colitis. For example, in a colitis disease model, it was found that an increase in the colonic synthesis of NPY was associated with decreased colonic Y1 receptor expression [208, 209]. Moreover, circulating levels of plasma PYY and NPY are also enhanced in patients with IBD, suggesting that the epithelial levels of NPY and PYY are exhausted by excessive release of these peptides from EECs and intestinal neurons [173]. In addition, NPY-depleted mice are resistant to developing colitis and the levels of colonic PYY are decreased in rats with chemically induced colitis or patients with IBS [24, 173, 208]. The effects of colitis on behavior are also modified by NPY or PYY. For example, depletion of NPY or PYY in mice prevents the anxiogenic and depressive-like behavior often promoted by colitis [210]. Moreover, the effect of increased expression of NPY on immune function, is mediated mainly via NPY receptors located on immune cells [173, 211], as well as through the modulatory role of TLR on PYY expression [201]. Crosstalk between the immune system and the brain neurocircuitry can drive the development of depression and raise anxiety [212]. Thus, it is likely that gut microbiota contributes indirectly to NPY release and signaling to affect its homeostatic role in balancing the brain’s response to stress in the brain. Recently, it was shown that the depletion of the gut microbiota in adult mice upregulated NPY expression within the amygdala and hypothalamus, followed by cognitive deficits [25], an effect also observed in rats housed in environmental enrichment [213]. However, anxiety- or depression-like behaviors were unaffected in these animals [25]. Interestingly, hypothalamic NPY was also found increased in germ-free mice [116]. Germ-free mice usually present an anxiolytic phenotype behaviorally [214]. In fact, the anxiolytic profile of NPY is associated with its central levels to promote a change in the anxiety phenotype of the animals [215]. Moreover, Schéle et al. [116] demonstrated the role of NPY in counteracting the increased plasma levels of corticosterone in germ-free rodents. Of note, germ-free rodents have an exaggerated release of corticosterone after acute restraint stress [27, 216]. In contrast, plasma NPY levels decreased after the introduction of conventional intestinal microbiota in germ-free mice [217], a shift also observed by Sudo et al. [216] with the corticosterone levels after recolonization of germ-free mice. Interestingly, a fiber-supplemented diet did not affect plasma PYY in germ-free rats, a result that was not observed in conventional mice [218]. Finally, NPY and PYY are also antimicrobial peptides, influencing the composition and function of the gut microbiota [219].
Although a mechanism pointing to how the gut microbiota influence the NPY system and then modulate brain function and behavior, it is worth noting that NPY receptors are expressed on immune cells, brain neurons, primary afferents in the gut, and sympathetic neurons. Thus, in addition to the literature mentioned above, this indicates that NPY, PYY, and PP somehow participate in the reciprocal interaction between the brain, the gut, and the immune system, therefore warranting investigation of their pathophysiological implications within the context of the microbiota–gut–brain axis.
GLP-1
GLP-1, an incretin hormone, is a 30 amino-acid peptide derived from the post-translational processing of preproglucagon, which is involved in the neural regulation of food intake, body weight, the modulation of the hypothalamic–pituitary–adrenal (HPA) axis, and overall response to stress via activation of the GLP-1 receptor (for review, see [220,221,222,223]).
In the brain, GLP-1 is present in the NTS and the olfactory bulb [5, 224]. In the NTS, GLP-1 fibers directly innervate hypophysiotrophic CRF neurons in the paraventricular nucleus (PVN), where GLP-1 activates the HPA axis response [225]. GLP-1 receptor is abundantly found in brain regions that are critical for the regulation of both metabolic and endocrine responses [226]. In the periphery, GLP-1 is secreted by L cells, which also secrete PYY in the ileum and the colon in response to neuronal and hormonal signals (Fig. 2) [5, 110], whereas the GLP-1 receptors are distributed in the gut, pancreatic β-cells, kidney, and in the vagus nerve [222, 227].
Following release into the bloodstream, GLP-1 is rapidly degraded, resulting in < 10% of secreted GLP-1 reaching systemic circulation as a result of enzymatic degradation [221]. Thus, it is likely that GLP-1 acts locally in a paracrine fashion in the intestine. Indeed, vagal afferent neurons express the GLP-1 receptor, activation of which induces firing of the respective vagal afferent neurons and most likely stimulation of NTS [228, 229]. As the NTS projects GLP-1 neurons directly to the paraventricular nucleus of the hypothalamus, it is plausible to affirm that GLP-1 restricts feeding behavior, which is dependent of the vagal pathway [230,231,232]. Although it is unlikely that nutrient-induced GLP-1 enters the circulation to act centrally to lower food intake, it has been proposed that rather than entering the systemic circulation, GLP-1 is released into the lymph [233,234,235]. GLP-1 levels in the lymph were found to be higher, lasting longer than in the bloodstream, supporting a plausible model where GLP-1 does reach the brain given its transport via the lymph [233]. If not, it is also known that intestinal intraepithelial lymphocytes can modulate energy availability, host microbial responses, and mucosal integrity through the activation of GLP-1 receptors in immune cells, and regulate the migration of lymphocytes and natural killer T cells [123, 236].
Gut Microbiota and GLP-1: Relevance to Anxiety and Depression
GLP-1 is widely recognized for its role in lowering postprandial blood glucose via augmentation of glucose-dependent insulin release and inhibition of glucagon secretion (for review, see [237]). Interestingly, brain abnormalities and cognitive deficits similar to those found in depressive patients are also frequently identified in individuals with metabolic disorders [238,239,240,241]. It is particularly relevant that patients with diabetes are twice as likely to suffer from anxiety and depression than the general population [242, 243]. Growing evidence supports the role of GLP-1 in the central effects of metabolic syndrome, as GLP-1 in anxiety appears to be related to the improvement of the glucoregulation in diabetic states, with subsequent reduction in proinflammatory cytokines and an increase in neuroprotection [244, 245]. For example, reduction of the phyla Firmicutes and Bacteroidetes in the gut ameliorates insulin resistance under conditions of a high-fat diet by stimulating GLP-1 secretion in mice [246]. Moreover, anxiety-like behavior in diabetic mice was blunted by a GLP-1 receptor agonist [247, 248], whereas in nondiabetic animals, the impact of a GLP-1 receptor agonist was less clear in reducing anxiety-like behavior [249,250,251]. However, chronic systemic stimulation of GLP-1 receptors promoted antidepressant-like effects in rodents, coupled with increasing hippocampal neurogenesis, suggesting plasticity-related effects could underpin such effects [252,253,254,255]. Furthermore, a GLP-1 receptor agonist was able to reverse LPS-induced depressive-like behavior without directly affecting inflammatory cytokines [256]. Taken together, it is likely that attenuation of glucose fluctuations by GLP-1 is responsible for its neuroprotective effects, especially during plasma hyperglycemia.
Evidence suggests a key role for SCFA in modulating GLP-1 expression through the stimulation of FFA2 and FFA3 receptors in rodents [83, 105, 257,258,259,260]. Moreover, butyrate-induced GLP-1 secretion is attenuated in FFA3 receptor-depleted mice [105], thereby supporting a role for the gut microbiota in GLP-1 release. Indeed, germ-free or antibiotic-treated mice have elevated plasma and intestinal GLP-1 levels, as well as increased proglucagon expression in the colon [118, 205, 246, 261,262,263]. The expression of GLP-1 in the hypothalamus of germ-free mice is higher than in conventionally colonized animals [116]. Probiotics such as Bifidobacterium animalis and Bifidobacterium bifidum increase intestinal and plasma GLP-1 levels in rodents [119, 202, 259]. GLP-1 is also upregulated in mice after treatment with the prebiotics GOS, oligofructose, and inulin-type fructans [127, 202, 264,265,266]. The depletion of GLP-1 in rodents alters the composition of the enteric microbiota [123] and abolishes the antidiabetic actions of oligofructose in mice [266]. Collectively, these findings suggest the gut microbiome influences central and peripheral GLP-1 production through L cells in the gut.
The effects observed in animals can also be found in humans. In healthy volunteers, a mixture of barley kernel bread and Bifidobacterium animalis, Lactobacillus reuteri, and Lactobacillus plantarum increased levels of plasma of GLP-1 without affecting metabolic outcomes [206]. The result was observed in glucose-tolerant volunteers, where the daily administration of Lactobacillus reuteri or the commercially available VSL#3 (a formulation with 8 strains of commensal bacteria, including different strains of Bifidobacterium and Lactobacillus) increased GLP-1 release into the blood, without a change in insulin sensitivity or body fat distribution [120, 124]. In patients with mood disorders and the presence of cognitive deficits, it was found that 4-week treatment with a GLP-1 agonist reverted the cognitive deficits without affecting metabolic parameters, suggesting GLP-1 target offers beneficial effects in some symptoms that can be overrepresented in psychiatric disorders [267].
Taken together, the localization of GLP-1 and the GLP-1 receptors in the gut–brain axis underscores the importance of the microbiota in the modulatory role for GLP-1 in the central regulation of energy homeostasis, which may influence depression and anxiety associated with metabolic dysfunctions.
CCK
CCK is derived from a 115 amino-acid precursor and is converted into multiple isoforms, which range from 4 to 58 amino acids in length, with a wide range of physiological effects such as the control of gastric emptying, gallbladder contraction, pancreatic enzyme release, and suppression of appetite (for review, see [268]). Generally, CCK acts through the G protein-coupled receptors CCK1 and CCK2 receptors to modulate other neurotransmitters, such as GABA, glutamate, dopamine, and acetylcholine.
CCK is also found abundantly in the peripheral and central nervous systems [23]. Within limbic areas, CCK is generally co-localized with 5-HT3 and CB1 receptors, which participate in regulation of mood (for review, see [269,270,271]). In the gut, CCK is released postprandially from I cells of the small intestine (Fig. 2), although mature EECs have the ability to co-express CCK with GLP-1 and PYY [5, 110]. Both CCK1 and CCK2 receptors have distinct patterns of expression. CCK1 is found mainly within the periphery, including vagal afferent terminals along the mucosal epithelium, whereas CCK2 is widely distributed in the brain, overlapping with CCK distribution [23, 272].
Gut Microbiota and CCK: Relevance to Anxiety and Depression
The role of CCK in emotional behaviors is seen primarily through the activation of the CCK2 receptors in limbic regions, mainly the basolateral amygdala and, to some extent, cortical areas and the hippocampus. Both human and rodents studies indicate a positive correlation of CCK levels and increased anxiety-like behavior [273,274,275]. Moreover, systemic administration of CCK-8 elevated the levels of brain-derived neurotrophic factor in the rat hippocampus, suggesting a neuroplasticity-related effect mediated by CCK [276].
CCK is the first peptide released postprandially to reduce food intake in both obese and lean individuals, remaining in the bloodstream for about 3 h [277, 278]. Then, CCK acts on its cognate receptors in the periphery located mainly on vagal afferent neurons that project to the NTS [2, 279, 280]. Indeed, vagotomy in mice attenuates the effects of CCK on gastric emptying, whereas the administration of LPS abolishes CCK-induced inhibition of food intake [281, 282]. Gut CCK has also been implicated in the pathophysiology of both IBS and IBD. The densities of CCK cells are reduced in IBD, which may lower the levels of gastric secretion, such as bicarbonate, pancreatic enzymes, and bile salts, resulting in the development of IBS [283, 284]. Interestingly, a potential role for CCK in regulating the immune system has also been suggested in mice. Mice lacking T-cell receptors, which develop inflammation on the colonic epithelium, have been associated with a decrease in the number of EEC cells specialized in secreting CCK [285]. As CCK has been reported to have anti-inflammatory effects, reduced expression of CCK in the gut may have an impact on gut inflammatory diseases, with subsequent consequences in IBD or IBS comorbidity.
CCK signaling is also modulated by the gut microbiota. Obese rodents, which have drastic changes in the composition of gut microbiota, exhibit reduced response to CCK in the nodose ganglia [286], as well as decreased CCK signaling in the brain [287, 288]. Moreover, depletion of CCK1 in mice reduces firing in NTS neurons, indicating that the activation of the vagal afferent neurons was inhibited [289]. However, germ-free mice have reduced concentrations of gut CCK and delayed intestinal transit [125, 126], whereas infection of germ-free rodents with Giardia increased colonic expression of CCK [283], corroborating previous studies showing that intestinal infection could facilitate CCK stimulation of vagal afferent neurons [290, 291].
Despite the changes observed in CCK levels in obese animals, plasma concentrations of CCK were not different in severely obese patients versus normal-weight individuals [292]. Federico et al. [292] did not observe changes in the gut microbiota after obese patients underwent bariatric surgery. Taken together, the results from this study show that circulating CCK may not be modulated by changes in the gut microbiome facilitated by alterations in energy metabolism.
The importance of the microbiota in the modulatory role for CCK in the central regulation of energy homeostasis may influence depression and anxiety associated with obesity and represent a potential target in this specific aspect of emotionality.
Ghrelin
Ghrelin is a 28 amino-acid stomach-derived peptide known for its powerful physiological orexigenic and adipogenic effects, as well as for its contribution toward the stress response, anxiety, and depression [293,294,295,296,297]. Ghrelin is the endogenous ligand of the growth hormone secretagogue receptor type 1a and was first discovered for its stimulation of growth hormone secretion from the pituitary [293]. The ghrelin receptor (growth hormone secretagogue receptor type 1a) is expressed in the brain predominantly in the anterior pituitary gland, arcuate nucleus, amygdala, substantia nigra, ventral tegmental area, and raphe nuclei, whereas peripheral expression is found in the pancreatic islets, adrenal gland, and thyroid [298, 299].
Ghrelin is secreted by A cells of the stomach (Fig. 2) [293, 299], although other peripheral organs such as the testis, placenta, kidney, small intestine, and pancreas also express low levels of ghrelin [28]. There is also evidence for the synthesis of ghrelin in the brain, albeit at a much lower level, in specific neuronal cells of the hypothalamus [28, 300]. Of note, central ghrelin expression remains highly controversial as no significant amounts could be detected in rodent neuronal cells, and ghrelin receptor-expressing neurons did not receive synaptic inputs from ghrelin-immunoreactive nerve terminals in these species, suggesting considerable inconsistency between different studies [299, 301].
Peripherally produced ghrelin exerts its appetite-inducing effects centrally after passing through the BBB [112, 302,303,304]. In addition, ghrelin axon terminals were found to innervate other hypothalamic peptidergic systems, such as agouti-related peptide- and proopiomelanocortin neurons in the arcuate nucleus and CRF in the PVN, suggesting that interactions between ghrelin and NPY/AGPR, proopiomelanocortin, and CRF circuits are critical for energy homeostasis and stress responses [305,306,307]. Nevertheless, it appears that in addition to its effects on feeding, ghrelin enhances the release of NPY onto GABAergic nerve terminals adjacent to CRH neurons, disinhibiting these neurons and stimulating CRF release into the pituitary–hypophyseal portal circulation, driving increased ACTH secretion from the pituitary [28, 308].
Gut Microbiota and Ghrelin: Relevance to Anxiety and Depression
Ghrelin crosses the BBB via active transport and direct diffusion, making the ghrelinergic system ideally placed to play an important role in the stress response [112, 297, 302]. Physiological state, such as fasting, strongly affects the transport of ghrelin into the brain [302]. However, different stressors, such as social defeat and restraint stress in rodents, can increase long-term ghrelin levels in the stomach [309,310,311]. Mice lacking endogenous ghrelin have a dampened circulating glucocorticoid response following an acute stressor due a deficit in the PVN response needed to stimulate ACTH from the pituitary [295, 311]. Moreover, mice lacking growth hormone secretagogue (GHS) receptors are more susceptible to stress and exhibit anxiety- and depressive-like behavior [309, 312]. Recently, some studies have demonstrated that hunger (which increases ghrelin levels) can instigate the search for food even under stressful and anxiogenic conditions, a sign of adaptation for survival [185, 313, 314]. In agreement with the survival theory, recent studies have shown that repeated injection of GHS receptor agonists into the amygdala increases stressor-inducing fear memory formation, whereas antagonists of GHS receptor administered during repeated stressors prevented fear memory formation, as well as anxiety and depression-like phenotypes in mice [296, 315]. Thus, ghrelin may regulate the network designed to enhance survival potential by increasing vigilance, fear, and controlling anxiety. This framework provides a model through which ghrelin could have myriad actions on mood, depending on contextual states and physiological feedback mechanisms.
Although released in the stomach, the regulatory role of ghrelin in appetite is mainly achieved centrally within the arcuate nucleus and the brainstem [316, 317]. Thus, the regulatory role of ghrelin in the CNS may be affected by the gut microbiota via vagus nerve to the NTS. Indeed, germ-free mice have lower levels of plasma ghrelin under basal conditions than conventional mice [128, 129]. However, after a period of fasting, germ-free mice have higher plasma ghrelin concentrations than conventionally colonized mice, which could reflect a direct effect of fasting and not the microbiota profile on ghrelin release [121]. Interestingly, serum ghrelin levels are negatively correlated with the abundance of certain gut bacteria, including the commensal Bifidobacterium and Lactobacillus strains [318]. In addition, chronically elevated plasma ghrelin, associated with changes in the gut microbiota, can prevent anxiety- and depression-like behavior through ghrelin receptors [309]. Ghrelin has also been shown to decrease in response to the prebiotic supplementation inulin and oligofructose uniquely in obese and lean rodents, with obese rats showing attenuated ghrelin levels [117, 122, 127]. However, oligofructose does not change plasma ghrelin levels in obese rats fed a high fat/high sucrose diet [203]. In addition, ingestion of conjugated linoleic acid induced changes in the gut microbiota, increasing Bacteroides/Prevotella species and ghrelin levels in the gastric mucosa of mice [319]. Increased ghrelin was also observed in rats after treatment with a diet containing 10% cocoa or cocoa fiber, which was negatively associated with Bifidobacterium and Streptococcus [320], whereas infection with pathogenic Toxoplasma gondii lowered ghrelin serum levels in rats [321]. A negative correlation between Firmicutes and ghrelin levels was observed both in humans and rodents [322, 323]. Moreover, the SCFA butyrate, as well as physical exercise, reduce serum ghrelin levels through involvement of FFA3 receptor [105, 318]. Of note, physical exercise alters the composition of the microbiota in the cecum and increases the concentration of butyrate in the cecal content of rats [324].
In humans, the relationship between the gut microbiota and ghrelin levels seems to mirror that in rodents where the relative abundance of bacterial taxa such as Bacteroides was negatively correlated with plasma ghrelin, which, in obese patients, was increased [130, 197, 292]. Interestingly, plasma ghrelin did not return to levels found in normal weight individuals after obese patients underwent bariatric surgery, despite a postoperative increase of Lactobacillus and Streptococcus [292]. The use of dietary capsaicin was shown to increase the Firmicutes/Bacteroidetes ratio, which was associated with decreased plasma ghrelin levels and an increase in butyrate [325]. Although dietary capsaicin was given to healthy subjects, this study may warrant future investigations into how the manipulation of the enteric microbiome may achieve lowered ghrelin levels in people. Reduced ghrelin levels could have implications in daily stress, minimizing incidence of psychiatric disorders. For example, injection of ghrelin in healthy humans increased plasma glucocorticoids levels [326].
Ghrelin has recently attracted attention not only as a hunger hormone, but also because of its role in modulating aspects of stress, including anxiety-like behavior and fear. In concert with other gut peptides, ghrelin signaling through the gut–brain axis may tune communication between the periphery and brain during daily life stressors. Nevertheless, a direct link between the gut microbiota and the modulation of ghrelin remains absent.
CRF
CRF is a 41 amino-acid residue peptide that plays a key role in orchestrating the endocrine, behavioral, and GI responses to stress (for review, see [327,328,329]). CRF is abundantly expressed in neurons of the PVN of the hypothalamus where it is released through effector neurons, which, in turn, mediate neural control of adrenocorticotropic hormone (ACTH) release from pituitary corticotrophs. Then, ACTH passes into systemic circulation to cause the release of cortisol (in humans) or corticosterone (in rodents) from the adrenal glands [328, 330]. Apart from hypothalamic CRF, this peptide and its receptors are also expressed throughout several other limbic regions and in the gut [331, 332]. In the gut, CRF is mainly localized in enterochromaffin cells of the colon [333, 334], and in submucosal and lamina propria cells, but not in epithelial cells in the ileum [335]. CRF can bind two G protein-coupled receptor subtypes, namely CRF1 and CRF2 receptors, with different affinities. Apart from receptor binding, CRF also binds to CRF-binding protein with high affinity. CRF-binding protein is thought to play an inhibitory role in which it binds CRF to prevent the activation of CRF receptors [336]. CRF is centrally expressed on GABAergic, glutamatergic, and dopaminergic neurons [337], and can modulate the catecholaminergic response to stress by activating the noradrenergic neurons in the locus coeruleus or regulating the synthesis and release of adrenaline in the adrenals [329, 330]. Bethin et al. [338] found that immune activation induced by endotoxin produced a robust rise in plasma corticosterone in CRF or CRF1 receptor knockout mice, comparable with wild-type mice. Since the gut microbiota strongly affects the immune system, intestinal diseases where the gut microbiota is changed, such as IBS and IBD, may induce the expression of inflammatory cells, which, in turn, can increase expression of glucocorticoids but not CRF.
Gut Microbiota and CRF: Relevance to Anxiety and Depression
Functional CRF1 receptors are essential for the activation of the endocrine response to stress [169]. However, when acute stress reactions become more frequent, it can lead to the development of stress-related disorders, including anxiety and depression (for review, see [329, 339]). Whereas the absence of CRF1 receptors impairs basal and stress-induced HPA axis response and induces an anxiolytic- and antidepressant-like effect [329, 340, 341], CRF1 overexpression results in an anxiogenic- and depressive-like profile in rodents and humans [342,343,344,345,346]. Interestingly, antidepressant treatment normalizes expression of CRF and CRF1 receptors in association with remission of depression [329, 344, 346].
The CRF system also induces functional changes within the gut, including slowed gastric emptying, colonic motile stimulation, and impairment of the intestinal epithelial barrier, each an effect independent of a stressful situation [347, 348]. Recently, a study has found that a single administration of CRF modified the gut microbiota in rats, with specific reductions in Lactobacillus [349]. Although the inhibitory effect of central CRF on gut motility and permeability does not change with peripheral injection of CRF or the removal of different HPA axis components [108, 350,351,352], acute stress in mice induced the release of intestinal CRF independently of the HPA axis [131]. Sun et al. [131] also observed changes in gut microbiota composition, which was reverted with a probiotic supplement. The central administration of CRF in rodents induces depressive and anxiolytic-like behavior, which was followed by changes in the intestinal microbial community [138]. Furthermore, the intestinal microbiota can interfere in host CRF signaling. Germ-free rodents have increased hypothalamic CRF gene expression, as well as circulating levels of ACTH and corticosterone to acute restraint stress in adulthood [27, 216]. These effects in the HPA axis are normalized in recolonized germ-free mice [216]. Taken together, such data indicate that CRF-mediated activation of the HPA axis depends on the gut microbiota. Interestingly, the depletion of the gut microbiota via antibiotic treatment in mice from weaning onward does not alter hypothalamic mRNA CRF expression in adulthood, even when mice underwent acute restraint stress [21], suggesting gut microbiota-mediated CRF signaling may occur during early life. Prenatal stress induced depressive-like behavior in adulthood mice, which led to a long-lasting change of the enteric microbiota composition associated with an exaggerated HPA axis response to stress [353,354,355]. Early-life stress such as maternal separation results in changes in the gut microbiota followed by an increase in stress reactivity, including a decrease in Lactobacillus in adult mice [137]. Lactobacillus also suppressed plasma corticosterone and hypothalamic CRF-expressing cells in rats that underwent water-avoidance stress [356]. Using a mixture of 8 bacterial strains, including Lactobacillus and Bifidobacterium, Abildgaard et al. [357] observed lowered hippocampal transcript levels of CRF receptors followed by marked reduction in depressive-like behavior in rats. Furthermore, social stress in mice also induced changes in the microbiota, which were accompanied by changes in cytokine and chemokine levels [149]. Taken together, these data support a role for the gut microbiota as a potential treatment intervention to blunt stress reactivity.
Preclinical data from prenatal stress studies have been confirmed in clinical trials. Infants of mothers with high self-reported stress and high levels of salivary cortisol during pregnancy had a significantly higher relative abundance of Proteobacteria and lower relative abundances of lactic acid bacteria (i.e., Lactobacillus, Lactoccus, Aerococcus) and Bifidobacteria [358]. Nonetheless, those infants with altered microbiota composition exhibited a higher level of infant GI symptoms and allergic reactions, highlighting the functional consequences of aberrant colonization patterns in early life, which can contribute to psychiatric comorbidities. Increased CRF concentration in the cerebrospinal fluid was also reported in depressive patients and victims of suicide [359, 360]. Interestingly, administration of Clostridium butyricum from the phylum Firmicutes can block increased CRF levels in patients before surgical procedures [361]. These patients also presented a reduced degree of anxiety before surgery, suggesting an interesting effect of probiotics in mediating CRF expression to modulate stress reactivity. However, how gut microbes initiate such a response through CRF signaling is still unknown. A better understanding of CRF–microbiota interactions may identify potential gut microbiota-targeted interventions to enhance resilience in stress situations.
Oxytocin
Although oxytocin facilitates parturition and lactation in females, decreased oxytocin levels have been linked with depression (not only postpartum depression) and maternal neglect [362, 363]. Oxytocin is synthesized by the magnocellular neurons in the supraoptic and paraventricular nucleus of the hypothalamus and secreted to the circulation by the posterior pituitary or to nerve terminals located mainly in the anterior pituitary, amygdala, hippocampus, and the bed nucleus of the stria terminalis (for review, see [364]). Peripherally, oxytocin and OXT receptors are mainly expressed in the uterus, with low expression in many other tissues and organs, including kidney, pancreas, adrenal, and adipose tissue [365]. Interestingly, oxytocin can be found in enteric neurons; however, gut OXT receptors are not only found in enteric neurons, but they are also found in enterocytes [366]. There is little evidence that oxytocin is modulated by gut bacteria, although gut microbiota seems to influence oxytocin in the brain [21, 26]. Moreover, there is cross reactivity in binding of oxytocin and vasopressin with its respective receptor, where oxytocin binds its receptor with only 10 times greater affinity than vasopressin [367].
Gut Microbiota and Oxytocin: Relevance to Anxiety and Depression
Oxytocin-expressing neurons within the hypothalamus are stimulated by stress-mediated noradrenaline release, which elevates oxytocin levels in mammals to counteract the physiologic actions of stressors (for review, see [368,369,370,371]). Oxytocin secretion is also directly modulated by various classical immune cytokines, such as inteleukin-1β and interleukin-10 [372, 373], an effect that minimizes immunologic insults and exerts protective effects by restoring host homeostasis. Moreover, the administration of oxytocin exerts an antidepressant-like effect in rodents [374,375,376,377], whereas lowered oxytocin levels observed in nonweaned rats induces a depressive-like phenotype, which correlates positively with changes in the gut bacterial taxa [378]. Experimental stress in rodents is associated with the downregulation of OXT receptors in the amygdala [378]. Since stress can alter the gut microbiota composition, it is not surprising that there is interaction between oxytocin and the gut microbiome. Interestingly, gut microbiota depletion from early adolescence impacts oxytocin signaling, reducing both hypothalamic oxytocin and vasopressin levels in mice subjected to stress [21, 379]. Normalized oxytocin levels in stressed animals was followed by a reduction of anxiety-like behavior and correction in cognitive deficits [21]. However, mice fed with L. reuteri restored hypothalamic oxytocin neurons, as well as social behaviors, in a study investigating the effects of maternal high-fat diet on the offspring [26]. Interestingly, the effects observed above are dependent on vagal signaling, which blunted the effects of Lactobacillus on host plasma and hypothalamic oxytocin [132, 133]. Oleoylethanolamide, a product of fat absorption, which has antidepressant-like and neuroimmune properties [380, 381], excites vagal afferent neurons and stimulates both axonal and somatodendritic oxytocin secretion [382,383,384]. Oleoylethanolamide-inducing activation of the NTS in rodents precedes the activation of oxytocinergic neurons in the PVN, and the latter response appears to be mediated by noradrenergic projections from the NTS to the PVN [383], suggesting that gut-signaling oxytocin may be mediated by oleoylethanolamide. Nonetheless, plasticity in the oxytocin system is likely a protective coping mechanism in response to stress shaped by the gut microbiota during the early life stage, mainly through maternal exchange, which can contribute to depressive phenotypes when offspring become adults. However, additional studies are needed to demonstrate a functional role of the gut microbiota in the host oxytocin system.
Psychobiotics: Towards a New Class of Psychiatric Treatments
Psychobiotics are defined as an exogenous intervention to effect changes in mental health via modulation of the gut microbiome (for review, see [54, 56]). This may include not only prebiotics and probiotics, but also diet and exercise that affect enteric bacterial communities [54]. Few studies have shown results that may support a clinical role for psychobiotic intervention. For example, changes in diet even for a short duration (24 h) can drastically alter the composition of the gut microbiota [385]. Moreover, the quality of the diet (i.e., a typical Western diet rich in fat and protein) drastically reduces Bifidobacteria and butyrate-producing bacteria [386, 387], whereas a Mediterranean diet, which is thought of as a healthy diet, shows significant increases in SCFAs [43]. An overview of epidemiological studies showing the impact of diet on SCFA produced by the gut microbiota can be found in Ríos-Covián et al. [388].
Regarding direct administration of probiotic or other commercially available formulations to manipulate the gut microbiota, a study found that students who ingested Lactobacillus spp. had lower plasma cortisol, which can be interpreted as a measure indicative of reduced anxiety during a stressful situation [389]. In a separate investigation, participants consumed a fermented milk product containing Lactobacillus casei self-reported the same as the control subjects [49]. However, when only participants whose baseline mood scores were lower (indicative of an antidepressive state), probiotic supplementation resulted in significantly more of this group of participants self-scoring as happy rather than depressed compared with placebo [49]. In yet another study, healthy participants who ingested Lactobacillus helveticus and Bifidobacterium longum showed less self-reported negative mood and decreased urinary cortisol [50, 390]. A similar effect was also observed in healthy participants who consumed a mixture of Bifidobacterium bifidum and Bifidobacterium lactis, and Lactobacillus acidophilus, Brevibacillus brevis, Brevibacterium casei, Bifidobacterium salivarius, and Lactococcus lactis [51]. Allen et al. [52] found that healthy individuals fed Bifidobacterium longum had attenuated levels of cortisol and reduced subjective anxiety in response to the socially evaluated cold stress pressor test. A similar effect was observed following consumption of a commercially available prebiotic Bimuno® galacto-oligosaccharides, where individuals showed a reduction in waking-cortisol response and decreased attentional vigilance to negative versus positive information, suggesting a role in modulating depression [391]. Schmidt et al. [391] found that Bimuno galacto-oligosaccharides intake decreased attentional vigilance to negative versus positive information. However, the probiotic Lactobacillus rhamnosus failed to alter mood, anxiety, and stress-related measures when subjects faced a socially evaluated cold pressor test [392], an effect previously observed in rodents [20]. Taken together, the above findings highlight the challenges associated with translating promising preclinical studies to healthy human subjects. Studies in populations with mental disorders are therefore required to assess the potential benefit of the manipulation of the gut microbiota as a therapeutic strategy. In recent studies investigating patients with depression and anxiety, a mixture of the probiotics Lactobacillus acidophilus, Lactobacillus casei, and Bifidobacterium bifidum found a significant decrease in depression scores [393], whereas a separate investigation of the effects of Bifidobacterium longum on anxiety and depression in patients with IBS demonstrated probiotic treatment reduced depression but not anxiety scores and increased the quality of life in patients with IBS [167], indicating that this probiotic may reduce limbic reactivity.
Psychobiotics: Targeting Gut Peptides
There are major challenges associated with the development of peptide therapeutics, including limited peptide stability, a short duration of action, and the ability to cross the BBB [394, 395]. Such limitations may reduce the potential of exogenous-delivered peptides to reach specific target sites, which may fail to reflect the effect of the endogenously derived peptide [396]. Although several approaches have been designed to address the efficacy of the peptide delivery, the majority are concerned with targeted modification of peptide chemistry.
Targeting the release of the gut peptides may be an approach to be considered in conjunction with future breakthroughs in psychobiotics. Advances in the field of pharmacomicrobiomics, which focuses on the interpretation of intraindividual human microbiomes stratified according to different lifestyles, may highlight how an improved understanding of the human microbiome may lead to the development of psychobiotic therapeutic agents [397, 398].
Thus, psychobiotics could be designed to target, for example, specific EECs to elicit the release of controlled endogenous peptides without upstream consequences. Personalized medicine may consider questions of the individual’s own microbiome, thereby necessitating the personalization of psychobiotics. This could focus potential peptide-mediated immune responses, improve vagal signaling, or modulate neuropeptide expression in brain regions associated with mental health.
Another interesting approach is the development of targeted antimicrobials. Since the nature of bacteria that occupy overlapping niches is to compete for the same nutrients, antimicrobials such as bacteriocins may help gain terrain over neighbor competitors. Bacteriocins are ribosomally synthesized peptides that are produced by bacteria to eliminate neighboring cells through toxic and immune functions, while protecting itself and its progeny [399,400,401,402]. Moreover, a recent paper described that commensal bacteria are able to produce G-protein-coupled receptor agonists to regulate the GI tract physiology, including metabolic hormones and glucose homeostasis [4].
Therefore, the manipulation of gut peptides via psychobiotics may represent a promising opportunity to target mental disorders from the site of the GI tract. Identification of the mechanism(s) underlying the activity of gut peptides and how might the gut microbiota modulate the peptide physiology may facilitate greater understanding of gut to brain communication and its role in the neurobiology of the anxiety and depression.
Conclusion
We are at the very early stages of understanding the complexities of communication along the microbiota–gut–brain axis. However, there is already strong evidence to support the influence of the enteric microbiome on brain function in health and disease, suggesting the gut microbiome plays a crucial role in normal brain development, as well as modulation of host physiological systems important in stress-related disorders. Among the diverse pathways by which the gut can signal the brain, the endocrine system seems to play an important role, as it is capable of modulating not only other endocrine functions, but also the neural and immune systems. As suggested here, peptides released by specialized cells in the gut participate in gut-to-brain communication. Peptides may be envisaged to orchestrate the molecular, functional, behavioral, and autonomic reactions that take place in response to alterations of the gut microbial community. There is significant anatomical and functional overlap of peptides released in the gut and brain, suggesting that these peptides exert common downstream effects on neural systems involved in mental health. Nevertheless, the dynamic changes in peptides and respective receptor expression in the gut and brain of rodents and humans with altered microbiota attests to a profound role of the gut bacteria on peptides. Understanding how the gut microbes might influence peptide physiology should be a central objective of future research in this field. The generation of specialized psychobiotics to modify gut hormone secretion from EECs may represent feasible and therapeutic microbiota-based strategies for the treatment of depression and anxiety disorders. Interestingly, microbiota-derived metabolites might be able directly to modify gut-peptide receptors, displaying gut-peptide mimicry. Rising interest in this area of research will no doubt lead to greater insights into the mechanism(s) underlying microbiome–gut–brain communication, and provide novel understanding of the potential for microbial-based therapeutic strategies that may aid in the treatment of mood disorders.
References
Dockray GJ. Gastrointestinal hormones and the dialogue between gut and brain. J Physiol 2014;592:2927–2941.
Dockray GJ. Enteroendocrine cell signalling via the vagus nerve. Curr Opin Pharmacol 2013;13:954–958.
Cani PD, Everard A, Duparc T. Gut microbiota, enteroendocrine functions and metabolism. Curr Opin Pharmacol 2013;13:935–940.
Cohen LJ, Esterhazy D, Kim S-H, et al. Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature 2017;549:48–53.
Latorre R, Sternini C, De Giorgio R, Greenwood-Van Meerveld B. Enteroendocrine cells: a review of their role in brain-gut communication. Neurogastroenterol Motil 2016;28:620–630.
Psichas A, Reimann F, Gribble F. Gut chemosensing mechanisms. J Clin Invest 2015;125:908–917.
Gribble FM, Reimann F. Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu Rev Physiol 2016;78:277–299.
De Palma G, Collins SM, Bercik P. The microbiota-gut-brain axis in functional gastrointestinal disorders. Gut Microbes 2014;5:419–429.
Dinan TG, Cryan JF. Melancholic microbes: a link between gut microbiota and depression? Neurogastroenterol Motil 2013;25:713–719.
Burcelin R. Gut microbiota and immune crosstalk in metabolic disease. Mol Metab 2016;5:771–781.
Geuking MB, Köller Y, Rupp S, McCoy KD. The interplay between the gut microbiota and the immune system. Gut Microbes 2014;5:411–418.
Patterson E, Ryan PM, Cryan JF, et al. Gut microbiota, obesity and diabetes. Postgrad Med J 2016;92:286–300.
van de Wouw M, Schellekens H, Dinan TG, Cryan JF. Microbiota-gut-brain axis: modulator of host metabolism and appetite. J Nutr 2017;147:727–745.
Arentsen T, Raith H, Qian Y, Forssberg H, Heijtz RD. Host microbiota modulates development of social preference in mice. Microb Ecol Health Dis 2015;26:29719.
Burokas A, Arboleya S, Moloney RD, et al. Targeting the microbiota-gut-brain axis: prebiotics have anxiolytic and antidepressant-like effects and reverse the impact of chronic stress in mice. Biol Psychiatry 2017;39:763–781.
Clarke G, Grenham S, Scully P, et al. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry 2013;18:666–673.
Heijtz RD, Wang S, Anuar F, et al. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U S A 2011;108:3047–3052.
Neufeld K-AM, Kang N, Bienenstock J, Foster JA. Effects of intestinal microbiota on anxiety-like behavior. Commun Integr Biol 2011;4:492–494.
Savignac HM, Kiely B, Dinan TG, Cryan JF. Bifidobacteria exert strain-specific effects on stress-related behavior and physiology in BALB/c mice. Neurogastroenterol Motil 2014;26:1615–1627.
Bravo JA, Forsythe P, Chew MV, et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci U S A 2011;108:16050–16055.
Desbonnet L, Clarke G, Traplin A, et al. Gut microbiota depletion from early adolescence in mice: implications for brain and behaviour. Brain Behav Immun 2015;48:165–173.
Wong M-L, Inserra A, Lewis MD, et al. Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Mol Psychiatry 2016;21:797–805.
McGonigle P. Peptide therapeutics for CNS indications. Biochem Pharmacol 2012;83:559–566.
Holzer P, Reichmann F, Farzi A. Neuropeptide Y, peptide YY and pancreatic polypeptide in the gut-brain axis. Neuropeptides 2012;46:261–274.
Fröhlich EE, Farzi A, Mayerhofer R, et al. Cognitive impairment by antibiotic-induced gut dysbiosis: analysis of gut microbiota-brain communication. Brain Behav Immun 2016;56:140–155.
Buffington SA, Di Prisco GV, Auchtung TA, Ajami NJ, Petrosino JF, Costa-Mattioli M. Microbial reconstitution reverses maternal diet-induced social and synaptic deficits in offspring. Cell 2016;165:1762–1775.
Crumeyrolle-Arias M, Jaglin M, Bruneau A, et al. Absence of the gut microbiota enhances anxiety-like behavior and neuroendocrine response to acute stress in rats. Psychoneuroendocrinology 2014;42:207–217.
Cowley MA, Smith RG, Diano S, et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003;37:649–661.
Joly-Amado A, Cansell C, Denis RGP, et al. The hypothalamic arcuate nucleus and the control of peripheral substrates. Best Pract Res Clin Endocrinol Metab 2014;28:725–737.
Valassi E, Scacchi M, Cavagnini F. Neuroendocrine control of food intake. Nutr Metab Cardiovasc Dis 2008;18:158–168.
Stanley S, Wynne K, McGowan B, Bloom S. Hormonal regulation of food intake. Physiol. Rev 2005;85.
Cummings DE, Overduin J. Gastrointestinal regulation of food intake. J Clin Invest 2007;117:13–23.
Gariepy G, Wang J, Lesage A, Schmitz N. The interaction of obesity and psychological distress on disability. Soc Psychiatry Psychiatr Epidemiol 2010;45:531–540.
Goldbacher EM, Matthews KA. Are psychological characteristics related to risk of the metabolic syndrome? A review of the literature. Ann Behav Med 2007;34:240–252.
Kloiber S, Ising M, Reppermund S, et al. Overweight and obesity affect treatment response in major depression. Biol Psychiatry 2007;62:321–326.
Marijnissen RM, Bus BAA, Holewijn S, et al. Depressive symptom clusters are differentially associated with general and visceral obesity. J Am Geriatr Soc 2011;59:67–72.
McElroy SL, Kotwal R, Malhotra S, Nelson EB, Keck PE, Nemeroff CB. Are mood disorders and obesity related? A review for the mental health professional. J Clin Psychiatry 2004;65:634–651.
Lang UE, Beglinger C, Schweinfurth N, Walter M, Borgwardt S. Nutritional aspects of depression. Cell Physiol Biochem 2015;37:1029–1043.
Steele CC, Pirkle JRA, Kirkpatrick K. Diet-induced impulsivity: effects of a high-fat and a high-sugar diet on impulsive choice in rats. PLOS ONE 2017;12.
Beilharz JE, Kaakoush NO, Maniam J, Morris MJ. Cafeteria diet and probiotic therapy: cross talk among memory, neuroplasticity, serotonin receptors and gut microbiota in the rat. Mol Psychiatry. 2017.
Ochoa-Repáraz J, Kasper LH. The second brain: is the gut microbiota a link between obesity and central nervous system disorders? Curr Obes Rep 2016;5:51–64.
Veniaminova E, Cespuglio R, Cheung CW, et al. Autism-like behaviours and memory deficits result from a Western diet in mice. Neural Plast 2017;2017:1–14.
De Filippis F, Pellegrini N, Vannini L, et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016;65:1812–1821.
Pratt LA, Brody DJ, Gu Q. Antidepressant use in persons aged 12 and over: United States, 2005–2008. NCHS Data Brief 2011;127:1–8.
Pratt LA, Brody DJ. Depression in the U.S. household population, 2009–2012. NCHS Data Brief 2014;1–8.
Pozzi M, Radice S, Clementi E, Molteni M, Nobile M. Antidepressants and, suicide and self-injury: causal or casual association? Int J Psychiatry Clin Pract. 2016;20:47–51.
Gartlehner G, Gaynes BN, Amick HR, et al. Comparative benefits and harms of antidepressant, psychological, complementary, and exercise treatments for major depression: an evidence report for a clinical practice guideline from the American College of Physicians. Ann Intern Med 2016;164:331–341.
Coupland C, Hill T, Morriss R, Moore M, Arthur A, Hippisley-Cox J. Antidepressant use and risk of cardiovascular outcomes in people aged 20 to 64: cohort study using primary care database. BMJ 2016;352:i1350.
Benton D, Williams C, Brown A. Impact of consuming a milk drink containing a probiotic on mood and cognition. Eur J Clin Nutr 2007;61:355–361.
Messaoudi M, Violle N, Bisson J-F, Desor D, Javelot H, Rougeot C. Beneficial psychological effects of a probiotic formulation (Lactobacillus helveticus R0052 and Bifidobacterium longum R0175) in healthy human volunteers. Gut Microbes 2011;2:256–261.
Steenbergen L, Sellaro R, van Hemert S, Bosch JA, Colzato LS. A randomized controlled trial to test the effect of multispecies probiotics on cognitive reactivity to sad mood. Brain Behav Immun 2015;48:258–264.
Allen AP, Hutch W, Borre YE, et al. Bifidobacterium longum 1714 as a translational psychobiotic: modulation of stress, electrophysiology and neurocognition in healthy volunteers. Transl Psychiatry 2016;6:e939.
Labus JS, Hollister EB, Jacobs J, et al. Differences in gut microbial composition correlate with regional brain volumes in irritable bowel syndrome. Microbiome 2017;5:49.
Sarkar A, Lehto SM, Harty S, Dinan TG, Cryan JF, Burnet PWJ. Psychobiotics and the manipulation of bacteria–gut–brain signals. Trends Neurosci 2016;39:763–781.
Allen AP, Dinan TG, Clarke G, Cryan JF. A psychology of the human brain-gut-microbiome axis. Soc Personal Psychol Compass 2017;11:e12309.
Dinan TG, Stanton C, Cryan JF. Psychobiotics: a novel class of psychotropic. Biol Psychiatry 2013;74:720–726.
Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature 2012;486:222–227.
Dominguez-Bello MG, Costello EK, Contreras M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A 2010;107:11971–11975.
Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med 2014;6:237ra65.
Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, Aagaard KM. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat Med 2017;23:314–326.
Jiménez E, Fernández L, Marín ML, et al. Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr Microbiol 2005;51:270–274.
Markenson GR, Adams LA, Hoffman DE, Reece MT. Prevalence of Mycoplasma bacteria in amniotic fluid at the time of genetic amniocentesis using the polymerase chain reaction. J Reprod Med 2003;48:775–779.
Charbonneau MR, Blanton LV, DiGiulio DB, et al. A microbial perspective of human developmental biology. Nature 2016;535:48–55.
Nicholson JK, Holmes E, Kinross J, et al. Host-gut microbiota metabolic interactions. Science 2012;336:1262–1267.
Ziętak M, Kovatcheva-Datchary P, Markiewicz LH, Ståhlman M, Kozak LP, Bäckhed F. Altered microbiota contributes to reduced diet-induced obesity upon cold exposure. Cell Metab 2016;23:1216–1223.
De Vadder F, Kovatcheva-Datchary P, Zitoun C, Duchampt A, Bäckhed F, Mithieux G. Microbiota-produced succinate improves glucose homeostasis via intestinal gluconeogenesis. Cell Metab 2016;24:151–157.
Desbonnet L, Garrett L, Clarke G, Kiely B, Cryan JF, Dinan TG. Effects of the probiotic Bifidobacterium infantis in the maternal separation model of depression. Neuroscience 2010;170:1179–1188.
Welly RJ, Liu TW, Zidon TM, et al. Comparison of diet versus exercise on metabolic function and gut microbiota in obese rats. Med Sci Sports Exerc 2016;48:1688-1698.
Campbell SC, Wisniewski PJ, Noji M, et al. The effect of diet and exercise on intestinal integrity and microbial diversity in mice. PLOS ONE 2016;11:e0150502.
Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010;464:59–65.
Hidalgo-Cantabrana C, Delgado S, Ruiz L, Ruas-Madiedo P, Sánchez B, Margolles A. Bifidobacteria and their health-promoting effects. Microbiol Spectr 2017;5:1–19.
Sánchez B, Delgado S, Blanco-Míguez A, Lourenço A, Gueimonde M, Margolles A. Probiotics, gut microbiota, and their influence on host health and disease. Mol Nutr Food Res 2017;61:1600240.
Swann JR, Want EJ, Geier FM, et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci U S A 2011;108:4523–4530.
Golubeva AV, Joyce SA, Moloney G, et al. Microbiota-related changes in bile acid & tryptophan metabolism are associated with gastrointestinal dysfunction in a mouse model of autism. EBioMedicine 2017;24:166-178.
Ostaff MJ, Stange EF, Wehkamp J. Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol Med 2013;5:1465–1483.
Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature 2016;535:75–84.
O’Mahony SM, Clarke G, McKernan DP, Bravo JA, Dinan TG, Cryan JF. Differential visceral nociceptive, behavioural and neurochemical responses to an immune challenge in the stress-sensitive Wistar Kyoto rat strain. Behav Brain Res 2013;253:310–317.
Olszak T, An D, Zeissig S, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science 2012;336:489–493.
Xu P, Hong F, Wang J, et al. DBZ is a putative PPARγ agonist that prevents high fat diet-induced obesity, insulin resistance and gut dysbiosis. Biochim Biophys Acta 2017;1861:2690–2701.
Lippert K, Kedenko L, Antonielli L, et al. Gut microbiota dysbiosis associated with glucose metabolism disorders and the metabolic syndrome in older adults. Benef Microbes 2017;8:545–556.
Liu X, Cao S, Zhang X. Modulation of gut microbiota-brain axis by probiotics, prebiotics, and diet. J Agric Food Chem 2015;63:7885–7895.
Wu G, Feder A, Wegener G, et al. Central functions of neuropeptide Y in mood and anxiety disorders. Expert Opin Ther Targets 2011;15:1317–1331.
Tolhurst G, Heffron H, Lam YS, et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012;61:364–371.
Kaji I, Karaki S, Kuwahara A. Short-chain fatty acid receptor and its contribution to glucagon-like peptide-1 release. Digestion 2014;89:31–36.
Psichas A, Sleeth ML, Murphy KG, et al. The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int J Obes 2015;39:424–429.
Simpson HL, Campbell BJ. Review article: dietary fibre-microbiota interactions. Aliment Pharmacol Ther 2015;42:158–179.
Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013;341:569–573.
Bellono NW, Bayrer JR, Leitch DB, et al. Enterochromaffin cells are gut chemosensors that couple to sensory neural pathways. Cell 2017;170:185–198.e16.
Carabotti M, Scirocco A, Maselli MA, Severi C. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol 2015;28:203–209.
Kentish SJ, Page AJ. Plasticity of gastro-intestinal vagal afferent endings. Physiol Behav 2014;136:170–178.
Duerkop BA, Vaishnava S, Hooper LV. Immune responses to the microbiota at the intestinal mucosal surface. Immunity 2009;31:368–376.
Fricker LD. Neuropeptides and other bioactive peptides: from discovery to function. Colloq Ser Neuropeptides 2012;1:1–122.
Côté CD, Zadeh-Tahmasebi M, Rasmussen BA, Duca FA, Lam TKT. Hormonal signaling in the gut. J Biol Chem 2014;289:11642–11649.
Mulvihill EE, Drucker DJ. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr Rev 2014;35:992–1019.
Grider JR. Neurotransmitters mediating the intestinal peristaltic reflex in the mouse. J Pharmacol Exp Ther 2003;307:460–467.
Johnson PJ, Bornstein JC. Neurokinin-1 and -3 receptor blockade inhibits slow excitatory synaptic transmission in myenteric neurons and reveals slow inhibitory input. Neuroscience 2004;126:137–147.
Spindel ER, Chin WW, Price J, Rees LH, Besser GM, Habener JF. Cloning and characterization of cDNAs encoding human gastrin-releasing peptide. Proc Natl Acad Sci U S A 1984;81:5699–5703.
Ebeid AM, Escourrou J, Fischer JE. Vasoactive intestinal peptide inhibition of stimulated gastric secretion. I. Inhibition of meat-stimulated gastric secretion. Am J Surg 1980;139:817–823.
Vosko AM, Schroeder A, Loh DH, Colwell CS. Vasoactive intestinal peptide and the mammalian circadian system. Gen Comp Endocrinol 2007; 152:165–175.
Zeng N, Athmann C, Kang T, et al. PACAP type I receptor activation regulates ECL cells and gastric acid secretion. J Clin Invest 1999;104:1383–1391.
Furness JB. The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol 2012;9:286–294.
Berg CJ, Kaunitz JD. Gut chemosensing: implications for disease pathogenesis. F1000Research 2016;5:2424.
Yoo BB, Mazmanian SK. The enteric network: interactions between the immune and nervous systems of the gut. Immunity 2017;46:910–926.
Mansouri A, Langhans W. Enterocyte-afferent nerve interactions in dietary fat sensing. Diabetes Obes Metab 2014;16:61–67.
Lin HV, Frassetto A, Kowalik EJ, Jr, et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLOS ONE 2012;7:e35240.
Gao Z, Yin J, Zhang J, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009;58:1509–1517.
Drazen DL, Vahl TP, D’Alessio DA, Seeley RJ, Woods SC. Effects of a fixed meal pattern on ghrelin secretion: evidence for a learned response independent of nutrient status. Endocrinology 2006;147:23–30.
Larauche M, Gourcerol G, Wang L, et al. Cortagine, a CRF1 agonist, induces stresslike alterations of colonic function and visceral hypersensitivity in rodents primarily through peripheral pathways. AJP Gastrointest Liver Physiol 2009;297:G215–G227.
Cho HJ, Kosari S, Hunne B, et al. Differences in hormone localisation patterns of K and L type enteroendocrine cells in the mouse and pig small intestine and colon. Cell Tissue Res 2015;359:693–698.
Egerod KL, Engelstoft MS, Grunddal KV, et al. A major lineage of enteroendocrine cells coexpress CCK, secretin, GIP, GLP-1, PYY, and neurotensin but not somatostatin. Endocrinology 2012;153:5782–5795.
Okano-Matsumoto S, McRoberts JA, Taché Y, Adelson DW. Electrophysiological evidence for distinct vagal pathways mediating CCK-evoked motor effects in the proximal versus distal stomach. J Physiol 2011;589:371–393.
Banks WA, Tschop M, Robinson SM, Heiman ML. Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. J Pharmacol Exp Ther 2002;302:822–827.
Whitcomb DC, Taylor IL, Vigna SR. Characterization of saturable binding sites for circulating pancreatic polypeptide in rat brain. Am J Physiol 1990;259:G687-G691.
Kastin AJ, Akerstrom V, Pan W. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J Mol Neurosci 2002;18:7–14.
Nonaka N, Shioda S, Niehoff ML, Banks WA. Characterization of blood-brain barrier permeability to PYY3-36 in the mouse. J Pharmacol Exp Ther 2003;306:948–953.
Schéle E, Grahnemo L, Anesten F, Halleń A, Backhed, F, Jansson JO. The gut microbiota reduces leptin sensitivity and the expression of the obesity-suppressing neuropeptides proglucagon (Gcg) and brain-derived neurotrophic factor (Bdnf) in the central nervous system. Endocrinology 2013;154:3643–3651.
Parnell JA, Reimer RA. Prebiotic fibres dose-dependently increase satiety hormones and alter Bacteroidetes and Firmicutes in lean and obese JCR:LA-cp rats. Br J Nutr 2012;107:601–613.
Wichmann A, Allahyar A, Greiner TU, et al. Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe 2013;14:582–590.
Stenman LK, Waget A, Garret C, et al. Probiotic B420 and prebiotic polydextrose improve efficacy of antidiabetic drugs in mice. Diabetol Metab Syndr 2015;7:75.
Yadav H, Lee JH, Lloyd J, Walter P, Rane SG. Beneficial metabolic effects of a probiotic via butyrate-induced GLP-1 hormone secretion. J Biol Chem 2013;288:25088–25097.
Khosravi Y, Seow SW, Amoyo AA, et al. Helicobacter pylori infection can affect energy modulating hormones and body weight in germ free mice. Sci Rep 2015;5:8731.
Cani PD, Dewever C, Delzenne NM. Inulin-type fructans modulate gastrointestinal peptides involved in appetite regulation (glucagon-like peptide-1 and ghrelin) in rats. Br J Nutr 2004;92:521–526.
Yusta B, Baggio LL, Koehler J, et al. GLP-1R agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte GLP-1R. Diabetes 2015;64:2537–2549.
Simon M-C, Strassburger K, Nowotny B, et al. Intake of Lactobacillus reuteri improves incretin and insulin secretion in glucose tolerant humans: a proof of concept. Diabetes Care 2015;38:dc142690.
Woods SE, Leonard MR, Hayden JA, et al. Impaired cholecystokinin-induced gallbladder emptying incriminated in spontaneous “black” pigment gallstone formation in germfree Swiss Webster mice. Am J Physiol Gastrointest Liver Physiol 2015;308:G335-G349.
Pen J, Welling GW. Influence of the microbial flora on the amount of CCK8- and secretin21-27-like immunoreactivity in the intestinal tract of mice. Comp Biochem Physiol B 1983;76:585–589.
Cani PD, Neyrinck AM, Maton N, Delzenne NM. Oligofructose promotes satiety in rats fed a high-fat diet: involvement of glucagon-like peptide-1. Obes Res 2005;13:1000–1007.
Duca FA, Swartz TD, Sakar Y, Covasa M. Increased oral detection, but decreased intestinal signaling for fats in mice lacking gut microbiota. PLOS ONE 2012;7:e39748.
Perry RJ, Peng L, Barry NA, et al. Acetate mediates a microbiome–brain–β-cell axis to promote metabolic syndrome. Nature 2016;534:213–217.
Glintborg D, Andersen M, Hagen C, et al. Evaluation of metabolic risk markers in polycystic ovary syndrome (PCOS). Adiponectin, ghrelin, leptin and body composition in hirsute PCOS patients and controls. Eur J Endocrinol 2006;155:337–345.
Sun Y, Zhang M, Chen CC, et al. Stress-induced corticotropin-releasing hormone-mediated NLRP6 inflammasome inhibition and transmissible enteritis in mice. Gastroenterology 2013;144:1478–1487.e8.
Poutahidis T, Kearney SM, Levkovich T, et al. Microbial symbionts accelerate wound healing via the neuropeptide hormone oxytocin. PLOS ONE 2013;8:e78898.
Varian BJ, Poutahidis T, DiBenedictis BT, et al. Microbial lysate upregulates host oxytocin. Brain Behav Immun 2017;61:36–49.
Nardone G, Compare D. The psyche and gastric functions. Dig Dis 2014;32:206–212.
Lach G, Morais LH, Costa APR, Hoeller AA. Envolvimento da flora intestinal na modulação de doenças psiquiátricas. Vittalle - Rev. Ciên. Saúde 2017;29:64–82.
Vuong HE, Yano JM, Fung TC, Hsiao EY. The microbiome and host behavior. Annu Rev Neurosci 2017;40:21–49.
Bailey MT, Coe CL. Maternal separation disrupts the integrity of the intestinal microflora in infant rhesus monkeys. Dev Psychobiol 1999;35:146–155.
Park AJ, Collins J, Blennerhassett PA, et al. Altered colonic function and microbiota profile in a mouse model of chronic depression. Neurogastroenterol Motil 2013;25:733-e575.
Rodes L, Paul A, Coussa-Charley M, et al. Transit time affects the community stability of Lactobacillus and Bifidobacterium species in an in vitro model of human colonic microbiotia. Artif Cells Blood Substit Immobil Biotechnol 2011;39:351–356.
O’Malley D, Julio-Pieper M, Gibney SM, Dinan TG, Cryan JF. Distinct alterations in colonic morphology and physiology in two rat models of enhanced stress-induced anxiety and depression-like behaviour. Stress 2010;13:114–122.
Kelly JR, Kennedy PJ, Cryan JF, Dinan TG, Clarke G, Hyland NP. Breaking down the barriers: the gut microbiome, intestinal permeability and stress-related psychiatric disorders. Front Cell Neurosci 2015;9:392.
Maes M, Kubera M, Leunis J-C, Berk M. Increased IgA and IgM responses against gut commensals in chronic depression: Further evidence for increased bacterial translocation or leaky gut. J Affect Disord 2012;141:55–62.
Maes M, Kubera M, Leunis J-C. The gut-brain barrier in major depression: intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro Endocrinol Lett 2008;29:117–124.
O’Brien SM, Scully P, Scott L V, Dinan TG. Cytokine profiles in bipolar affective disorder: Focus on acutely ill patients. J Affect Disord 2006;90:263–267.
Jiang H, Ling Z, Zhang Y, et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun 2015;48:186–194.
Naseribafrouei A, Hestad K, Avershina E, et al. Correlation between the human fecal microbiota and depression. Neurogastroenterol Motil 2014;26:1155–1162.
Kelly JR, Borre Y, O’Brien C, et al. Transferring the blues: Depression-associated gut microbiota induces neurobehavioural changes in the rat. J Psychiatr Res 2016;82:109–118.
Yu M, Jia H, Zhou C, et al. Variations in gut microbiota and fecal metabolic phenotype associated with depression by 16S rRNA gene sequencing and LC/MS-based metabolomics. J. Pharm Biomed Anal 2017;138:231–239.
Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun 2011;25:397–407.
De Palma G, Blennerhassett P, Lu J, et al. Microbiota and host determinants of behavioural phenotype in maternally separated mice. Nat Commun 2015;6:7735.
O’Mahony CM, Clarke G, Gibney S, Dinan TG, Cryan JF. Strain differences in the neurochemical response to chronic restraint stress in the rat: relevance to depression. Pharmacol Biochem Behav 2011;97:690–699.
Duncan SH, Louis P, Thomson JM, Flint HJ. The role of pH in determining the species composition of the human colonic microbiota. Environ Microbiol 2009;11:2112–2122.
El-Zaatari M, Chang Y-M, Zhang M, et al. Tryptophan catabolism restricts IFN-γ–expressing neutrophils and Clostridium difficile immunopathology. J Immunol 2014;193:807–816.
El Aidy S, Ramsteijn AS, Dini-Andreote F, et al. Serotonin transporter genotype modulates the gut microbiota composition in young rats, an effect augmented by early life stress. Front Cell Neurosci 2017;11:222.
Yang M, Fukui H, Eda H, et al. Involvement of gut microbiota in the association between gastrointestinal motility and 5-HT expression/M2 macrophage abundance in the gastrointestinal tract. Mol Med Rep 2017;16:3482–3488.
Saraf MK, Piccolo BD, Bowlin AK, et al. Formula diet driven microbiota shifts tryptophan metabolism from serotonin to tryptamine in neonatal porcine colon. Microbiome 2017;5:77.
Hata T, Asano Y, Yoshihara K, et al. Regulation of gut luminal serotonin by commensal microbiota in mice. PLOS ONE 2017;12:e0180745.
Ge X, Ding C, Zhao W, et al. Antibiotics-induced depletion of mice microbiota induces changes in host serotonin biosynthesis and intestinal motility. J Transl Med 2017;15:13.
Lieb J. The immunostimulating and antimicrobial properties of lithium and antidepressants. J Infect 2004;49:88–93.
Munoz-Bellido JL, Munoz-Criado S, Garcìa-Rodrìguez JA. Antimicrobial activity of psychotropic drugs. Selective serotonin reuptake inhibitors. Int J Antimicrob Agents 2000;14:177–180.
Ferreira Mello BS, Monte AS, McIntyre RS, et al. Effects of doxycycline on depressive-like behavior in mice after lipopolysaccharide (LPS) administration. J Psychiatr Res 2013;47:1521–1529.
Miyaoka T, Wake R, Furuya M, et al. Minocycline as adjunctive therapy for patients with unipolar psychotic depression: an open-label study. Prog Neuropsychopharmacol Biol Psychiatry 2012;37:222–226.
Ahmed AIA, van der Heijden FMMA, van den Berkmortel H, Kramers K. A man who wanted to commit suicide by hanging himself: an adverse effect of ciprofloxacin. Gen Hosp Psychiatry 2011;33:82.e5-e7.
Grassi L, Biancosino B, Pavanati M, Agostini M, Manfredini R. Depression or hypoactive delirium? A report of ciprofloxacin-induced mental disorder in a patient with chronic obstructive pulmonary disease. Psychother Psychosom 2001;70:58–59.
Kaur K, Fayad R, Saxena A, et al. Fluoroquinolone-related neuropsychiatric and mitochondrial toxicity: a collaborative investigation by scientists and members of a social network. J Community Support Oncol 2016;14:54–65.
Rollof J, Vinge E. Neurologic adverse effects during concomitant treatment with ciprofloxacin, NSAIDS, and chloroquine: possible drug interaction. Ann Pharmacother 1993;27:1058–1059.
Pinto-Sanchez MI, Hall GB, Ghajar K, et al. Probiotic Bifidobacterium longum NCC3001 reduces depression scores and alters brain activity: a pilot study in patients with irritable bowel syndrome. Gastroenterology 2017;153:448–459.e8.
Lindner D, Stichel J, Beck-Sickinger AG. Molecular recognition of the NPY hormone family by their receptors. Nutrition 2008;24:907–917.
Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol 2011;164:S1-S324.
Chen X, DiMaggio DA, Han SP, Westfall TC. Autoreceptor-induced inhibition of neuropeptide Y release from PC-12 cells is mediated by Y2 receptors. Am J Physiol 1997;273:H1737-H1744.
Greber S, Schwarzer C, Sperk G. Neuropeptide Y inhibits potassium-stimulated glutamate release through Y2 receptors in rat hippocampal slices in vitro. Br J Pharmacol 1994;113:737–740.
Wood J, Verma D, Lach G, et al. Structure and function of the amygdaloid NPY system: NPY Y2 receptors regulate excitatory and inhibitory synaptic transmission in the centromedial amygdala. Brain Struct Funct 2016;221:3373–3391.
El-Salhy M, Hausken T. The role of the neuropeptide Y (NPY) family in the pathophysiology of inflammatory bowel disease (IBD). Neuropeptides 2016;55:137–144.
Tasan RO, Lin S, Hetzenauer A, Singewald N, Herzog H, Sperk G. Increased novelty-induced motor activity and reduced depression-like behavior in neuropeptide Y (NPY)-Y4 receptor knockout mice. Neuroscience 2009;158:1717–1730.
Kask A, Harro J, von Hörsten S, Redrobe JP, Dumont Y, Quirion R. The neurocircuitry and receptor subtypes mediating anxiolytic-like effects of neuropeptide Y. Neurosci Biobehav Rev 2002;26:259–283.
Cox HM. Neuropeptide Y receptors; antisecretory control of intestinal epithelial function. Auton Neurosci 2007;133:76–85.
Ekblad E, Sundler F. Distribution of pancreatic polypeptide and peptide YY. Peptides 2002;23:251–261.
Dumont Y, Moyse E, Fournier A, Quirion R. Distribution of peripherally injected peptide YY ([125I] PYY (3-36)) and pancreatic polypeptide ([125I] hPP) in the CNS: enrichment in the area postrema. J Mol Neurosci 2007;33:294–304.
Koda S, Date Y, Murakami N, et al. The role of the vagal nerve in peripheral PYY3-36-induced feeding reduction in rats. Endocrinology 2005;146:2369–2375.
Ueno H, Yamaguchi H, Mizuta M, Nakazato M. The role of PYY in feeding regulation. Regul Pept 2008;145:12–16.
Farzi A, Reichmann F, Holzer P. The homeostatic role of neuropeptide Y in immune function and its impact on mood and behaviour. Acta Physiol 2015;213:603–627.
Reichmann F, Holzer P. Neuropeptide Y: a stressful review. Neuropeptides 2016;55:99–109.
Malva JO, Xapelli S, Baptista S, et al. Multifaces of neuropeptide Y in the brain – Neuroprotection, neurogenesis and neuroinflammation. Neuropeptides 2012;46:299–308.
dos Santos VV, Santos DB, Lach G, et al. Neuropeptide Y (NPY) prevents depressive-like behavior, spatial memory deficits and oxidative stress following amyloid-β (Aβ1–40) administration in mice. Behav Brain Res 2013;244:107–115.
Verma D, Wood J, Lach G, Herzog H, Sperk G, Tasan R. Hunger promotes fear extinction by activation of an amygdala microcircuit. Neuropsychopharmacology 2016;41:431–439.
Painsipp E, Wultsch T, Edelsbrunner ME, et al. Reduced anxiety-like and depression-related behavior in neuropeptide Y Y4 receptor knockout mice. Genes Brain Behav 2008;7:532–542.
Field BCT, Chaudhri OB, Bloom SR. Bowels control brain: gut hormones and obesity. Nat Rev Endocrinol 2010;6:444–453.
Fujimiya M, Inui A. Peptidergic regulation of gastrointestinal motility in rodents. Peptides 2000;21:1565–1582.
Yang H, Li WP, Reeve JR, Rivier J, Taché Y. PYY-preferring receptor in the dorsal vagal complex and its involvement in PYY stimulation of gastric acid secretion in rats. Br J Pharmacol 1998;123:1549–1554.
Tough IR, Holliday ND, Cox HM. Y(4) receptors mediate the inhibitory responses of pancreatic polypeptide in human and mouse colon mucosa. J Pharmacol Exp Ther 2006;319:20–30.
Verma D, Hörmer B, Bellmann-Sickert K, et al. Pancreatic polypeptide and its central Y 4 receptors are essential for cued fear extinction and permanent suppression of fear. Br J Pharmacol 2016;173:1925–1938.
De Lartigue G, Lur G, Dimaline R, Varro A, Raybould H, Dockray GJ. EGR1 is a target for cooperative interactions between cholecystokinin and leptin, and inhibition by ghrelin, in vagal afferent neurons. Endocrinology 2010;151:3589–3599.
Charlot K, Faure C, Antoine-Jonville S. Influence of hot and cold environments on the regulation of energy balance following a single exercise session: a mini-review. Nutrients 2017;9:592.
Schubert MM, Sabapathy S, Leveritt M, Desbrow B. Acute exercise and hormones related to appetite regulation: a meta-analysis. Sport Med 2014;44:387–403.
Moloney RD, Desbonnet L, Clarke G, Dinan TG, Cryan JF. The microbiome: stress, health and disease. Mamm Genome 2014;25:49–74.
Clark A, Mach N. Exercise-induced stress behavior, gut-microbiota-brain axis and diet: a systematic review for athletes. J Int Soc Sports Nutr 2016;13:43.
Liu R, Zhang C, Shi Y, et al. Dysbiosis of gut microbiota associated with clinical parameters in polycystic ovary syndrome. Front Microbiol 2017;8:324.
Cox HM. Peptide YY: a neuroendocrine neighbor of note. Peptides 2007;28:345–351.
Fu-Cheng X, Anini Y, Chariot J, Voisin T, Galmiche JP, Rozé C. Peptide YY release after intraduodenal, intraileal, and intracolonic administration of nutrients in rats. Pflugers Arch 1995;431:66–75.
Brooks L, Viardot A, Tsakmaki A, et al. Fermentable carbohydrate stimulates FFAR2-dependent colonic PYY cell expansion to increase satiety. Mol Metab 2017;6:48–60.
Larraufie P, Doré J, Lapaque N, Blottière HM. TLR ligands and butyrate increase Pyy expression through two distinct but inter-regulated pathways. Cell Microbiol 2017;19:e12648.
Hong KB, Kim JH, Kwon HK, Han SH, Park Y, Suh HJ. Evaluation of prebiotic effects of high-purity galactooligosaccharides in vitro and in vivo. Food Technol Biotechnol 2016;54:156–163.
Cluny NL, Eller LK, Keenan CM, Reimer RA, Sharkey KA. Interactive effects of oligofructose and obesity predisposition on gut hormones and microbiota in diet-induced obese rats. Obesity 2015;23:769–778.
Steensels S, Cools L, Avau B, et al. Supplementation of oligofructose, but not sucralose, decreases high-fat diet induced body weight gain in mice independent of gustducin-mediated gut hormone release. Mol Nutr Food Res 2017;61:1600716.
Breton J, Tennoune N, Lucas N, et al. Gut commensal E. coli proteins activate host satiety pathways following nutrient-induced bacterial growth. Cell Metab 2016;23:324–334.
Nilsson A, Johansson-Boll E, Sandberg J, Björck I. Gut microbiota mediated benefits of barley kernel products on metabolism, gut hormones, and inflammatory markers as affected by co-ingestion of commercially available probiotics: a randomized controlled study in healthy subjects. Clin Nutr ESPEN 2016;15:49–56.
Rajpal DK, Klein JL, Mayhew D, et al. Selective spectrum antibiotic modulation of the gut microbiome in obesity and diabetes rodent models. PLOS ONE 2015;10:e0145499.
Chandrasekharan B, Bala V, Kolachala VL, et al. Targeted deletion of neuropeptide Y (NPY) modulates experimental colitis. PLOS ONE 2008;3:e3304.
Pang XH, Li TK, Xie Q, et al. Amelioration of dextran sulfate sodium-induced colitis by neuropeptide Y antisense oligodeoxynucleotide. Int J Colorectal Dis 2010;25:1047–1053.
Painsipp E, Herzog H, Sperk G, Holzer P. Sex-dependent control of murine emotional-affective behaviour in health and colitis by peptide YY and neuropeptide Y. Br J Pharmacol 2011;163:1302–1314.
Dimitrijević M, Stanojević S. The intriguing mission of neuropeptide Y in the immune system. Amino Acids 2013;45:41–53.
Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci 2017;20:136–144.
Lach G, Bicca MA, Hoeller AA, da Silva Santos EC, Costa APR, de Lima TCM. Short-term enriched environment exposure facilitates fear extinction in adult rats: the NPY-Y1 receptor modulation. Neuropeptides 2016;55:73–78.
Luczynski P, McVey Neufeld K-A, Oriach CS, Clarke G, Dinan TG, Cryan JF. Growing up in a bubble: using germ-free animals to assess the influence of the gut microbiota on brain and behavior. Int J Neuropsychopharmacol 2016;19:1–7.
Lach G, de Lima TCM. Role of NPY Y1 receptor on acquisition, consolidation and extinction on contextual fear conditioning: dissociation between anxiety, locomotion and non-emotional memory behavior. Neurobiol Learn Mem 2013;103:26–33.
Sudo N, Chida Y, Aiba Y, et al. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol 2004;558:263–275.
Husebye E, Hellström PM, Sundler F, Chen J, Midtvedt T. Influence of microbial species on small intestinal myoelectric activity and transit in germ-free rats. Am J Physiol Gastrointest Liver Physiol 2001;280:G368-G380.
Goodlad RA, Ratcliffe B, Fordham JP, et al. Plasma enteroglucagon, gastrin and peptide YY in conventional and germ-free rats refed with a fibre-free or fibre-supplemented diet. Q J Exp Physiol 1989;74:437–442.
El Karim IA, Linden GJ, Orr DF, Lundy FT. Antimicrobial activity of neuropeptides against a range of micro-organisms from skin, oral, respiratory and gastrointestinal tract sites. J Neuroimmunol 2008;200:11–16.
Ghosal S, Myers B, Herman JP. Role of central glucagon-like peptide-1 in stress regulation. Physiol Behav 2013;122:201–207.
Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev 2007;87:1409–1439.
Marathe CS, Rayner CK, Jones KL, Horowitz M. Glucagon-like peptides 1 and 2 in health and disease: a review. Peptides 2013;44:75–86.
Willard FS, Sloop KW. Physiology and emerging biochemistry of the glucagon-like peptide-1 receptor. Exp Diabetes Res 2012;2012:470851.
Vrang N, Larsen PJ. Preproglucagon derived peptides GLP-1, GLP-2 and oxyntomodulin in the CNS: role of peripherally secreted and centrally produced peptides. Prog Neurobiol 2010;92:442–462.
Rinaman L. Interoceptive stress activates glucagon-like peptide-1 neurons that project to the hypothalamus. Am J Physiol 1999;277:R582-R590.
Dickson SL, Shirazi RH, Hansson C, Bergquist F, Nissbrandt H, Skibicka KP. The glucagon-like peptide 1 (GLP-1) analogue, exendin-4, decreases the rewarding value of food: a new role for mesolimbic GLP-1 receptors. J Neurosci 2012;32:4812–4820.
Nakagawa A, Satake H, Nakabayashi H, et al. Receptor gene expression of glucagon-like peptide-1, but not glucose-dependent insulinotropic polypeptide, in rat nodose ganglion cells. Auton Neurosci 2004;110:36–43.
Kakei M, Yada T, Nakagawa A, Nakabayashi H. Glucagon-like peptide-1 evokes action potentials and increases cytosolic Ca2+ in rat nodose ganglion neurons. Auton Neurosci 2002;102:39–44.
Katsurada K, Maejima Y, Nakata M, et al. Endogenous GLP-1 acts on paraventricular nucleus to suppress feeding: projection from nucleus tractus solitarius and activation of corticotropin-releasing hormone, nesfatin-1 and oxytocin neurons. Biochem Biophys Res Commun 2014;451:276–281.
Abbott CR, Monteiro M, Small CJ, et al. The inhibitory effects of peripheral administration of peptide YY(3-36) and glucagon-like peptide-1 on food intake are attenuated by ablation of the vagal-brainstem-hypothalamic pathway. Brain Res 2005;1044:127–131.
Secher A, Jelsing J, Baquero AF, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest 2014;124:4473–4488.
Krieger JP, Arnold M, Pettersen KG, Lossel P, Langhans W, Lee SJ. Knockdown of GLP-1 receptors in vagal afferents affects normal food intake and glycemia. Diabetes 2016;65:34–43.
D’Alessio D, Lu W, Sun W, et al. Fasting and postprandial concentrations of GLP-1 in intestinal lymph and portal plasma: evidence for selective release of GLP-1 in the lymph system. Am J Physiol Regul Integr Comp Physiol 2007;293:R2163-R2169.
Kohan A, Yoder S, Tso P. Lymphatics in intestinal transport of nutrients and gastrointestinal hormones. Ann N Y Acad Sci 2010;1207:E44-E51.
Ohlsson L, Kohan AB, Tso P, Ahrén B. GLP-1 released to the mesenteric lymph duct in mice: effects of glucose and fat. Regul Pept 2014;189:40–45.
Hogan AE, Tobin AM, Ahern T, et al. Glucagon-like peptide-1 (GLP-1) and the regulation of human invariant natural killer T cells: lessons from obesity, diabetes and psoriasis. Diabetologia 2011;54:2745–2754.
Zietek T, Rath E. Inflammation meets metabolic disease: gut feeling mediated by GLP-1. Front Immunol 2016;7:154.
Bornstein NM, Brainin M, Guekht A, Skoog I, Korczyn AD. Diabetes and the brain: issues and unmet needs. Neurol Sci 2014;35:995–1001.
Seto SW, Yang GY, Kiat H, Bensoussan A, Kwan YW, Chang D. Diabetes mellitus, cognitive impairment, and traditional Chinese medicine. Int J Endocrinol 2015;2015:1–14.
Rivera HM, Christiansen KJ, Sullivan EL. The role of maternal obesity in the risk of neuropsychiatric disorders. Front Neurosci 2015;9:194.
den Heijer T, Vermeer SE, van Dijk EJ, et al. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia 2003;46:1604–1610.
Anderson RJ, Freedland KE, Clouse RE, Lustman PJ. The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care 2001;24:1069–1078.
Collins MM, Corcoran P, Perry IJ. Anxiety and depression symptoms in patients with diabetes. Diabet Med 2009;26:153–161.
Lee C-H, Jeon SJ, Cho KS, et al. Activation of glucagon-like peptide-1 receptor promotes neuroprotection in experimental autoimmune encephalomyelitis by reducing neuroinflammatory responses. Mol Neurobiol 2017 29.
Harkavyi A, Abuirmeileh A, Lever R, Kingsbury AE, Biggs CS, Whitton PS. Glucagon-like peptide 1 receptor stimulation by exendin-4 reverses key deficits in distinct rodent models of Parkinson’s disease. J Neuroinflammation 2008;5:19.
Hwang I, Park YJ, Kim Y-R, et al. Alteration of gut microbiota by vancomycin and bacitracin improves insulin resistance via glucagon-like peptide 1 in diet-induced obesity. FASEB J 2015;29:2397–2411.
Komsuoglu Celikyurt I, Mutlu O, Ulak G, et al. Exenatide treatment exerts anxiolytic- and antidepressant-like effects and reverses neuropathy in a mouse model of type-2 diabetes. Med Sci Monit Basic Res 2014;20:112–117.
Sharma AN, Pise A, Sharma JN, Shukla P. Glucagon-like peptide-1 (GLP-1) receptor agonist prevents development of tolerance to anti-anxiety effect of ethanol and withdrawal-induced anxiety in rats. Metab Brain Dis 2015;30:719–730.
Kinzig KP, D’Alessio DA, Herman JP, et al. CNS glucagon-like peptide-1 receptors mediate endocrine and anxiety responses to interoceptive and psychogenic stressors. J Neurosci 2003;23:6163–6170.
Krass M, Volke A, Rünkorg K, et al. GLP-1 receptor agonists have a sustained stimulatory effect on corticosterone release after chronic treatment. Acta Neuropsychiatr 2015;27:25–32.
Möller C, Sommer W, Thorsell A, Rimondini R, Heilig M. Anxiogenic-like action of centrally administered glucagon-like peptide-1 in a punished drinking test. Prog Neuropsychopharmacol Biol Psychiatry 2002;26:119–122.
Isacson R, Nielsen E, Dannaeus K, et al. The glucagon-like peptide 1 receptor agonist exendin-4 improves reference memory performance and decreases immobility in the forced swim test. Eur J Pharmacol 2011;650:249–255.
Anderberg RH, Richard JE, Hansson C, Nissbrandt H, Bergquist F, Skibicka KP. GLP-1 is both anxiogenic and antidepressant; divergent effects of acute and chronic GLP-1 on emotionality. Psychoneuroendocrinology 2016;65:54–66.
DellaValle B, Brix GS, Brock B, et al. Glucagon-like peptide-1 analog, liraglutide, delays onset of experimental autoimmune encephalitis in Lewis rats. Front Pharmacol 2016;7:433.
Gejl M, Rungby J, Brock B, Gjedde A. At the centennial of Michaelis and Menten, competing Michaelis-Menten steps explain effect of GLP-1 on blood-brain transfer and metabolism of glucose. Basic Clin Pharmacol Toxicol 2014;115:162–171.
Ventorp F, Bay-Richter C, Nagendra AS, et al. Exendin-4 treatment improves LPS-induced depressive-like behavior without affecting pro-inflammatory cytokines. J Parkinsons Dis 2017;7:263–273.
Cherbut C, Ferrier L, Rozé C, et al. Short-chain fatty acids modify colonic motility through nerves and polypeptide YY release in the rat. Am J Physiol 1998;275:G1415-G1422.
Margolskee RF, Dyer J, Kokrashvili Z, et al. T1R3 and gustducin in gut sense sugars to regulate expression of Na+-glucose cotransporter 1. Proc Natl Acad Sci U S A 2007;104:15075–15080.
Aoki R, Kamikado K, Suda W, et al. A proliferative probiotic Bifidobacterium strain in the gut ameliorates progression of metabolic disorders via microbiota modulation and acetate elevation. Sci Rep 2017;7:43522.
Everard A, Cani PD. Gut microbiota and GLP-1. Rev Endocr Metab Disord 2014;15:189–196.
Selwyn FP, Csanaky IL, Zhang Y, Klaassen CD. Importance of large intestine in regulating bile acids and GLP-1 in germ-free mice. Drug Metab Dispos 2015;5:1544–1556.
Yu Y, Wang X, Liu C, et al. Combined contributions of over-secreted glucagon-like peptide 1 and suppressed insulin secretion to hyperglycemia induced by gatifloxacin in rats. Toxicol Appl Pharmacol 2013;266:375–384.
Grasset E, Puel A, Charpentier J, et al. A specific gut microbiota dysbiosis of type 2 diabetic mice induces GLP-1 resistance through an enteric NO-dependent and gut-brain axis mechanism. Cell Metab 2017;25:1075–1090.
Bomhof MR, Saha DC, Reid DT, Paul HA, Reimer RA. Combined effects of oligofructose and Bifidobacterium animalis on gut microbiota and glycemia in obese rats. Obesity 2014;22:763–771.
Cani PD, Lecourt E, Dewulf EM, et al. Gut microbiota fermentation of prebiotics increases satietogenic and incretin gut peptide production with consequences for appetite sensation and glucose response after a meal. Am J Clin Nutr 2009;90:1236–1243.
Delzenne NM, Cani PD, Neyrinck AM. Modulation of glucagon-like peptide 1 and energy metabolism by inulin and oligofructose: experimental data. J Nutr 2007;137:2547S–2551S.
Mansur RB, Ahmed J, Cha DS, et al. Liraglutide promotes improvements in objective measures of cognitive dysfunction in individuals with mood disorders: a pilot, open-label study. J Affect Disord 2017;207:114–120.
Wang H, Wong PT-H, Spiess J, Zhu YZ. Cholecystokinin-2 (CCK2) receptor-mediated anxiety-like behaviors in rats. Neurosci Biobehav Rev 2005;29:1361–1373.
Freund TF. Interneuron diversity series: rhythm and mood in perisomatic inhibition. Trends Neurosci 2003;26:489–495.
McLaughlin RJ, Hill MN, Gorzalka BB. A critical role for prefrontocortical endocannabinoid signaling in the regulation of stress and emotional behavior. Neurosci Biobehav Rev 2014;42:116–131.
Park SM, Williams CL. Contribution of serotonin type 3 receptors in the successful extinction of cued or contextual fear conditioned responses: interactions with GABAergic signaling. Rev Neurosci 2012;23:555–569.
Schäfer U, Harhammer R, Boomgaarden M, et al. Binding of cholecystokinin-8 (CCK-8) peptide derivatives to CCKA and CCKB receptors. J Neurochem 1994;62:1426–1431.
Del Boca C, Lutz PE, Le Merrer J, Koebel P, Kieffer BL. Cholecystokinin knock-down in the basolateral amygdala has anxiolytic and antidepressant-like effects in mice. Neuroscience 2012;218:185–195.
Rezayat M, Roohbakhsh A, Zarrindast M-R, Massoudi R, Djahanguiri B. Cholecystokinin and GABA interaction in the dorsal hippocampus of rats in the elevated plus-maze test of anxiety. Physiol Behav 2005;84:775–782.
Desai SJ, Borkar CD, Nakhate KT, Subhedar NK, Kokare DM. Neuropeptide Y attenuates anxiety- and depression-like effects of cholecystokinin-4 in mice. Neuroscience 2014;277:818–830.
Tirassa P, Costa N. CCK-8 induces NGF and BDNF synthesis and modulates TrkA and TrkB expression in the rat hippocampus and septum: effects on kindling development. Neurochem Int 2007;50:130–138.
Mishra AK, Dubey V, Ghosh AR. Obesity: an overview of possible role(s) of gut hormones, lipid sensing and gut microbiota. Metabolism 2016;65:48–65.
Rehfeld JF. The predominant cholecystokinin in human plasma and intestine is cholecystokinin-33. J Clin Endocrinol Metab 2001;86:251–258.
Baptista V, Zheng ZL, Coleman FH, Rogers RC, Travagli RA. Cholecystokinin octapeptide increases spontaneous glutamatergic synaptic transmission to neurons of the nucleus tractus solitarius centralis. J Neurophysiol 2005;94:2763–2771.
Burdyga G, de Lartigue G, Raybould HE, et al. Cholecystokinin regulates expression of Y2 receptors in vagal afferent neurons serving the stomach. J Neurosci 2008;28:11583–11592.
de La Serre CB, de Lartigue G, Raybould HE. Chronic exposure to low dose bacterial lipopolysaccharide inhibits leptin signaling in vagal afferent neurons. Physiol Behav 2015;139:188–194.
Bauer P V., Hamr SC, Duca FA. Regulation of energy balance by a gut–brain axis and involvement of the gut microbiota. Cell Mol Life Sci 2016;73:737–755.
Hsu L-T, Hung K-Y, Wu H-W, et al. Gut-derived cholecystokinin contributes to visceral hypersensitivity via nerve growth factor-dependent neurite outgrowth. J Gastroenterol Hepatol 2016;31:1594–1603.
El-Salhy M, Hatlebakk JG, Hausken T. Reduction in duodenal endocrine cells in irritable bowel syndrome is associated with stem cell abnormalities. World J Gastroenterol 2015;21:9577–9587.
Rubin DC, Zhang H, Qian P, Lorenz RG, Hutton K, Peters MG. Altered enteroendocrine cell expression in T cell receptor alpha chain knock-out mice. Microsc Res Tech 2000;51:112–120.
Daly DM, Park SJ, Valinsky WC, Beyak MJ. Impaired intestinal afferent nerve satiety signalling and vagal afferent excitability in diet induced obesity in the mouse. J Physiol 2011;589:2857–2870.
Anini Y, Brubaker PL. Role of leptin in the regulation of glucagon-like peptide-1 secretion. Diabetes 2003;52:252–259.
Covasa M, Grahn J, Ritter RC. High fat maintenance diet attenuates hindbrain neuronal response to CCK. Regul Pept 2000;86:83–88.
Whited KL, Thao D, Lloyd KCK, Kopin AS, Raybould HE. Targeted disruption of the murine CCK1 receptor gene reduces intestinal lipid-induced feedback inhibition of gastric function. Am J Physiol Gastrointest Liver Physiol 2006;291:G156-G162.
Leslie FC, Thompson DG, McLaughlin JT, Varro A, Dockray GJ, Mandal BK. Plasma cholecystokinin concentrations are elevated in acute upper gastrointestinal infections. QJM An Int J Med 2003;96:870–871.
McDermott JR, Leslie FC, D’Amato M, Thompson DG, Grencis RK, McLaughlin JT. Immune control of food intake: enteroendocrine cells are regulated by CD4+ T lymphocytes during small intestinal inflammation. Gut 2006;55:492–497.
Federico A, Dallio M, Tolone S, et al. Gastrointestinal hormones, intestinal microbiota and metabolic homeostasis in obese patients: effect of bariatric surgery. In Vivo 2016;30:321–330.
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999;402:656–660.
Nakazato M, Murakami N, Date Y, et al. A role for ghrelin in the central regulation of feeding. Nature 2001;409:194–198.
Spencer SJ, Xu L, Clarke MA, et al. Ghrelin regulates the hypothalamic-pituitary-adrenal axis and restricts anxiety after acute stress. Biol Psychiatry 2012;72:457–465.
Huang H-JJ, Zhu X-CC, Han Q-QQ, et al. Ghrelin alleviates anxiety- and depression-like behaviors induced by chronic unpredictable mild stress in rodents. Behav Brain Res 2017;326:33–43.
Schellekens H, Finger BC, Dinan TG, Cryan JF. Ghrelin signalling and obesity: at the interface of stress, mood and food reward. Pharmacol Ther 2012;135:316–326.
Alvarez-Crespo M, Skibicka KP, Farkas I, et al. The amygdala as a neurobiological target for ghrelin in rats: neuroanatomical, electrophysiological and behavioral evidence. PLOS ONE 2012;7:e46321.
Furness JB, Hunne B, Matsuda N, et al. Investigation of the presence of ghrelin in the central nervous system of the rat and mouse. Neuroscience 2011;193:1–9.
Hou Z, Miao Y, Gao L, Pan H, Zhu S. Ghrelin-containing neuron in cerebral cortex and hypothalamus linked with the DVC of brainstem in rat. Regul Pept 2006;134:126–131.
Sakata I, Nakano Y, Osborne-Lawrence S, et al. Characterization of a novel ghrelin cell reporter mouse. Regul Pept 2009;155:91–98.
Schaeffer M, Langlet F, Lafont C, et al. Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc Natl Acad Sci U S A 2013;110:1512–1517.
Cabral A, Fernandez G, Perello M. Analysis of brain nuclei accessible to ghrelin present in the cerebrospinal fluid. Neuroscience 2013;253:406–415.
Banks WA. The blood-brain barrier: connecting the gut and the brain. Regul Pept 2008;149:11–14.
Kalra SP, Dube MG, Pu S, Xu B, Horvath TL, Kalra PS. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight 1. Endocr Rev 1999;20:68–100.
Müller TD, Nogueiras R, Andermann ML, et al. Ghrelin. Mol Metab 2015;4:437–460.
Willesen MG, Kristensen P, Rømer J. Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology 1999;70:306–316.
Arvat E, Maccario M, Di Vito L, et al. Endocrine activities of ghrelin, a natural growth hormone secretagogue (GHS), in humans: comparison and interactions with hexarelin, a nonnatural peptidyl GHS, and GH-releasing hormone. J Clin Endocrinol Metab 2001;86:1169–1174.
Lutter M, Sakata I, Osborne-Lawrence S, et al. The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci 2008;11:752–753.
Patterson ZR, Ducharme R, Anisman H, Abizaid A. Altered metabolic and neurochemical responses to chronic unpredictable stressors in ghrelin receptor-deficient mice. Eur J Neurosci 2010;32:632–639.
Spencer SJ, Emmerzaal TL, Kozicz T, Andrews ZB. Ghrelin’s role in the hypothalamic-pituitary-adrenal axis stress response: implications for mood disorders. Biol Psychiatry 2015;78:19–27.
Walker AK, Rivera PD, Wang Q, et al. The P7C3 class of neuroprotective compounds exerts antidepressant efficacy in mice by increasing hippocampal neurogenesis. Mol Psychiatry 2015;20:500–508.
Hirano Y, Masuda T, Naganos S, et al. Fasting launches CRTC to facilitate long-term memory formation in Drosophila. Science 2013;339:443–446.
Plaçais P-Y, Preat T, Ghalambor C, et al. To favor survival under food shortage, the brain disables costly memory. Science 2013;339:440–442.
Meyer RM, Burgos-Robles A, Liu E, Correia SS, Goosens KA. A ghrelin-growth hormone axis drives stress-induced vulnerability to enhanced fear. Mol Psychiatry 2014;19:1284–1294.
Date Y, Murakami N, Toshinai K, et al. The role of the gastric afferent vagal nerve in Ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 2002;123:1120–1128.
Howick K, Griffin B, Cryan J, Schellekens H. From belly to brain: targeting the ghrelin receptor in appetite and food intake regulation. Int J Mol Sci 2017;18:273.
Queipo-Ortuño MI, Seoane LM, Murri M, et al. Gut microbiota composition in male rat models under different nutritional status and physical activity and its association with serum leptin and ghrelin levels. PLOS ONE 2013;8:e65465.
Chaplin A, Parra P, Serra F, Palou A. Conjugated linoleic acid supplementation under a high-fat diet modulates stomach protein expression and intestinal microbiota in adult mice. PLOS ONE 2015;10:e0125091.
Massot-Cladera M, Mayneris-Perxachs J, Costabile A, et al. Association between urinary metabolic profile and the intestinal effects of cocoa in rats. Br J Nutr 2017;117:623–634.
Parvin Z, Iraj MD, Minoo S, Fatemeh K. Effects of Toxoplasma gondii infection on anxiety, depression and ghrelin level in male rats. J Parasit Dis 2016;40:688–693.
Nilaweera KN, Cabrera-Rubio R, Speakman JR, et al. Whey protein effects on energy balance link the intestinal mechanisms of energy absorption with adiposity and hypothalamic neuropeptide gene expression. Am J Physiol Endocrinol Metab 2017;313:E1–E11.
Tubbs E, Theurey P, Vial G, et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014;63:3279–3294.
Matsumoto M, Inoue R, Tsukahara T, et al. Voluntary running exercise alters microbiota composition and increases n-butyrate concentration in the rat cecum. Biosci Biotechnol Biochem 2008;72:572–576.
Kang C, Zhang Y, Zhu X, et al. Healthy subjects differentially respond to dietary capsaicin correlating with specific gut enterotypes. J Clin Endocrinol Metab 2016;101:4681–4689.
Schmid DA, Held K, Ising M, Uhr M, Weikel JC, Steiger A. Ghrelin stimulates appetite, imagination of food, GH, ACTH, and cortisol, but does not affect leptin in normal controls. Neuropsychopharmacology 2005;30:1187–1192.
Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 1981;213:1394–1397.
Turnbull A V, Rivier C. Corticotropin-releasing factor (CRF) and endocrine responses to stress: CRF receptors, binding protein, and related peptides. Proc Soc Exp Biol Med 1997;215:1–10.
Aubry J-M. CRF system and mood disorders. J Chem Neuroanat 2013;54:20–24.
Tsatsanis C, Dermitzaki E, Venihaki M, et al. The corticotropin-releasing factor (CRF) family of peptides as local modulators of adrenal function. Cell Mol Life Sci 2007;64:1638–1655.
Muramatsu Y, Fukushima K, Iino K, et al. Urocortin and corticotropin-releasing factor receptor expression in the human colonic mucosa. Peptides 2000;21:1799–1809.
Tache Y, Perdue MH. Role of peripheral CRF signalling pathways in stress-related alterations of gut motility and mucosal function. Neurogastroenterol Motil 2004;16:137–142.
Kawahito Y, Sano H, Kawata M, et al. Local secretion of corticotropin-releasing hormone by enterochromaffin cells in human colon. Gastroenterology 1994;106:859–865.
Liu S, Chang J, Long N, et al. Endogenous CRF in rat large intestine mediates motor and secretory responses to stress. Neurogastroenterol Motil 2016;28:281–291.
Wlk M, Wang CC, Venihaki M, et al. Corticotropin-releasing hormone antagonists possess anti-inflammatory effects in the mouse ileum. Gastroenterology 2002;123:505–515.
Seasholtz AF, Valverde RA, Denver RJ. Corticotropin-releasing hormone-binding protein: Biochemistry and function from fishes to mammals. J Endocrinol 2002;175:89–97.
Refojo D, Schweizer M, Kuehne C, et al. Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science 2011;333:1903–1907.
Bethin KE, Vogt SK, Muglia LJ. Interleukin-6 is an essential, corticotropin-releasing hormone-independent stimulator of the adrenal axis during immune system activation. Proc Natl. Acad Sci U S A 2000;97:9317–9322.
Fox JH, Lowry CA. Corticotropin-releasing factor-related peptides, serotonergic systems, and emotional behavior. Front Neurosci 2013;7:169.
Habib KE, Weld KP, Rice KC, et al. Oral administration of a corticotropin-releasing hormone receptor antagonist significantly attenuates behavioral, neuroendocrine, and autonomic responses to stress in primates. Proc Natl Acad Sci U S A 2000;97:6079–6084.
Smith GW, Aubry JM, Dellu F, et al. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron 1998;20:1093–1102.
Kasahara M, Groenink L, Breuer M, Olivier B, Sarnyai Z. Altered behavioural adaptation in mice with neural corticotrophin-releasing factor overexpression. Genes Brain Behav 2007;6:598–607.
Timpl P, Spanagel R, Sillaber I, et al. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet 1998;19:162–166.
Fernández Macedo GV, Cladouchos ML, Sifonios L, Cassanelli PM, Wikinski S. Effects of fluoxetine on CRF and CRF1 expression in rats exposed to the learned helplessness paradigm. Psychopharmacology (Berl) 2013;225:647–659.
Merali Z, Kent P, Du L, et al. Corticotropin-releasing hormone, arginine vasopressin, gastrin-releasing peptide, and neuromedin B alterations in stress-relevant brain regions of suicides and control subjects. Biol Psychiatry 2006;59:594–602.
Stout SC, Owens MJ, Nemeroff CB. Regulation of corticotropin-releasing factor neuronal systems and hypothalamic-pituitary-adrenal axis activity by stress and chronic antidepressant treatment. J Pharmacol Exp Ther 2002;300:1085–1092.
Rodiño-Janeiro BK, Alonso-Cotoner C, Pigrau M, Lobo B, Vicario M, Santos J. Role of corticotropin-releasing factor in gastrointestinal permeability. J Neurogastroenterol Motil 2015;21:33–50.
Galley JD, Bailey MT. Impact of stressor exposure on the interplay between commensal microbiota and host inflammation. Gut Microbes 2014;5:390–296.
Murakami T, Kamada K, Mizushima K, et al. Changes in intestinal motility and gut microbiota composition in a rat stress model. Digestion 2017;95:55–60.
Nozu T, Martinez V, Rivier J, Taché Y. Peripheral urocortin delays gastric emptying: role of CRF receptor 2. Am J Physiol 1999;276:G867–G874.
Bueno L, Fioramonti J. Effects of corticotropin-releasing factor, corticotropin and cortisol on gastrointestinal motility in dogs. Peptides 1986;7:73–77.
Stengel A, Taché Y. Neuroendocrine control of the gut during stress: corticotropin-releasing factor signaling pathways in the spotlight. Annu Rev Physiol 2009;71:219–239.
Jašarević E, Howerton CL, Howard CD, Bale TL. Alterations in the vaginal microbiome by maternal stress are associated with metabolic reprogramming of the offspring gut and brain. Endocrinology 2015;156:3265–3276.
Golubeva AV, Crampton S, Desbonnet L, et al. Prenatal stress-induced alterations in major physiological systems correlate with gut microbiota composition in adulthood. Psychoneuroendocrinology 2015;60:58–74.
Abe H, Hidaka N, Kawagoe C, et al. Prenatal psychological stress causes higher emotionality, depression-like behavior, and elevated activity in the hypothalamo-pituitary-adrenal axis. Neurosci Res 2007;59:145–151.
Takada M, Nishida K, Kataoka-Kato A, et al. Probiotic Lactobacillus casei strain Shirota relieves stress-associated symptoms by modulating the gut–brain interaction in human and animal models. Neurogastroenterol Motil 2016;28:1027–1036.
Abildgaard A, Elfving B, Hokland M, Wegener G, Lund S. Probiotic treatment reduces depressive-like behaviour in rats independently of diet. Psychoneuroendocrinology 2017;79:40–48.
Zijlmans MAC, Korpela K, Riksen-Walraven JM, de Vos WM, de Weerth C. Maternal prenatal stress is associated with the infant intestinal microbiota. Psychoneuroendocrinology 2015;53:233–245.
Sanders J, Nemeroff C. The CRF system as a therapeutic target for neuropsychiatric disorders. Trends Pharmacol Sci 2016;37:1045–1054.
Wasserman D, Wasserman J, Sokolowski M. Genetics of HPA-axis, depression and suicidality. Eur Psychiatry 2010;25:278–280.
Yang H, Zhao XX, Tang S, et al. Probiotics reduce psychological stress in patients before laryngeal cancer surgery. Asia Pac J Clin Oncol 2016;12:e92–e96.
Leng G, Ludwig M. Intranasal oxytocin: myths and delusions. Biol Psychiatry 2015;79:243–250.
Neumann ID, Landgraf R. Balance of brain oxytocin and vasopressin: implications for anxiety, depression, and social behaviors. Trends Neurosci 2012;35:649–659.
Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev 2001;81:629–683.
Baribeau DA, Anagnostou E. Oxytocin and vasopressin: linking pituitary neuropeptides and their receptors to social neurocircuits. Front Neurosci 2015;9:335.
Welch MG, Tamir H, Gross KJ, Chen J, Anwar M, Gershon MD. Expression and developmental regulation of oxytocin (OT) and oxytocin receptors (OTR) in the enteric nervous system (ENS) and intestinal epithelium. J Comp Neurol 2009;512:256–270.
Kimura T, Makino Y, Saji F, et al. Molecular characterization of a cloned human oxytocin receptor. Eur J Endocrinol 1994;131:385–390.
Olff M, Frijling JL, Kubzansky LD, et al. The role of oxytocin in social bonding, stress regulation and mental health: an update on the moderating effects of context and interindividual differences. Psychoneuroendocrinology 2013;38:1883–1894.
Amico JA, Mantella RC, Vollmer RR, Li X. Anxiety and stress responses in female oxytocin deficient mice. J Neuroendocrinol 2004;16:319–324.
DeVries AC, Young WS, Nelson RJ. Reduced aggressive behaviour in mice with targeted disruption of the oxytocin gene. J Neuroendocrinol 1997;9:363–368.
MacDonald K, Feifel D. Oxytocin’s role in anxiety: a critical appraisal. Brain Res 2014;1580:22–56.
Landgraf R, Neumann I, Holsboer F, Pittman QJ. Interleukin-1β stimulates both central and peripheral release of vasopressin and oxytocin in the rat. Eur J Neurosci 1995;7:592–598.
Erdman SE, Poutahidis T. Probiotic “glow of health”: it’s more than skin deep. Benef Microbes 2014;5:109–119.
Cohen H, Kaplan Z, Kozlovsky N, Gidron Y, Matar MA, Zohar J. Hippocampal microinfusion of oxytocin attenuates the behavioural response to stress by means of dynamic interplay with the glucocorticoid-catecholamine responses. J Neuroendocrinol 2010;22:889–904.
Landgraf R, Wigger A. Born to be anxious: neuroendocrine and genetic correlates of trait anxiety in HAB rats. Stress 2003;6:111–119.
Slattery DA, Neumann ID. Chronic icv oxytocin attenuates the pathological high anxiety state of selectively bred Wistar rats. Neuropharmacology 2010;58:56–61.
Norman GJ, Karelina K, Morris JS, Zhang N, Cochran M, Courtney DeVries A. Social interaction prevents the development of depressive-like behavior post nerve injury in mice: a potential role for oxytocin. Psychosom Med 2010;72:519–526.
Farshim P, Walton G, Chakrabarti B, et al. Maternal weaning modulates emotional behavior and regulates the gut-brain axis. Sci Rep 2016;6:21958.
Desbonnet L, Clarke G, Shanahan F, Dinan TG, Cryan JF. Microbiota is essential for social development in the mouse. Mol Psychiatry 2014;19:146–148.
Jin P, Yu H-L, Tian-Lan, Zhang F, Quan Z-S. Antidepressant-like effects of oleoylethanolamide in a mouse model of chronic unpredictable mild stress. Pharmacol Biochem Behav 2015;133:146–154.
Yu HL, Sun LP, Li MM, Quan ZS. Involvement of norepinephrine and serotonin system in antidepressant-like effects of oleoylethanolamide in the mice models of behavior despair. Neurosci Lett 2015;593:24–28.
Gaetani S, Fu J, Cassano T, et al. The fat-induced satiety factor oleoylethanolamide suppresses feeding through central release of oxytocin. J Neurosci 2010;30:8096–8101.
Romano A, Cassano T, Tempesta B, et al. The satiety signal oleoylethanolamide stimulates oxytocin neurosecretion from rat hypothalamic neurons. Peptides 2013;49:21–26.
Wang X, Miyares RL, Ahern GP. Oleoylethanolamide excites vagal sensory neurones, induces visceral pain and reduces short-term food intake in mice via capsaicin receptor TRPV1. J Physiol 2005;564:541–547.
David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014;505:559–563.
Brinkworth GD, Noakes M, Buckley JD, Keogh JB, Clifton PM. Long-term effects of a very-low-carbohydrate weight loss diet compared with an isocaloric low-fat diet after 12 mo. Am J Clin Nutr 2009;90:23–32.
Russell WR, Gratz SW, Duncan SH, et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr 2011;93:1062–1072.
Ríos-Covián D, Ruas-Madiedo P, Margolles A, Gueimonde M, de los Reyes-Gavilán CG, Salazar N. Intestinal short chain fatty acids and their link with diet and human health. Front Microbiol 2016;7:185.
Kato-Kataoka A, Nishida K, Takada M, et al. Fermented milk containing Lactobacillus casei strain shirota preserves the diversity of the gut microbiota and relieves abdominal dysfunction in healthy medical students exposed to academic stress. Appl Environ Microbiol 2016;82:3649–3658.
Messaoudi M, Lalonde R, Violle N, et al. Assessment of psychotropic-like properties of a probiotic formulation (Lactobacillus helveticus R0052 and Bifidobacterium longum R0175) in rats and human subjects. Br J Nutr 2010;105:755–764.
Schmidt K, Cowen PJ, Harmer CJ, Tzortzis G, Errington S, Burnet PWJ. Prebiotic intake reduces the waking cortisol response and alters emotional bias in healthy volunteers. Psychopharmacology (Berl) 2015;232:1793–1801.
Kelly JR, Allen AP, Temko A, et al. Lost in translation? The potential psychobiotic Lactobacillus rhamnosus (JB-1) fails to modulate stress or cognitive performance in healthy male subjects. Brain Behav Immun 2017;61:50–59.
Akkasheh G, Kashani-Poor Z, Tajabadi-Ebrahimi M, et al. Clinical and metabolic response to probiotic administration in patients with major depressive disorder: a randomized, double-blind, placebo-controlled trial. Nutrition 2016;32:315–320.
Rafferty J, Nagaraj H, McCloskey AP, et al. Peptide therapeutics and the pharmaceutical industry: barriers encountered translating from the laboratory to patients. Curr Med Chem 2016;23:4231–4259.
Yin L, Yuvienco C, Montclare JK. Protein based therapeutic delivery agents: contemporary developments and challenges. Biomaterials 2017;134:91–116.
Pillai O, Dhanikula AB, Panchagnula R. Drug delivery: an odyssey of 100 years. Curr Opin Chem Biol 2001;5:439–446.
ElRakaiby M, Dutilh BE, Rizkallah MR, Boleij A, Cole JN, Aziz RK. Pharmacomicrobiomics: the impact of human microbiome variations on systems pharmacology and personalized therapeutics. OMICS 2014;18:402–414.
Maldonado-Gómez MX, Martínez I, Bottacini F, et al. Stable engraftment of Bifidobacterium longum AH1206 in the human gut depends on individualized features of the resident microbiome. Cell Host Microbe 2016;20:515–526.
Walsh CJ, Guinane CM, Hill C, Ross RP, O’Toole PW, Cotter PD. In silico identification of bacteriocin gene clusters in the gastrointestinal tract, based on the Human Microbiome Project’s reference genome database. BMC Microbiol 2015;15:183.
Zheng X, Zhang X, Kang A, Ran C, Wang G, Hao H. Thinking outside the brain for cognitive improvement: Is peripheral immunomodulation on the way? Neuropharmacology 2015;96:94–104.
Kommineni S, Bretl DJ, Lam V, et al. Bacteriocin production augments niche competition by enterococci in the mammalian gastrointestinal tract. Nature 2015;526:719–722.
Corr SC, Li Y, Riedel CU, O’Toole PW, Hill C, Gahan CGM. Bacteriocin production as a mechanism for the antiinfective activity of Lactobacillus salivarius UCC118. Proc Natl Acad Sci U S A 2007;104:7617–7621.
Acknowledgment
This work was supported by Science Foundation Ireland in the form of a center grant (APC Microbiome Institute; Grant No. SFI/12/RC/2273). The advice of Dr. Joshua M. Lyte, Dr. Kiran Sandhu and Simon Spichak is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Electronic supplementary material
ESM 1
(PDF 486 kb)
Rights and permissions
About this article

Cite this article
Lach, G., Schellekens, H., Dinan, T.G. et al. Anxiety, Depression, and the Microbiome: A Role for Gut Peptides. Neurotherapeutics 15, 36–59 (2018). https://doi.org/10.1007/s13311-017-0585-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-017-0585-0