
ABSTRACT
Macroautophagy is an evolutionarily conserved intracellular degradation system used by life ranging from yeasts to mammals. The core autophagic machinery is composed of ATG (autophagy-related) protein constituents. One particular member of the ATG protein family, Atg7, has been the focus of recent research. Atg7 acts as an E1-like activating enzyme facilitating both microtubule-associated protein light chain 3 (LC3)-phosphatidylethanolamine and ATG12 conjugation. Thus, Atg7 stands at the hub of these two ubiquitin-like systems involving LC3 and Atg12 in autophagic vesicle expansion. In this review, I focus on the pleiotropic function of Atg7 in development, maintenance of health, and alternations of such control in disease.
Similar content being viewed by others
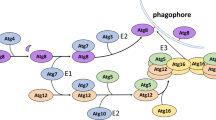

INTRODUCTION
As a cellular scavenger, autophagy is a fundamental catabolic process consisting of three primary classes of autophagy: macroautophagy (the most prevalent form of autophagy and hereafter referred to as autophagy), microautophagy, and chaperone-mediated autophagy (Feng et al., 2015). Central to the sequential events of autophagy is de novo formation of cup-shaped isolation membranes (also known as phagophores) to sequester cytoplasmic components, expansion of this membrane to create a seal for a double membrane-bound vesicles called an autophagosome, and fusion of the autophagosome with a lysosome membrane to generate an autolysosome allowing degradation and recycling of the cargoes (Nakatogawa et al., 2009). Eukaryotic cells have evolved a well-organized autophagic machinery to adapt to and survive adverse microenvironmental conditions, including dwindling nutrient supplies (Galluzzi et al., 2014). Genetic screening of autophagy-deficient mutants in yeast provides us with almost 40 ATG (autophagy-related) genes, among which approximately 18 genes possess orthologues in higher eukaryotes. These ATG-encoded products act as the core autophagy machinery and contribute to the sequential steps of autophagosome formation including (I) induction of autophagosome formation by Atg1 complex, (II) phagophore expansion by Atg9-related cycling system, (III) vesicle nucleation by the phosphatidylinositol 3-kinase complex, and (IV) vesicle expansion by two ubiquitin-like conjugation systems. One such protein is Atg7, which is uniquely shared by, and plays crucial roles in, the two ubiquitin-like conjugation systems of microtubule-associated protein light chain 3 (LC3, a mammalian homologue of Atg8) and Atg12 respectively (Feng et al., 2015).
The ubiquitin-like conjugation system of LC3 involves Atg3, Atg4, Atg7, and LC3 for LC3-phosphatidylethanolamine production, while the ubiquitin-like conjugation system of Atg12 involves the Atg5, Atg7, Atg10, Atg12, and Atg16 for Atg12-Atg5-Atg16 production. The common ubiquitin E1-like activating enzyme, Atg7, is essential for the assembly and function of these two conjugates in the expansion of autophagosomal membranes (Nakatogawa et al., 2009; Feng et al., 2015). Substantial progress has been made during the past decade revealing the pivotal roles of Atg7 in autophagy-related cell homeostasis. Recent studies have unveiled the diverse and complex autophagy-dependent function of the evolutionarily conserved Atg7 in varying species, especially plants and animals. This review focuses on how this dynamic function is achieved and discusses the implications of altered Atg7-mediated autophagic activities in molecular, cellular, and organismal levels.
FUNCTION OF ATG7 IN PLANTS
Phenotypic analyses of Atg7 mutants indicates that Atg7 disruption renders Arabidopsis (Arabidopsis thaliana) cells hypersensitive to a shortage of nutrients with features of premature leaf senescence, though the mutant is otherwise normal (Doelling et al., 2002). Increased expression of multiple LC3 isoforms are observed in Atg7 mutants due to impaired control of the two ubiquitin-like conjugation systems of LC3 and Atg12 (Thompson et al., 2005). Deletion of Atg7 in Nicotiana benthamiana and Arabidopsis leads to unrestricted hypersensitive responses during plant innate immunity (Liu et al., 2005; Hofius et al., 2009). Arabidopsis Atg7 mutant is also more susceptible to fungal infection (Lenz et al., 2011). Minina and colleagues showed that the autotroph Arabidopsis can benefit from caloric restriction-induced lifespan extension via Atg7-regulated autophagy (Minina et al., 2013).
FUNCTION OF ATG7 IN INVERTEBRATES
Nematode
A similar role of Atg7-regulated autophagy in dietary restriction-induced lifespan extension exists in Caenorhabditis elegans (C. elegans) (Jia and Levine, 2007). In addition, genetic inactivation of C. elegans Atg7 exacerbates accumulation of toxic polyglutamine expansion protein aggregates and accelerates progress of neurodegenerative disorders (Jia et al., 2007).
Fruit fly
Steroid- and radiation-triggered programmed cell death accompanies increased Atg7 transcripts in Drosophila cells (Lee et al., 2003). Normal levels of Atg7-modulated autophagy, albeit dispensable for metamorphosis, seem to be critical for preventing neurodegeneration, resisting stresses, and promoting longevity in Drosophila (Juhasz et al., 2007; Juhasz and Neufeld, 2008). Using the Drosophila eye as a model system, Chen et al. described Atg7 as a downstream effector of heat shock protein 27, and as a participant in the regulation of normal eye development, neuronal homeostasis, and lifespan (Chen et al., 2012). Such critical roles of fruit fly Atg7 has been reported not only in development but also in infection. Mycobacterium marinum infection is sufficient to counteract the effectiveness of antimycobacterial treatment, and thereby drastically affects the survival rate in Atg7 mutant Drosophila (Kim et al., 2012). In addition, a recent study reveals a novel example of Atg7-independent autophagy during the developmental shortening of Drosophila intestine (Chang et al., 2013).
FUNCTION OF ATG7 IN ZEBRAFISH AND RATS
In zebrafish, approximately one third of Atg7-knockdown morphants had ectopic expression of essential transcription factors and severe developmental defects in cardiac morphology encompassing heart looping, pericardial edema, and malformation of chamber and valve (Lee et al., 2014). Gain- and loss-of-function of Atg7 studies in the αB-crystallin R120G mutation (CryABR120G) model of rat desmin-related cardiomyopathy reveal the significant ability of ATG7 in reversing autophagic deficiency and maintaining physiological levels of basal autophagy (Pattison et al., 2011). As a consequence of Cathepsin B treatment, stimulated ATG7-mediated autophagy aggravates lipotoxicity via induction of nod-like receptor 3 proinflammatory response in rat insulinoma cells (Li et al., 2013).
FUNCTION OF ATG7 IN MICE
To investigate the in vivo function of ATG7 in mammals, Komatsu et al. generated Atg7-deficient mice (Atg7−/−). As anticipated, Atg7−/− mice exhibit impaired constitutive and starvation-induced autophagy; however, they die soon after birth (Komatsu et al., 2005). Therefore, Ubc-CreERT2 mice were crossed with Atg7-floxed (Atg7flox/flox) mice for the generation of tamoxifen-inducible whole body Atg7 knockout mice. Karsli-Uzunbas et al. further reported that acute systemic deletion of Atg7 in adult mice leads to perturbed glucose metabolism, but blocks the progression of non-small cell lung cancer (NSCLC) in vivo (Karsli-Uzunbas et al., 2014). Thus, cells from embryo/fetus of Atg7−/− mice and certain cell/tissue-specific Atg7-knockout postnatal mice were extensively employed in the quest for understanding the mechanisms underlying the pleiotropic effects of ATG7 in development, physiology, and pathology (Table 1).
Embryonic fibroblasts
Wild-type (WT) mouse embryonic fibroblasts (MEFs) were used to recapitulate robust autophagy-mediated capability of bacteria clearance, which is absent in Atg7−/− MEFs (Sun et al., 2008). Using WT and Atg7−/− MEFs and small interference RNA (siRNA)-mediated silencing of Atg7 in BAX/BAK-knockout MEFs, it has been demonstrated that Atg7-regulated autophagy is dispensable for obatoclax-induced toxicity (McCoy et al., 2010). Subsequently, Lee et al. found a novel function of Atg7, independent of its E1-like enzymatic activity. Briefly, ATG7 coordinates tumor suppressor p53-mediated cell division cycle and cell apoptosis via physical interaction with p53 under limited nutrients, providing an explanation for the simultaneous or sequential metabolic stress-induced events, including exit from cell cycle, induction of autophagy, and activation of cell death signaling. In addition, the augmented genomic instability in Atg7−/− mice may be a reason for its postnatal death (Lee et al., 2012). To characterize the regulatory network of autophagy, quantitative iTRAQ labeling coupled with on-line 2D LC/MS/MS proteomics analysis was performed in WT and Atg7−/− MEFs. The result implied that basal and starvation-induced autophagy depends on an intact cytoskeletal protein filamentous actin network (Zhuo et al., 2013).
Liver cells
Mx1-Cre transgenic mice were crossed with Atg7-floxed (Atg7flox/flox) mice for the generation of hepatocyte-specific polyinosinic acid-polycytidylic acid-inducible Atg7 knockout (iMx1-Atg7−/−) mice (Komatsu et al., 2005; Matsumoto et al., 2008). iMx1-Atg7−/− mice present hepatomegaly with malformations of organelles and ubiquitin-positive protein aggregates (Komatsu et al., 2005). Alb-Cre mice were crossed with Atg7flox/flox mice for the generation of hepatocyte-specific Atg7 knockout (Alb-Atg7−/−) mice (Matsumoto et al., 2008; Singh et al., 2009a). Comprehensive proteomics analyses of iMx1-Atg7−/− and Alb-Atg7−/− mice and their controls suggest that autophagy-deficient hepatic cells exert oxidative stress with increased total protein mass, specifically glutathione S-transferase families, protein disulfide isomerase, and glucose-regulated proteins (Matsumoto et al., 2008). Alb-Atg7−/− mice also showed higher triglyceride storage in lipid droplets during nutrient deprivation than controls, providing evidence that lipolysis and autophagy are interrelated through macrolipophagy (Singh et al., 2009a). GFAP (glial fibrillary acid protein)-Cre mice were crossed with Atg7flox/flox mice for the generation of hepatic stellate cell-specific Atg7 knockout (GFAP-Atg7−/−) mice. A surprising detrimental consequence of autophagy in deteriorating hepatic fibrogenesis through release of lipids from activated stellate cells, has been established in GFAP-Atg7−/− mice in vivo and the mouse immortalized stellate cell line JS1 in vitro (Hernandez-Gea et al., 2012).
Pancreatic β cells and skeletal muscle cells
RIP-Cre mice were crossed with Atg7flox/flox mice for the generation of pancreatic β cell-specific Atg7 knockout (β cell-Atg7−/−) mice (Ebato et al., 2008; Jung et al., 2008; Wu et al., 2009). These mice display impaired glucose tolerance and degenerated islets accompanied by reduced β cell mass and insulin secretion levels. A series of morphological malformations occur in Atg7 mutant β cells, including accumulation of ubiquitinated inclusions, enlargement of mitochondria, and distension of the endoplasmic reticulum (Ebato et al., 2008; Jung et al., 2008). MCK-Cre mice were crossed with Atg7flox/flox mice for the generation of skeletal muscle cell-specific Atg7 knockout (SMC-Atg7−/−) mice. Furthermore, Wu et al. observed a decrease of mitochondrial oxidation consumption and an increase of compensatory basal glycolytic rates and reactive oxygen species levels in cells derived from β cell-Atg7−/− and SMC-Atg7−/− mice (Wu et al., 2009).
Endothelial cells and vascular smooth muscle cells
VE-cadherin-Cre transgenic mice were crossed with Atg7flox/flox mice for the generation of endothelial cell-specific Atg7 knockout (EC-Atg7−/−) mice (Torisu et al., 2013; Singh et al., 2015). Compared to WT littermate controls, EC-Atg7−/− mice have impaired von Willebrand factor (VWF) release elicited by epinephrine, implying a promising strategy for transient prevention of thrombosis (Torisu et al., 2013). Moreover, Atg7-null endothelial cells also confer susceptibility to bleomycin-induced pulmonary fibrosis in vivo by endothelial-to-mesenchymal transition (EndMT) (Singh et al., 2015).
SM22α-Cre transgenic mice were crossed with Atg7flox/flox mice for the generation of vascular smooth muscle cell-specific Atg7 knockout mice. Vascular smooth muscle cell-specific Atg7 deletion leads to sarcoplasmic reticulum swelling and imbalanced Ca2+ homeostasis, resulting in altered contractility (Michiels et al., 2015).
Fat cells and mammary gland cells
Fab4 (aP2)-Cre mice were crossed with Atg7flox/flox mice for the generation of adipocyte-specific Atg7 knockout (FC-Atg7−/−) mice. Targeted deletion of Atg7 in adipose tissues leads to a lean body mass with an elevated rate of β-oxidation and a low rate of lipolysis. The white adipose tissue in FC-Atg7−/− mice acquired more features of brown adipose tissue, and its mass diminished (Zhang et al., 2009a; Singh et al., 2009b). Strikingly, disruption of ATG7 confers sensitivity to insulin stimuli (Zhang et al., 2009a). Additional evidence that ATG7 plays a vital role in adipogenesis has been obtained in 3T3-L1 preadipocytes, wherein inhibition of ATG7 hampered adipocyte differentiation and lipid accumulation (Singh et al., 2009b).
WAP-Cre mice were crossed with Atg7flox/flox mice for the generation of mammary gland cell-specific Atg7 knockout (MGC-Atg7−/−) mice (Kongara et al., 2010; Teplova et al., 2013). Using MGC-Atg7−/− mice, Kongara et al. linked ATG7-regulated autophagy to limiting ER and oxidative stress and orchestrating keratin 8 homeostasis in mammary cells (Kongara et al., 2010). Besides this phenotype, MGC-Atg7−/− mice undergo defective phagocytosis, compromised dead cell clearance, and enhanced inflammatory responses in mammary involution, reminiscent of tumor-modulating niche and ductal ectasia. Consistent with these observations, specific knockdown of Atg7 in immortalized mouse mammary epithelial cells strengthened the conclusion that ATG7 is needed for effective dead cell engulfment (Teplova et al., 2013).
Neurons
(I) Nestin-Cre transgenic mice were crossed with Atg7flox/flox mice for the generation of neuron-specific Atg7 knockout (nestin-Atg7−/−) mice (Komatsu et al., 2006a). Consistent with the findings from invertebrates (Jia et al., 2007; Juhasz et al., 2007; Juhasz and Neufeld, 2008; Chen et al., 2012), nestin-Atg7−/− mice lacking autophagy in the central nervous system displayed a broad range of neurodegenerative symptoms, including accumulation of inclusion bodies in Atg7-deletion neurons, loss of massive neurons in the cerebral and cerebellar cortices, and defects of behavioral coordination (Komatsu et al., 2006a). (II) Pcp2-Cre transgenic mice were crossed with Atg7flox/flox mice for the generation of Purkinje cell-specific Atg7 knockout mice. Similar to nestin-Atg7−/− mice, Purkinje cell-specific loss of Atg7 function impeded autophagy-related membrane trafficking and turnover resulting in axonal dystrophy, a sign of axonopathy associated with neurodegenerative disease (Komatsu et al., 2007). (III) POMC (pro-opiomelanocortin)-Cre transgenic mice were crossed with Atg7flox/flox mice for the generation of POMC neuron-specific Atg7 knockout (POMC-Atg7−/−) mice (Kaushik et al., 2012; Coupe et al., 2012). In POMC-Atg7−/− mice, Kaushik et al. drew a consistent conclusion by a previous study in Alb-Atg7−/− mice that autophagy negatively regulates lipolysis (Singh et al., 2009a; Kaushik et al., 2012). Moreover, direct genetic evidence was obtained that ATG7 participates in normal development and metabolic modulation in POMC neurons, indicating potential roles of Atg7 deficiency in the pathogenesis of obesity and aging-related metabolic syndrome (Kaushik et al., 2012; Coupe et al., 2012). (IV) CamKII-Cre transgenic mice were crossed with Atg7flox/flox mice for the generation of forebrain neuron-specific Atg7 knockout (CamKII-Atg7−/−) mice. Remarkably, protective roles of Atg7 in neurodegeneration of forebrain neurons have been elucidated in CamKII-Atg7−/− mice (Inoue et al., 2012a; Nilsson et al., 2013). Atg7 ablation correlates with the progression of age-dependent neurodegeneration via tau phosphorylation pathway (Inoue et al., 2012a). By breeding CamKII-Atg7−/− mice with amyloid precursor protein transgenic mice, Nilsson et al. found that autophagy deficiency led to reduced amyloid beta (Aβ) secretion and concurrent accumulation of intracellular Aβ peptide, indicative of Alzheimer’s disease (Nilsson et al., 2013). (V) Atg7 knockout astrocytes failed to orchestrate intricate mitochondria network for normal inflammation responses (Motori et al., 2013). In addition, VAChT-Cre mice were crossed with Atg7flox/flox mice for the generation of motor neuron-specific Atg7 knockout (VAChT-Atg7−/−) mice. Using VAChT-Atg7−/− mice, Tashiro et al. exclude the potential involvement of autophagy in the pathogenesis of amyotrophic lateral sclerosis (Tashiro et al., 2012). In contrast, ATG7-mediated autophagy as an upstream cell death driver controls lysosomal dysfunction-induced cell apoptosis in mouse C17.2 neural stem cells (Walls et al., 2010).
Interestingly, seemingly opposing effects of ATG7 in dopamine neurons have been delineated by different groups (Cheng et al., 2011; Inoue et al., 2012a; Hernandez et al., 2012). DAT-Cre mice were crossed with Atg7flox/flox mice for the generation of dopamine neuron (enriched in the substantia nigra pars compacta)-specific Atg7 knockout (DAT-Atg7−/−) mice (Inoue et al., 2012a; Hernandez et al., 2012). Inoue et al. observed that DAT-Atg7−/− exhibits an even more severe phenotype of age-dependent neurodegeneration than CamKII-Atg7−/− mice (Inoue et al., 2012a). Conversely, conditional deletion of Atg7 in substantia nigra dopaminergic neurons of adult mice by intranigral injection of adeno-associated virus-Cre achieves unexpected protection in retrograde degeneration of dopaminergic axons. Additionally, this process is tightly controlled by Akt/Rheb/the kinase mammalian target of rapamycin (mTOR) signaling pathways (Cheng et al., 2011). Likewise, using DAT-Atg7−/− mouse model, Hernandez et al. revealed that mTOR inhibitor rapamycin decreases evoked dopamine secretion and decelerates recovery in Dopamine neurons, which is an autophagy-dependent regulation of presynaptic neurotransmission (Hernandez et al., 2012).
Hematopoietic cells
(I) EIIa-Cre mice were bred with Atg7flox/flox mice, and the progeny heterozygous Atg7+/− mice were intercrossed for the generation of E13.5 homozygous Atg7−/− mice. Then, transplantation of E13.5 Atg7−/− fetal liver cells into H2K-GFP mice was carried out to examine the hematopoietic lineages. A novel finding was observed that Atg7-dependent and independent mechanisms contribute to mitochondrial clearance during reticulocyte maturation (Zhang et al., 2009b). (II) Lck-Cre mice were crossed with Atg7flox/flox mice for the generation of T cell-specific Atg7 knockout (Lck-Atg7−/−) mice (Hubbard et al., 2010; Jia et al., 2011). Based on analyses of Lck-Atg7−/− mice, it has been reported that that autophagy is responsible for maintenance of normal production of IL-2 and IFN-γ, stimulated proliferation, endoplasmic reticulum homeostasis, and calcium mobilization, in T lymphocytes; nevertheless, T cells derived from Lck-Atg7−/− mice had no detectable increased apoptosis (Hubbard et al., 2010; Jia et al., 2011). The very slow activation-induced proliferation makes it difficult to differentiate polarized Th1 cell populations. To this end, Cre-ER mice were crossed with Atg7flox/flox mice for the generation of tamoxifen-inducible Atg7 knockout (ER-Atg7−/−) mice. Unsurprisingly, deletion of Atg7 in isolated T cells from ER-Atg7−/− mice resulted in decreased activation-induced cytokine production (Hubbard et al., 2010). (III) Vav-iCre mice were crossed with Atg7flox/flox mice for the generation of hematopoietic system-specific Atg7 knockout (Vav-Atg7−/−) mice (Mortensen et al., 2010, 2011; Rasmussen et al., 2011; Jacquel et al., 2012). Loss of Atg7-mediated autophagy hampered mitochondria removal and erythroid development, as well as proliferation and genomic integrity of hematopoietic stem cells, giving rise to severe and fatal anemia and myeloproliferation in Vav-Atg7−/− mice (Mortensen et al., 2010; 2011). In addition to these functions, bone marrow-derived dendritic cells from Vav-Atg7−/− mice mitigated the response to α-herpesvirus infection and viral DNA recognition due to reduction of ATG7-dependent IFN-β expression (Rasmussen et al., 2011). Ex vivo assessment of monocytes from Vav-Atg7−/− mice indicates that macrophagic differentiation induction and function acquisition could be attributed to ATG7-mediated autophagy (Jacquel et al., 2012).
Intestinal cells
Villi-Cre (or Villi-CreER) transgenic mice were crossed with Atg7flox/flox mice for the generation of intestinal epithelium-specific (tamoxifen-inducible) Atg7 knockout (Villi-Atg7−/−) mice (Cadwell et al., 2009; Fujishima et al., 2011; Wittkopf et al., 2012; Inoue et al., 2012b; Nishiumi et al., 2012; Adolph et al., 2013). Like its orthologues Atg16L1 and Atg5, Atg7 aids in the normal morphology, and granule formation and exocytosis of Paneth cells, as suggested by analyses of this mouse model (Cadwell et al., 2009; Wittkopf et al., 2012). Villi-Atg7−/− mice displayed upregulated gene expression associated with inflammation and, thereby, endotoxin or Citrobacter rodentium-induced inflammatory responses via NF-κB inactivation (Cadwell et al., 2009; Fujishima et al., 2011; Inoue et al., 2012b). These observations are underscored by another seminal mouse genetic work showing that Villi-Atg7−/− mice synergistically with intestinal epithelium-specific Xbp1-deficient mice recapitulates features of Crohn’s disease, as a specific type of Paneth cell disease (Adolph et al., 2013). Nonetheless, thus far, no overt evidence has been obtained from Villi-Atg7−/− mice that Atg7 is implicated in the pathogenesis of intestinal tumors and maintenance of gut immune homeostasis (Nishiumi et al., 2012; Wittkopf et al., 2012).
Skin cells, kidney cells, and cardiomyocytes
K14-Cre mice were crossed with Atg7flox/flox mice for the generation of epidermal keratinocyte-specific Atg7 knockout mice (Zhao et al., 2013; Rossiter et al., 2013). Using this mouse model, Zhao et al. highlighted the importance of ATG7 for the removal of reactive oxidized phospholipids and damaged protein aggregates in the epidermis exposed to environmental insults (Zhao et al., 2013). However, ATG7-mediated autophagy appears to be nonessential to execute skin barrier function (Rossiter et al., 2013). ATG7 has also been documented as a core regulator in caspase-8 inhibition-induced autophagic cell death in mouse L929 skin fibroblast cells (Yu et al., 2004).
PEPCK-Cre mice were crossed with Atg7flox/flox mice for the generation of kidney proximal tubular cell-specific Atg7 knockout (PEPCK-Atg7−/−) mice. These autophagy-deficient PEPCK-Atg7−/− mice are particularly vulnerable to cisplatin- and ischemia-reperfusion induced acute renal injury, suggesting potent renal protection by ATG7-mediated autophagy (Jiang et al., 2012).
To test whether autophagy can ameliorate or restore proteinopathy in CryABR120G cardiac model of cardiomyopathy, Bhuiyan et al. crossed ATG7-expressing mice and CryABR120G mice to generate Atg7-crossed CryABR120G mice. Indeed, the entire cohort of Atg7-crossed CryABR120G mice acquire relatively sustained autophagy, leading to improved cardiac function (Bhuiyan et al., 2013).
FUNCTION OF ATG7 IN HUMAN
Prompted by the clues from model organisms, the architecture of the functional ATG7-mediated regulatory network has been explored in the settings of human biology and disease, such as cancer, infectious disease, and neurodegenerative diseases (Fig. 1).

Schematic illustration of physiopathological roles of ATG7 in human
Cancer
As implied earlier, autophagy has been considered as both a pro-survival pathway and type 2 cell death (Kroemer and Levine, 2008). Two main hallmarks of cancer cells are unrestricted proliferation and suppressed cell death (Hanahan and Weinberg, 2011), raising the possibility of ATG7 as both an oncogene and a tumor suppressor. On one hand, ATG7 suppresses resistance of human breast cancer cells to photodynamic therapy (Xue et al., 2010). It also facilitates the anti-tumor actions of cytosolic FoxO1 and obatoclax in human NSCLC cells (Zhao et al., 2010; McCoy et al., 2010), compound 2-Methoxyestradiol in human osteosarcoma (Yang et al., 2013), and tetrandrine in human hepatocellular carcinoma (Gong et al., 2012). Moreover, the caspase-8 inhibition-initiated autophagic cell death program requires ATG7 via activation of receptor-interacting protein/c-Jun N-terminal kinase signaling in human U937 monocyte lymphoma cells (Yu et al., 2004). Additionally, two microRNAs, miR-17 and miR-137, have been shown to target ATG7 for acquisition of resistance to anticancer drugs and low-dose ionizing radiation treatments in human glioma cells (Comincini et al., 2013; Zeng et al., 2015). On the other hand, ATG7 can also serve as an oncogene. Heat shock factor 1-controlled transcriptional expression of ATG7 is inversely correlated with the chemotherapeutic prognosis of breast cancer patients (Desai et al., 2013). Inhibition of redundant Atg7-mediated lysosome-autophagy pathway augments the anti-cancer effects of a proteasome inhibitor in some human prostate cancer cells (Zhu et al., 2010), epidermal growth factor receptor-tyrosine kinase inhibitors in human lung cancer cells (Han et al., 2011), and cisplatin in human esophageal squamous cell carcinoma cells (Zhu et al., 2013). Through reciprocal mechanical interaction, ATG7, rather than ATG5 and Beclin-1, represses caspase-9-mediated apoptosis in human colon and cervical cancer cells. Caspase-9 promotes Atg7-mediated autophagy (Han et al., 2014). Notably, ATG7 has a relatively higher expression in human THP1 acute monocytic leukemia cells than a panel of human immune and epithelial cells (Rioux et al., 2007). Importantly, two physical interactions between ATG7 and acetyltransferase p300 and between ATG7 and transcription factor p53 have been depicted in human HeLa cervical and HCT116 colon cancer cells, respectively, in the context of limited nutrient availability (Lee and Finkel, 2009; Lee et al., 2012).
Infectious disease
Dual effects of ATG7-mediated autophagy intersection with human immunodeficiency virus (HIV) biogenesis fuel the viral yields as they do in human U937 monocytoid cells and in primary human macrophages (Kyei et al., 2009). A battery of morphological and biochemical assays have been conducted showing that hepatitis C virus (HCV) causes an unfolded protein response-dependent incomplete ATG7-mediated autophagy during pathogenesis in human hepatoma cells (Sir et al., 2008). Also, it has been documented that disruption of ATG7-mediated autophagy can evoke the interferon signaling pathway resulting in apoptosis of HCV-infected immortalized human hepatocytes (Shrivastava et al., 2011), and dramatically enhanced infectivity of human papillomavirus in primary human keratinocytes (Griffin et al., 2013). Knockdown of Atg7 also interferes with the elimination of intracellular pathogen Mycobacterium tuberculosis by human immunity-related GTPase family M protein in U937 cells (Singh et al., 2006). ATG7-mediated autophagy is also involved in the constricting activity of HIV infection via release of the HIV-1 transactivator Tat in human embryonic kidney 293 cells and MAGIC5B cells (i.e., HeLa cells modified to express CD4 and CXCR4 together with β-galactosidase under the control of the HIV LTR promoter) (Sagnier et al., 2015). Silencing of ATG7 may delay the progression of Chikungunya virus-induced caspase-dependent cell death in human fibroblast cells and HeLa cells (Joubert et al., 2012), and suppress the colony stimulating factor-1-induced differentiation of human peripheral blood monocytes into macrophages (Jacquel et al., 2012).
Neurodegenerative disease
It has been shown that downregulation of ATG7 can compensate the loss of mitochondria in PTEN-induced kinase 1 (PINK1) deficient dopaminergic human neuroblastoma cells, likely supporting the potential role of ATG7 in PINK1 mutation-related familial Parkinson’s disease (Dagda et al., 2009). By analyzing a large number of European Huntington disease patients, Metzger et al. found that the V471A polymorphism in ATG7 was significantly associated with the age at onset. More specifically, the V471A polymorphism in ATG7 correlates with an earlier disease onset of 4 years in a mixed group of Huntington disease populations (Metzger et al., 2010, 2013). In five patients with Parkinson’s disease, four novel genetic variants including 11313449G>A, 11313811T>C, 11313913G>A, and 11314041G>A, were identified on the ATG7 gene promoter, implying the altered transcriptional activity of the ATG7 may be a risk factor (Chen et al., 2013).
Miscellaneous
Together with FOXO3-ATG101 complex, coupling of acetylated FOXO1 with ATG7, upon stimulation with prosecretory mitogen lacritin, can rescue the metabolic homeostasis in human corneal epithelial cells (Wang et al., 2013). In a similar manner, acetylated FOXO1 and ATG7 can preserve human umbilical vein endothelial cells (HUVECs) viability under circumstances of oxidative stress (Han et al., 2012). ATG7 is also essential for normal secretion of VWF in HUVECs (Torisu et al., 2013), and EndMT in both HUVECs and human pulmonary aortic endothelial cells (Singh et al., 2015).
CONCLUDING REMARKS
The word “autophagy”, literally auto-, meaning “self”, and phagein, meaning “to eat”, in Greek, was originally coined by Belgian cytologist Christian de Duve in 1963 (Klionsky, 2008). More than 50 years have passed since autophagy was defined as a core mechanism underlying both elimination and recycling of intracellular materials in normal development and diverse disease categories (Mizushima and Komatsu, 2011; Choi et al., 2013; Murrow and Debnath, 2013). These include, but are not limited to, immunity (Virgin and Levine, 2009), metabolism (Codogno and Meijer, 2010; Rabinowitz and White, 2010), aging (Madeo et al., 2010), and the cardiovascular (De Meyer et al., 2015; Nussenzweig et al., 2015), and nervous system (Komatsu et al., 2006b). Although still in the early stages, it appears to be almost clear how core machinery plays in Atg7-dependent and -independent autophagosome biogenesis (Nishida et al., 2009; Lamb et al., 2013). No less important than this ATG7-mediated autophagic assembly is the function and regulation of Atg7 in natural and stressed pathophysiological conditions. Thanks to the dedication and contribution of numerous laboratories over the years, recent exciting findings on ATG7 have caused a paradigm shift in the field of ATG7-mediated autophagic regulation. A brief historical overview of select prior landmark investigations has been summarized in the timeline of Fig. 2. Given that these studies reflect the nature of ATG7’s intrinsic double-edged sword in development and disease, it is likely that excessive or deficient Atg7-mediated autophagy is harmful. Following advances in therapeutic manipulation of autophagy (Kroemer, 2015), it will be important to determine the specific and safe methods for pharmacologic fine-tuning of ATG7 activity.

Retrospective analyses of major events of Atg7 research in development and disease
Another cardinal question concerns personal medicine for accurate and rapid diagnosis of ATG7-related disease. The complex role of ATG7 seems to be highly structured in a spatiotemporal fashion rather than ad libitum (Behrends et al., 2010). The diverse roles of Atg7 in different settings summarized by this review may be attributed to selective Atg7-mediated autophagy at distinct organelle, cell, tissue, organ, and organism levels. An accurate understanding of the specified and delicate roles of Atg7-autophagy requires the in-depth knowledge of both the contextual extracellular cues and the intracellular responses. Thus, it will be interesting to search for the exact niches responsible for how Atg7 activity is encoded. It is also plausible that certain intricate forms of crosstalk interactions between Atg7-mediated autophagy and other autophagy-dependent or -independent pathways are responsible for shaping the versatile functions of Atg7. Therefore, it might be essential to identify and characterize the key coordinators of Atg7-mediated autophagy and other regulatory networks. Despite these challenges to be faced, academic and industry’s research progress in Atg7 offers new avenues towards refined autophagic mechanism and Atg7-based clinical treatment.
Abbreviations
- 2D LC:
-
two-dimensional liquid chromatography
- Aβ:
-
amyloid beta
- ATG:
-
autophagy-related
- Atg7−/− :
-
Atg7-deficient mice
- Atg7flox/flox :
-
Atg7-floxed mice
- CryABR120G :
-
αB-crystallin R120G mutation
- EndMT:
-
endothelial-to-mesenchymal transition
- FOXO:
-
forkhead box O
- HCV:
-
hepatitis C virus
- HIV:
-
human immunodeficiency virus
- HUVECs:
-
human umbilical vein endothelial cells
- iTRAQ:
-
isobaric tags for relative and absolute quantitation
- LC3:
-
light chain 3
- MEF:
-
mouse embryonic fibroblast
- MS/MS:
-
tandem mass spectrometry
- mTOR:
-
mammalian target of rapamycin
- NIH:
-
National Institutes of Health
- NSCLC:
-
non-small cell lung cancer
- PINK1:
-
PTEN-induced kinase 1
- siRNA:
-
small interference RNA
- VWF:
-
Von Willebrand factor
- WT:
-
wild-type
REFERENCES
Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, Glickman JN, Tschurtschenthaler M, Hartwig J, Hosomi S et al (2013) Paneth cells as a site of origin for intestinal inflammation. Nature 503:272–276
Behrends C, Sowa ME, Gygi SP, Harper JW (2010) Network organization of the human autophagy system. Nature 466:68–76
Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, Hill JA, Sadoshima J, Robbins J (2013) Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Investig 123:5284–5297
Cadwell K, Patel KK, Komatsu M, Virgin HWt, Stappenbeck TS (2009) A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 5:250–252
Chang TK, Shravage BV, Hayes SD, Powers CM, Simin RT, Wade Harper J, Baehrecke EH (2013) Uba1 functions in Atg7- and Atg3-independent autophagy. Nat Cell Biol 15:1067–1078
Chen SF, Kang ML, Chen YC, Tang HW, Huang CW, Li WH, Lin CP, Wang CY, Wang PY, Chen GC et al (2012) Autophagy-related gene 7 is downstream of heat shock protein 27 in the regulation of eye morphology, polyglutamine toxicity, and lifespan in Drosophila. J Biomed Sci 19:52
Chen D, Pang S, Feng X, Huang W, Hawley RG, Yan B (2013) Genetic analysis of the ATG7 gene promoter in sporadic Parkinson’s disease. Neurosci Lett 534:193–198
Cheng HC, Kim SR, Oo TF, Kareva T, Yarygina O, Rzhetskaya M, Wang C, During M, Talloczy Z, Tanaka K et al (2011) Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J Neurosci 31:2125–2135
Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368:651–662
Codogno P, Meijer AJ (2010) Autophagy: a potential link between obesity and insulin resistance. Cell Metab 11:449–451
Comincini S, Allavena G, Palumbo S, Morini M, Durando F, Angeletti F, Pirtoli L, Miracco C (2013) microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol Ther 14:574–586
Coupe B, Ishii Y, Dietrich MO, Komatsu M, Horvath TL, Bouret SG (2012) Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab 15:247–255
Dagda RK, Cherra SJ III, Kulich SM, Tandon A, Park D, Chu CT (2009) Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284:13843–13855
De Meyer GR, Grootaert MO, Michiels CF, Kurdi A, Schrijvers DM, Martinet W (2015) Autophagy in vascular disease. Circ Res 116:468–479
Desai S, Liu Z, Yao J, Patel N, Chen J, Wu Y, Ahn EE, Fodstad O, Tan M (2013) Heat shock factor 1 (HSF1) controls chemoresistance and autophagy through transcriptional regulation of autophagy-related protein 7 (ATG7). J Biol Chem 288:9165–9176
Doelling JH, Walker JM, Friedman EM, Thompson AR, Vierstra RD (2002) The APG8/12-activating enzyme APG7 is required for proper nutrient recycling and senescence in Arabidopsis thaliana. J Biol Chem 277:33105–33114
Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E et al (2008) Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 8:325–332
Feng Y, Yao Z, Klionsky DJ (2015) How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol 25:354–363
Fujishima Y, Nishiumi S, Masuda A, Inoue J, Nguyen NM, Irino Y, Komatsu M, Tanaka K, Kutsumi H, Azuma T et al (2011) Autophagy in the intestinal epithelium reduces endotoxin-induced inflammatory responses by inhibiting NF-kappaB activation. Arch Biochem Biophys 506:223–235
Galluzzi L, Pietrocola F, Levine B, Kroemer G (2014) Metabolic control of autophagy. Cell 159:1263–1276
Gong K, Chen C, Zhan Y, Chen Y, Huang Z, Li W (2012) Autophagy-related gene 7 (ATG7) and reactive oxygen species/extracellular signal-regulated kinase regulate tetrandrine-induced autophagy in human hepatocellular carcinoma. J Biol Chem 287:35576–35588
Griffin LM, Cicchini L, Pyeon D (2013) Human papillomavirus infection is inhibited by host autophagy in primary human keratinocytes. Virology 437:12–19
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, Ge W, Feng L, Lin X, Wang X et al (2011) EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One 6:e18691
Han J, Pan XY, Xu Y, Xiao Y, An Y, Tie L, Pan Y, Li XJ (2012) Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy 8:812–825
Han J, Hou W, Goldstein LA, Stolz DB, Watkins SC, Rabinowich H (2014) A complex between Atg7 and caspase-9: a novel mechanism of cross-regulation between autophagy and apoptosis. J Biol Chem 289:6485–6497
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hernandez D, Torres CA, Setlik W, Cebrian C, Mosharov EV, Tang G, Cheng HC, Kholodilov N, Yarygina O, Burke RE et al (2012) Regulation of presynaptic neurotransmission by macroautophagy. Neuron 74:277–284
Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, Friedman SL (2012) Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142:938–946
Hofius D, Schultz-Larsen T, Joensen J, Tsitsigiannis DI, Petersen NH, Mattsson O, Jorgensen LB, Jones JD, Mundy J, Petersen M (2009) Autophagic components contribute to hypersensitive cell death in Arabidopsis. Cell 137:773–783
Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, Macian F (2010) Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol 185:7349–7357
Inoue K, Rispoli J, Kaphzan H, Klann E, Chen EI, Kim J, Komatsu M, Abeliovich A (2012a) Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol Neurodegener 7:48
Inoue J, Nishiumi S, Fujishima Y, Masuda A, Shiomi H, Yamamoto K, Nishida M, Azuma T, Yoshida M (2012b) Autophagy in the intestinal epithelium regulates Citrobacter rodentium infection. Arch Biochem Biophys 521:95–101
Jacquel A, Obba S, Boyer L, Dufies M, Robert G, Gounon P, Lemichez E, Luciano F, Solary E, Auberger P (2012) Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood 119:4527–4531
Jia K, Levine B (2007) Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy 3:597–599
Jia K, Hart AC, Levine B (2007) Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy 3:21–25
Jia W, Pua HH, Li QJ, He YW (2011) Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol 186:1564–1574
Jiang M, Wei Q, Dong G, Komatsu M, Su Y, Dong Z (2012) Autophagy in proximal tubules protects against acute kidney injury. Kidney Int 82:1271–1283
Joubert PE, Werneke SW, de la Calle C, Guivel-Benhassine F, Giodini A, Peduto L, Levine B, Schwartz O, Lenschow DJ, Albert ML (2012) Chikungunya virus-induced autophagy delays caspase-dependent cell death. J Exp Med 209:1029–1047
Juhasz G, Neufeld TP (2008) Drosophila Atg7: required for stress resistance, longevity and neuronal homeostasis, but not for metamorphosis. Autophagy 4:357–358
Juhasz G, Erdi B, Sass M, Neufeld TP (2007) Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev 21:3061–3066
Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW et al (2008) Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 8:318–324
Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD et al (2014) Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 4:914–927
Kaushik S, Arias E, Kwon H, Lopez NM, Athonvarangkul D, Sahu S, Schwartz GJ, Pessin JE, Singh R (2012) Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep 13:258–265
Kim JJ, Lee HM, Shin DM, Kim W, Yuk JM, Jin HS, Lee SH, Cha GH, Kim JM, Lee ZW et al (2012) Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 11:457–468
Klionsky DJ (2008) Autophagy revisited: a conversation with Christian de Duve. Autophagy 4:740–743
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y et al (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169:425–434
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E et al (2006a) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884
Komatsu M, Kominami E, Tanaka K (2006b) Autophagy and neurodegeneration. Autophagy 2:315–317
Komatsu M, Wang QJ, Holstein GR, Friedrich VL Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z (2007) Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci USA 104:14489–14494
Kongara S, Kravchuk O, Teplova I, Lozy F, Schulte J, Moore D, Barnard N, Neumann CA, White E, Karantza V (2010) Autophagy regulates keratin 8 homeostasis in mammary epithelial cells and in breast tumors. Mol Cancer Res 8:873–884
Kroemer G (2015) Autophagy: a druggable process that is deregulated in aging and human disease. J Clin Investig 125:1–4
Kroemer G, Levine B (2008) Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 9:1004–1010
Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A et al (2009) Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol 186:255–268
Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14:759–774
Lee IH, Finkel T (2009) Regulation of autophagy by the p300 acetyltransferase. J Biol Chem 284:6322–6328
Lee CY, Clough EA, Yellon P, Teslovich TM, Stephan DA, Baehrecke EH (2003) Genome-wide analyses of steroid- and radiation-triggered programmed cell death in Drosophila. Curr Biol 13:350–357
Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, Cao L, Finkel T (2012) Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 336:225–228
Lee E, Koo Y, Ng A, Wei Y, Luby-Phelps K, Juraszek A, Xavier RJ, Cleaver O, Levine B, Amatruda JF (2014) Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy 10:572–587
Lenz HD, Vierstra RD, Nurnberger T, Gust AA (2011) ATG7 contributes to plant basal immunity towards fungal infection. Plant Signal Behav 6:1040–1042
Li S, Du L, Zhang L, Hu Y, Xia W, Wu J, Zhu J, Chen L, Zhu F, Li C et al (2013) Cathepsin B contributes to autophagy-related 7 (Atg7)-induced nod-like receptor 3 (NLRP3)-dependent proinflammatory response and aggravates lipotoxicity in rat insulinoma cell line. J Biol Chem 288:30094–30104
Liu Y, Schiff M, Czymmek K, Talloczy Z, Levine B, Dinesh-Kumar SP (2005) Autophagy regulates programmed cell death during the plant innate immune response. Cell 121:567–577
Madeo F, Tavernarakis N, Kroemer G (2010) Can autophagy promote longevity? Nat Cell Biol 12:842–846
Matsumoto N, Ezaki J, Komatsu M, Takahashi K, Mineki R, Taka H, Kikkawa M, Fujimura T, Takeda-Ezaki M, Ueno T et al (2008) Comprehensive proteomics analysis of autophagy-deficient mouse liver. Biochem Biophys Res Commun 368:643–649
McCoy F, Hurwitz J, McTavish N, Paul I, Barnes C, O’Hagan B, Odrzywol K, Murray J, Longley D, McKerr G et al (2010) Obatoclax induces Atg7-dependent autophagy independent of beclin-1 and BAX/BAK. Cell Death Dis 1:e108
Metzger S, Saukko M, Van Che H, Tong L, Puder Y, Riess O, Nguyen HP (2010) Age at onset in Huntington’s disease is modified by the autophagy pathway: implication of the V471A polymorphism in Atg7. Hum Genet 128:453–459
Metzger S, Walter C, Riess O, Roos RA, Nielsen JE, Craufurd D, Network RIotEHsD, Nguyen HP (2013) The V471A polymorphism in autophagy-related gene ATG7 modifies age at onset specifically in Italian Huntington disease patients. PLoS One 8:e68951
Michiels CF, Fransen P, De Munck DG, De Meyer GR, Martinet W (2015) Defective autophagy in vascular smooth muscle cells alters contractility and Ca(2)(+) homeostasis in mice. Am J Physiol Heart Circ Physiol 308:H557–H567
Minina EA, Sanchez-Vera V, Moschou PN, Suarez MF, Sundberg E, Weih M, Bozhkov PV (2013) Autophagy mediates caloric restriction-induced lifespan extension in Arabidopsis. Aging Cell 12:327–329
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147:728–741
Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, Simon AK (2010) Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci USA 107:832–837
Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, Stranks AJ, Glanville J, Knight S, Jacobsen SE et al (2011) The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med 208:455–467
Motori E, Puyal J, Toni N, Ghanem A, Angeloni C, Malaguti M, Cantelli-Forti G, Berninger B, Conzelmann KK, Gotz M et al (2013) Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab 18:844–859
Murrow L, Debnath J (2013) Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol 8:105–137
Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y (2009) Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 10:458–467
Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC (2013) Abeta secretion and plaque formation depend on autophagy. Cell Rep 5:61–69
Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S (2009) Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461:654–658
Nishiumi S, Fujishima Y, Inoue J, Masuda A, Azuma T, Yoshida M (2012) Autophagy in the intestinal epithelium is not involved in the pathogenesis of intestinal tumors. Biochem Biophys Res Commun 421:768–772
Nussenzweig SC, Verma S, Finkel T (2015) The role of autophagy in vascular biology. Circ Res 116:480–488
Pattison JS, Osinska H, Robbins J (2011) Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res 109:151–160
Rabinowitz JD, White E (2010) Autophagy and metabolism. Science 330:1344–1348
Rasmussen SB, Horan KA, Holm CK, Stranks AJ, Mettenleiter TC, Simon AK, Jensen SB, Rixon FJ, He B, Paludan SR (2011) Activation of autophagy by alpha-herpes viruses in myeloid cells is mediated by cytoplasmic viral DNA through a mechanism dependent on stimulator of IFN genes. J Immunol 187:5268–5276
Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW et al (2007) Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 39:596–604
Rossiter H, Konig U, Barresi C, Buchberger M, Ghannadan M, Zhang CF, Mlitz V, Gmeiner R, Sukseree S, Fodinger D et al (2013) Epidermal keratinocytes form a functional skin barrier in the absence of Atg7 dependent autophagy. J Dermatol Sci 71:67–75
Sagnier S, Daussy CF, Borel S, Robert-Hebmann V, Faure M, Blanchet FP, Beaumelle B, Biard-Piechaczyk M, Espert L (2015) Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J Virol 89:615–625
Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB (2011) Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 53:406–414
Singh SB, Davis AS, Taylor GA, Deretic V (2006) Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313:1438–1441
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009a) Autophagy regulates lipid metabolism. Nature 458:1131–1135
Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ (2009b) Autophagy regulates adipose mass and differentiation in mice. J Clin Investig 119:3329–3339
Singh KK, Lovren F, Pan Y, Quan A, Ramadan A, Matkar PN, Ehsan M, Sandhu P, Mantella LE, Gupta N et al (2015) The essential autophagy gene ATG7 modulates organ fibrosis via regulation of endothelial-to-mesenchymal transition. J Biol Chem 290:2547–2559
Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH (2008) Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48:1054–1061
Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q (2008) Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA 105:19211–19216
Tashiro Y, Urushitani M, Inoue H, Koike M, Uchiyama Y, Komatsu M, Tanaka K, Yamazaki M, Abe M, Misawa H et al (2012) Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem 287:42984–42994
Teplova I, Lozy F, Price S, Singh S, Barnard N, Cardiff RD, Birge RB, Karantza V (2013) ATG proteins mediate efferocytosis and suppress inflammation in mammary involution. Autophagy 9:459–475
Thompson AR, Doelling JH, Suttangkakul A, Vierstra RD (2005) Autophagic nutrient recycling in Arabidopsis directed by the ATG8 and ATG12 conjugation pathways. Plant Physiol 138:2097–2110
Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs CA, Wu XS, Rovira II, Fergusson MM, Weigert R et al (2013) Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med 19:1281–1287
Virgin HW, Levine B (2009) Autophagy genes in immunity. Nat Immunol 10:461–470
Walls KC, Ghosh AP, Franklin AV, Klocke BJ, Ballestas M, Shacka JJ, Zhang J, Roth KA (2010) Lysosome dysfunction triggers Atg7-dependent neural apoptosis. J Biol Chem 285:10497–10507
Wang N, Zimmerman K, Raab RW, McKown RL, Hutnik CM, Talla V, Tyler MFt, Lee JK, Laurie GW (2013) Lacritin rescues stressed epithelia via rapid forkhead box O3 (FOXO3)-associated autophagy that restores metabolism. J Biol Chem 288:18146–18161
Wittkopf N, Gunther C, Martini E, Waldner M, Amann KU, Neurath MF, Becker C (2012) Lack of intestinal epithelial atg7 affects Paneth cell granule formation but does not compromise immune homeostasis in the gut. Clin Dev Immunol 2012:278059
Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, Rovira II, Gutkind S, Daniels MP, Komatsu M et al (2009) Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY) 1:425–437
Xue LY, Chiu SM, Oleinick NL (2010) Atg7 deficiency increases resistance of MCF-7 human breast cancer cells to photodynamic therapy. Autophagy 6:248–255
Yang C, Shogren KL, Goyal R, Bravo D, Yaszemski MJ, Maran A (2013) RNA-dependent protein kinase is essential for 2-methoxyestradiol-induced autophagy in osteosarcoma cells. PLoS One 8:e59406
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304:1500–1502
Zeng Y, Huo G, Mo Y, Wang W, Chen H (2015) MIR137 regulates starvation-induced autophagy by targeting ATG7. J Mol Neurosci 56:815–821
Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S (2009a) Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci USA 106:19860–19865
Zhang J, Randall MS, Loyd MR, Dorsey FC, Kundu M, Cleveland JL, Ney PA (2009b) Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood 114:157–164
Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG (2010) Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol 12:665–675
Zhao Y, Zhang CF, Rossiter H, Eckhart L, Konig U, Karner S, Mildner M, Bochkov VN, Tschachler E, Gruber F (2013) Autophagy is induced by UVA and promotes removal of oxidized phospholipids and protein aggregates in epidermal keratinocytes. J Investig Dermatol 133:1629–1637
Zhu K, Dunner K Jr, McConkey DJ (2010) Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 29:451–462
Zhu L, Du H, Shi M, Chen Z, Hang J (2013) ATG7 deficiency promote apoptotic death induced by cisplatin in human esophageal squamous cell carcinoma cells. Bull Cancer 100:15–21
Zhuo C, Ji Y, Chen Z, Kitazato K, Xiang Y, Zhong M, Wang Q, Pei Y, Ju H, Wang Y (2013) Proteomics analysis of autophagy-deficient Atg7−/− MEFs reveals a close relationship between F-actin and autophagy. Biochem Biophys Res Commun 437:482–488
ACKNOWLEDGEMENTS
The author is grateful for the support from the National Institutes of Health (NIH) Intramural Program and the Leducq Foundation. The author thanks the NIH Fellows Editorial Board and Cindy Clark, NIH Library Writing Center, for manuscript editing assistance, and apologizes to colleagues whose work could not be cited or fully discussed due to space limitations.
COMPLIANCE WITH ETHICS GUIDELINES
Jianhua Xiong declares that he has no conflict of interest. This article does not contain any studies with human or animal subjects performed by the author.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Xiong, J. Atg7 in development and disease: panacea or Pandora’s Box?. Protein Cell 6, 722–734 (2015). https://doi.org/10.1007/s13238-015-0195-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-015-0195-8