
Abstract
Medical or surgical castration serves as the backbone of systemic therapy for advanced and metastatic prostate cancer, taking advantage of the importance of androgen signaling in this disease. Unfortunately, resistance to castration emerges almost universally. Despite the development and approval of new and more potent androgen synthesis inhibitors and androgen receptor (AR) antagonists, prostate cancers continue to develop resistance to these therapeutics, while often maintaining their dependence on the AR signaling axis. This highlights the need for innovative therapeutic approaches that aim to continue disrupting AR downstream signaling but are orthogonal to directly targeting the AR itself. In this review, we discuss the preclinical research that has been done, as well as clinical trials for prostate cancer, on inhibiting several important families of AR-interacting proteins, including chaperones (such as heat shock protein 90 (HSP90) and FKBP52), pioneer factors (including forkhead box protein A1 (FOXA1) and GATA-2), and AR transcriptional coregulators such as the p160 steroid receptor coactivators (SRCs) SRC-1, SRC-2, SRC-3, as well as lysine deacetylases (KDACs) and lysine acetyltransferases (KATs). Researching the effect of—and developing new therapeutic agents that target—the AR signaling axis is critical to advancing our understanding of prostate cancer biology, to continue to improve treatments for prostate cancer and for overcoming castration resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite recent advances in treatment, prostate cancer—the second most commonly diagnosed cancer in men worldwide—remains a leading cause of cancer-related death in men, with over 220,800 new cases and 27,540 deaths reported in the USA annually [1–3]. Prostate adenocarcinoma (PCa), as a primarily endocrine disease, is frequently dependent (at least initially) on androgens to maintain growth and viability of the tumor. This dependence on androgens provides a therapeutic opportunity. Blocking androgen production or action in PCa has served as the basis for PCa therapeutics for over 70 years [4]. In the current standard of treatment, PCa that has recurred following surgical resection or radiation therapy is initially treated via reduction of gonadal androgen secretion, which has been well-documented to have clinical activity [4]. This is accomplished either by surgical castration (orchiectomy) or by chemical castration through the use of gonadotropin-releasing hormone (GnRH) analogs. Despite an initial response to gonadal hormone suppression, PCa cells almost inevitably overcome this approach, recurring as what had been previously called a “hormone-refractory” or “androgen-independent” disease. These terms have largely fallen out of use in favor of “castration-resistant prostate cancer” (CRPC), as extensive studies have demonstrated that, frequently, these recurring cancers are neither hormone-refractory nor androgen-independent. Instead, many cancers have adapted to castrate conditions but—through a number of possible mechanisms—still largely depend on the androgen receptor (AR) signaling axis and in some cases can still respond to androgens [5].
Androgens, including androstenedione, but especially testosterone and dihydrotestosterone (DHT), are particularly important for the maintenance of the normal prostate and, by extension, for the proliferation of PCa (especially early PCa). Androgens, similar to other steroid hormones, are initially derived from cholesterol and are gradually converted by enzymes into their chemically distinct structures. Testosterone, which is synthesized primarily in the testes but can also be synthesized from adrenal precursors, circulates in serum until it diffuses into cells where it is converted by 5α-reductase into its significantly more active derivative, DHT [6]. Of the three isoforms, type II 5α-reductase is the dominant isoform in normal prostate and benign prostate hyperplasia (BPH), while type I 5α-reductase is increased in PCa [7]. Current therapeutic approaches include depriving PCa of testosterone and DHT by blocking 5α-reductase via finasteride (which inhibits isoforms II and III) and dutasteride (which inhibits isoforms I and II). Although long-term studies demonstrate that these inhibitors can significantly decrease PCa incidence [8], their preventative effect is limited to low-grade cancers, thus decreasing the value of these agents for averting PCa deaths [9–11].
Structure and Function of AR and Its Role in PCa
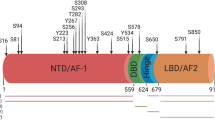
The full-length AR protein can be divided into several functional domains (Fig. 1): the amino-terminal domain, which contains an AR transcriptional activation function; the DNA-binding domain (DBD); a hinge region; and the carboxy-terminal ligand-binding domain (LBD). The AR signaling axis primarily follows the classical mechanism of nuclear receptor signaling (Fig. 2), which relies upon a large network of interacting proteins—and thus offers a number of potential targets beyond AR itself for therapeutic intervention. When not bound to its ligand, the AR protein is primarily localized in the cytosol, sequestered, and stabilized by chaperone proteins. Upon binding of ligand, the AR undergoes a conformational change and translocates to the nucleus (likely via a dynein microtubule-depended fashion) [12]. In the nucleus, pioneer factors and histone modifiers prime the chromatin for transcription factor binding, allowing the AR to bind as a dimer to androgen response elements (AREs) in the promoter or enhancer regions of its target genes such as transmembrane protease serine 2 (TMPRSS2), prostate-specific antigen (PSA), and many others. Once bound to chromatin, the AR protein recruits additional coregulators and transcription machinery components that ultimately lead to activation of a transcriptional network that promotes growth and survival.

Comparison of the full-length AR and two AR variants. The full-length AR is comprised of eight exons (a) which make up the amino-terminal domain (NTD), the DNA-binding domain (DBD), a hinge region, and the carboxy-terminal ligand-binding domain (LBD). The ARv7—also known as AR3—contains exons 1–3 (b), comprising the NTD and the DBD, followed by a short cryptic exon encoding 16 amino acids which are not present in the full-length AR. The LBD is missing in the ARv7, resulting in a constitutively active variant. The ARv567es (c) contains exons 1–4 but is missing exons 5–7. In addition, exon 8 has a translational frameshift

Representative cartoon of the canonical AR signaling axis in a normal prostate epithelial cell. Androgens diffuse through the plasma membrane into the cytosol, where they bind to the chaperone-sequestered AR and induce a conformational change, resulting in its nuclear translocation and dimerization. In the nucleus, pioneer factors such as GATA-2 and FoxA1 make the chromatin more accessible to other transcription factors and allow dimerized AR to bind to its hormone response elements on chromatin. Once bound, AR recruits coregulator proteins that help assemble the transcriptional machinery and result in the transcription of the AR-target genes
In addition to the full-length protein, AR variants (ARVs) have also been described (Fig. 1). One of the first alternative forms of the AR was observed in 1996, and—through antibody mapping—it was established that this form lacked the LBD [13, 14]. Although this form was proposed to be a proteolytic fragment of full-length AR, studies have established that ARVs can also arise through alternative messenger RNA (mRNA)—either derived from alternative splicing [15, 16] or chromosomal rearrangements [17]. Through the characterization of a PCa cell line that harbors one of these variants—the CWR22 PCa xenograft-derived line—it was discovered that ARVs lacking the LBD are constitutively nuclear and are able to restore part of the AR signaling activity in a ligand-independent manner [18]. In 2009, Hu et al. reported four cryptic exons (one of which had been discovered previously by Dehm et al. [16]). Hu et al. further reported that among the four cryptic exons, alternative splicing led to seven AR splice variants, all lacking the LBD. In addition to these seven variants, other groups—utilizing technologies such as 3′ and 5′ RACE—have identified several alternative splice variants, some containing the LBD [19, 20] and others lacking the LBD [21].
In contrast to its reliance on the AR signaling axis, PCa is resistant to many of the standard treatment modalities utilized against other solid malignancies. The notable exception to this is the taxane family of chemotherapeutics, which are active against several cancers, primarily through the inhibition of microtubule function in the cell. Taxane treatment has been observed to suppress nuclear translocation of the AR and, thus, its transcriptional activity [22, 23]. Importantly, it has been reported that some ARVs may elude this effect of microtubule inhibitors, as they may lack the microtubule-binding domain present in full-length AR [24, 25]. Historically, docetaxel and cabazitaxel have been used to treat advanced stage PCas [26, 27] after hormonal therapies have lost their activity. However, very recent data suggest that early addition of docetaxel with gonadal suppression may significantly increase patient survival compared to later use of the same chemotherapy [28]. It remains to be determined whether this synergistic effect is due to a more potent suppression of AR signaling achieved by the combination regimen [29].
Diagnosis, Treatment, and Current Understanding of Resistance Mechanisms
Patients diagnosed with localized PCa will generally undergo surgical resection or radiation therapy, upon which serum levels of PSA (or gene name KLK3)—a gene product directly induced by active AR—will be monitored as a biochemical marker for PCa recurrence. Rising level of PSA after prostatectomy or irradiation can serve as an early indicator of recurrence. Once PCa becomes locally advanced or metastatic, the next stage in treatment is to start the patient on GnRH analogs to suppress gonadal androgen synthesis. The goal of first-line hormonal treatment is the suppression of gonadal androgens (with a target level of serum testosterone below 50 ng/dL). Despite this suppression of serum testosterone levels, some cancers maintain intratumoral androgen levels via local synthesis and metabolism [7, 30, 31]. Alternatively, some PCas have been observed to overexpress AR mRNA (frequently via amplification of the AR gene) and are sensitized to lower levels of androgens. These molecular alterations in the AR axis increase sensitivity to hormone and allow resumption of PCa cell proliferation despite suboptimal levels of androgen in the serum [32, 33]. Once patient exhibits resistance to initial androgen suppression therapies, AR antagonists are given to directly inhibit the AR. Flutamide, followed by bicalutamide (Casodex), are among the first-generation AR antagonists which inhibit AR by competitively binding to its LBD and disrupting activation [34, 35]. Sadly, restoration of PCa growth and AR signaling still occur (Fig. 3).

Representative cartoon of currently FDA-approved prostate cancer therapies, their mechanisms of action, and proposed mechanisms of resistance. Therapies include GnRH analogs that suppress gonadal androgen synthesis, small molecule androgen synthesis inhibitors (e.g., abiraterone) that can block enzymatic steps required for androgen steroidogenesis, androgen receptor antagonists (e.g., enzalutamide) that bind to the AR LBD and prevent activation, and microtubule inhibitors that can disrupt the cell cycle and also may prevent AR translocation into the nucleus. Mechanisms of resistance include intratumoral androgen production to locally generate ligand for the AR, overexpression of AR (frequently through amplification of the AR gene) to enhance sensitivity to low levels of ligand, AR LBD mutations to relax stringency for the activating ligand, ARVs which are constitutively active, and other nuclear receptors substituting for the AR
In some patients, withdrawing treatment of the first-generation AR antagonist results in a decrease of circulating PSA [36], suggesting that the drug was functioning as an AR agonist. Point mutations in the LBD of the AR have been reported to confer promiscuity or relaxed stringency for ligands [37], for example, in AR LBD mutant W741L, bicalutamide switches from acting as an AR antagonist to acting as an agonist and driving AR signaling [38, 39]. In humans, the AR gene is located on the X-chromosome (Xq12); thus, there is only one copy of the AR gene in the male germline. As previously mentioned, AR functions as a homodimer. Consequently, in cells that manage to acquire an AR gene mutation in the single AR gene copy, every molecule of AR will harbor this mutation, resulting in homodimers of mutant AR and greater oncogenic potential. Alternatively and in addition to point mutations, overexpression of AR (frequently via gene amplification) can also result in first-generation AR antagonist-to-agonist conversion [40].
Unfortunately, in patients with metastatic PCa, the reduction in serum PSA after first-line hormonal therapy is almost inevitably short-lived. When PSA levels begin to rise, castration-resistant disease has emerged and needs to be treated with second-line hormonal therapies. Currently, these include (1) abiraterone acetate (approved by FDA in 2011)—a selective and irreversible inhibitor of 17-α-hydroxylase/C17-20-lyase (CYP17A1), an enzyme that catalyzes the sequential reactions of the conversion of pregnenolone and progesterone to their 17-α-hydroxy derivatives and the subsequent formation of dehydroepiandrosterone (DHEA) and androstenedione [41–43], and (2) enzalutamide (MDV3100) (approved by FDA in 2012)—a second-generation AR antagonist that can prevent AR from binding its natural ligands: testosterone or 5α-dihydrotestosterone [44]. Although abiraterone is able to suppress circulating androgen to near undetectable levels [45, 46] and enzalutamide is probably the most potent FDA approved anti-androgen currently available (as it is clearly active in bicalutamide-refractory patients), there is significant evidence that many PCas remain dependent on the AR signaling axis for growth even after treatment with these agents.
Recently, mechanisms of resistance to second-generation AR antagonists, such as enzalutamide, have also been described. Stemming from the experiences with resistance to bicalutamide, efforts were made to screen for potential mutations that might confer resistance to enzalutamide. In 2013, an in vitro screen for potential resistance discovered a point mutation in the LBD of the AR (F876L) that blunts the effect of enzalutamide and allows proliferation of cancer cells [47]. Two studies have shown this same acquired somatic point mutation in PCa cells from patients treated with different AR antagonists: one who was treated with enzalutamide [48] and the other with a different second-generation AR antagonist—ARN-509 [49]. In addition to point mutations, an AR-independent mechanism for resistance has also been described: a cellular switch by which the AR signaling axis is maintained by an upregulation of the glucocorticoid receptor—which recognizes and binds the same hormone response element DNA motif as AR [50, 51]. Resistance to CYP17A1 inhibitors, such as abiraterone, has also been reported in CRPC patient-derived xenograft (PDX) models, through increased expression of the CYP17A1 enzyme, as well as through induction of ARVs [52].
ARVs (Fig. 1) are a critical mechanism of resistance observed in PCa [53]; they can occur in response to a variety of treatments and pose a challenge for the management of PCa [54, 55]. Transgenic mice engineered to express ARV in the prostate exhibit epithelial hyperplasia by 16 weeks and invasive PCa by 1 year of age, as well as expression of genes critical for tumor initiation and progression [56]. Regardless of the origin and heterogeneity of these splice variants, they provide a hurdle for our existing therapies: Because many of these ARVs lack a LBD, neither treatments targeting hormone production nor AR antagonists targeted toward the LBD are effective against these ARVs. In fact, several studies have noted increased presence of ARVs in PCa cells resistant to enzalutamide [57] and subsequently was associated with shorter PSA progression-free as well as less overall survival in patient samples with PCa resistant to abiraterone or enzalutamide [58–60].
Due to the transient success of abiraterone and enzalutamide, efforts to develop newer and more potent AR antagonists and androgen synthesis inhibitors are ongoing. Novel AR-directed approaches include ARN-509 [49] and galeterone (a novel CYP17 inhibitor [61] which is in clinical trials for CRPC, in particular AR variant-expressing CRPC); their ability to further advance PCa treatment remains to be seen. In addition, AKR1C3, an enzyme that is overexpressed and implicated in driving androgen synthesis in advanced PCa, has been studied for potential therapeutic value [62, 63]. One attractive feature of inhibition of AKR1C3 over abiraterone is that inhibition of AKR1C3 does not block glucocorticoid synthesis. Small molecule inhibitors for AKR1C3 have already been developed, but their effect remains to be tested in clinical trials to assess for clinical benefit in abiraterone-refractory patients [64].
Future Direction of PCa Molecular Target Discovery and Drug Design
AR is an excellent molecule to target therapeutically, as its inhibition in PCa patients has limited risk of morbidity (although there are some serious considerations with regard to quality of life) and the presence of a defined LBD has allowed the development and FDA approval of first- and now second-generation AR antagonists. Unfortunately, as outlined above, mechanisms of resistance to therapies targeting the AR LBD are commonly observed, suggesting that new strategies are needed. Several newer studies aim to design inhibitors capable of blocking required conformational changes of the AR or protein-protein interactions between the AR and coregulators [65].
It is worth contemplating what might constitute an “ideal” candidate molecule to target therapeutically. A good candidate for development of a small molecule inhibitor should exhibit favorable biological activity and favorable technical feasibility [66]. In Oncology, biological activity should be assessed through assays that clearly indicate that disruption or degradation of the target slows or kills cancer cells and that disruption or degradation of the target will yield favorable results in patients. In addition to promising data for the biological importance of the target, the technical feasibility of drugging the molecule (“druggability” of the target) must be considered. Traditionally, this has focused on taking advantage of ligand-binding pockets (like the LBD in the AR), especially if structural data—such as X-ray crystals or NMR—is available to help identify targetable regions and guide development [66]. Recently, however, much focus has been placed on the development of inhibitors of protein-protein interactions. With increased understanding of macromolecular binding energetics and advances in assessing structural data, protein surfaces that were previously considered ill-fit for small molecule binding have been reassessed [67, 68]. Furthermore, research into targetable delivery systems increases the feasibility of using small molecule inhibitors against candidate targets that pose challenges such as toxicity in other tissues, or less favorable bioavailability. One such approach in prostate cancer takes advantage of the prostate-specific expression of prostate-specific membrane antigen (PSMA). PSMA has been successfully utilized to image PCa in mice [69] and has been coupled with nanoparticles to deliver targeted therapeutics in animal models [70], potentially providing a mechanism to limit off-target effects of small molecule inhibitors targeting AR-interacting proteins.
Disrupting or inhibiting the AR signaling axis—either mediated by full-length AR or ARVs—by targeting the AR-interacting proteins may prove to be a viable strategy to supplement traditional therapeutics in order to enhance their activity, delay the emergence of resistance, or even treat resistant disease. The numerous escape and reactivation mechanisms observed in PCa cells allow them to maintain the benefit of a functioning AR signaling axis and utilize it for cell growth. It follows then that new treatments will continue to target this signaling axis, while aiming to overcome the currently known routes to de novo or acquired resistance. Although efforts are being made to target other previously considered “less druggable” domains of the AR, including the N-terminal domain antagonist EPI-001 [65, 71], the shifting views on what constitutes druggability of proteins, technological advances in viewing and using structural data for inhibitor development, along with added specificity and delivery mechanisms from targeted therapies such as PSMA nanoparticles provide the opportunity to expand and capitalize on the dependence of AR for other proteins, in addition to directly targeting androgens and the AR itself.
Chaperones
In theory, targeting chaperones or heat shock proteins (including heat shock protein 90 (HSP90), HSP70, FKBP52, and p23) seems to have high therapeutic potential, as successful inhibition would result in misfolding or degradation of their client proteins. Although early biochemical studies in yeast suggested that inhibiting chaperone proteins as a way to target the AR axis might hold promise [72], so far this approach has been unsuccessful in PCa either due to unacceptable toxicity of these inhibitors or emergence of novel ARVs.
For over 30 years, chaperone proteins have been known to regulate maturation, activation, and stability of ligand-free steroid receptors such as AR [73]. Although HSP90 was the first identified chaperone protein for the AR, additional chaperone or cochaperone proteins have since been reported, including HSP70, FKBP52 (HSP56), and p23 [74, 75]. Stabilization by chaperones is vital to the ability of the AR to respond to ligand, as evidenced by observations in yeast that mutant forms of the HSP90 yeast homolog do not decrease overall levels of the AR but inhibit hormone-dependent transactivation, even in the presence of the potent synthetic androgen metribolome (R1881) [72]. This basic mechanism of action presents two general strategies for inhibition of chaperone activity: (1) inhibit the protein-protein interacting domains of the client protein with the chaperone and (2) inhibit the ATP-binding pocket to prevent release of the client protein. Of these, most work with chaperone inhibitors has followed the latter approach.
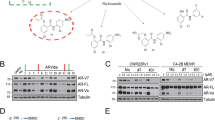
Given the evidence of Hsp90-mediated transactivation of AR, the feasibility of targeting HSP90 with small molecules has been examined extensively. And at least in vitro, some success has been observed. Geldanamycin, a well-established HSP90 inhibitor (via competitively interacting with its ATP-binding pocket), has been observed to decrease AR protein levels (Fig. 4a), as well as mRNA for the AR target gene PSA, even in the presence of R1881 in LNCaP PCa cells [76]. Despite this, geldanamycin and another major HSP90 inhibitor, 17-allylamino-17-demethoxygeldanamycin (17-AAG), have been investigated in vivo, and disappointingly, studies have so far failed to establish any clinically meaningful benefit. Geldanamycin was shown to result in unacceptable liver toxicity in animal models [77], while 17-AAG was discontinued in phase II clinical trials due to a failure to reduce serum PSA levels [78].

a Chaperone (e.g., Hsp90) inhibitors can disrupt AR protein stabilization or proper folding and result in degradation and/or dysfunction of the AR protein. b Pioneer factor (e.g., FoxA1, GATA2) inhibitors may prevent the AR from accessing response elements on chromatin or may interfere with the ability of AR to recruit coregulators needed for transcriptional activity. c p160 steroid receptor coactivator (SRC) inhibitors may prevent the recruitment of the transcriptional machinery components required for efficient transcription. d KAT and KDAC inhibitors may directly modulate post-translational modifications on the AR itself, reducing its activity or may interfere with chromatin remodeling to make the chromatin less amenable for active transcription
Despite these setbacks, interest in HSP90 inhibition remains, and several new inhibitors are being tested. One of these, ganetespib (STA-9090), was shown to have therapeutic activity against a number of PCa cell lines, while exhibiting significantly better safety and pharmacological profiles [79]. In clinical trials, single-agent use of ganetespib was unable to prolong progression-free survival in metastatic CRPC patients—however, combination therapy remains an option for future studies [80]. As highlighted earlier, ARVs has been established as a major mechanism for CRPC. Two inhibitors, geldanamycin and MJC13 (which block HSP90 and its cochaperone FKBP52, respectively), were demonstrated to be effective at blocking ligand-dependent AR activity but were unable to modulate ARv7 protein stability or transcriptional activity [81], suggesting that ARVs may contribute to resistance to HSP90 inhibitors in clinical settings.
Although inhibition of chaperone proteins with regards to their classical function may not prove to be an effective strategy, interest in the field remains as new functions of chaperones have emerged. Chaperones have been shown to have a potentially more direct role in the AR transcriptional program. Live cell imaging experiments have illustrated that chaperones, including HSP90, may help mediate the nuclear translocation and subnuclear localization of nuclear receptors [82]. Furthermore, it has been suggested that chaperones of the AR might also participate in the regulation of transcriptional complexes on chromatin [83] and that cochaperone p23 may actually help stabilize AR on its hormone response elements, potentiating AR-mediated transcription [84].
When taking into consideration some of the other key cancer-associated proteins and pathways (such as AKT [85]) that are stabilized by HSP90 and other chaperones, it is even more remarkable that HSP inhibitors failed to have effect in PCa clinical trials. However, the functions of chaperones for AR stabilization as well as subcellular trafficking and their potential roles as part of the transcriptional machinery highlight their importance and potential as therapeutic targets. New efforts at targeting chaperones may enhance specificity for the cancer and ameliorate toxicity by focusing on inhibition of the protein-protein interaction between AR and specific chaperones or by focusing on inhibiting the AR axis by attacking the transcriptional or trafficking roles of chaperones. It remains to be seen if these strategies prove to be more selective or less amenable to ARV-mediated resistance.
Pioneer Factors
Pioneer factors are critical for priming the chromatin to allow AR to bind and become transcriptionally active, and thus, targeting pioneer factors could have high therapeutic potential in PCa. Two classes of proteins have been identified as key pioneer factors for AR recruitment to chromatin: forkhead box A (FOXAs) (specifically FOXA1) and GATAs (especially GATA-2). These factors are the earliest chromatin-binding factors, and unlike other transcription factors, they are able to bind condensed chromatin [86]. FOXA1 contains a winged-helix structure (similar to the DNA-binding domain of linker histone, H1) that allows it to bind the major groove of DNA [87] and makes the chromatin more accessible to other transcription factors to initiate productive transcription [86]. Several mechanisms for inhibition can be envisioned to target their role in the AR signaling axis: Inhibitors could degrade FOXA1 and/or GATA-2, prevent interaction of FOXA1 and/or GATA-2 with chromatin, or interfere with the ability of FOXA1/GATA-2 to interact with the AR. Few small molecule inhibitors for these proteins have been investigated; therefore, all of these avenues continue to have potential.
The importance of FOXA1 in allowing the AR to bind chromatin and initiate transcription was first described in 2003, when experiments showed that FOXA1 binding to the core KLK3 enhancer preceded AR binding and that overexpressing mutant forms of FOXA1 significantly ablated KLK3 transcription [88]. Evidence for the relationship and dependence of the AR on FOXA1 was further supported by experiments demonstrating the colocalization of the two proteins on chromatin [89], including in castration-resistant PCa cells that exhibit a novel AR transcriptional program [90]. Elevated FOXA1 expression levels were associated with poor prognostic features (higher Gleason scores) and clinical outcomes (early biochemical recurrence) in three studies [91–93]. In one study, overexpression of FOXA1 resulted in broad increases in AR binding on chromatin, and that these new AR binding sites are primarily unresponsive to DHT stimulation [93]. These data raise the hypothesis that patients with CRPC may also benefit from inhibiting FOXA1 levels and/or activity.
However, the pioneering effect of FOXA1 for AR is far more complex, and important questions about the clinical value of inhibiting FOXA1 to treat PCa have been raised by other studies reporting somewhat contradicting results. Specifically, it has been proposed that FOXA1 is not required for AR-chromatin interaction at canonical AREs but is instrumental in recruiting AR to low-affinity half-AREs by opening local chromatin [94]. In the presence of high levels of FOXA1, these open chromatin regions serve as reservoirs that retain AR via abundant half-AREs and decrease availability of AR for full-ARE sites. Thus, suppressing FOXA1 levels can release AR to bind new full-AREs across the genome, resulting in substantial reprogramming of the AR cistrome and AR-dependent gene expression, even in the absence of androgen [94, 95]. As a result of the above AR reprogramming and alternative hormonal response, these studies have also reported that FOXA1 downregulation activates a castration-resistant AR transcriptional program [94], while lower FoxA1 levels are significantly associated with increased metastatic potential and poor prognosis [95], and that silencing FOXA1, surprisingly, results in increased cell progression to S phase [95]. These data contradict, to some degree, the other studies discussed in the previous paragraph and raise some concern about the potential usefulness of FOXA1 inhibition for targeting the AR signaling axis and inhibiting PCa growth. To further complicate matters, the AR and the ARv7 exhibit, at least partially, different propensities for FOXA1 dependency, suggesting that targeting FOXA1 may not overcome resistance mediated by AR splice variants [96].
Another pioneer factor that can serve as a candidate target for the inhibition of the AR signaling axis is GATA-2. Similar to FOXA1, GATA-2 has also been shown to colocalize with the AR on chromatin, with enhanced binding of GATA-2 at AREs in response to androgen [89]. Increased GATA-2 expression has also been correlated with poor prognosis and more aggressive cancers [97, 98] and has been implicated as a potential driving gene in metastatic PCa, as evidenced in patient expression data as well as cell phenotypic assessments, which demonstrated that silencing GATA-2 decreases cell migration, tissue invasion, and focal adhesion [99]. Recently, GATA-2 has been garnering more attention as a key player in AR-mediated transcription.
GATA-2 is another pioneer factor that regulates AR function and, unlike FOXA1, also promotes AR expression. GATA-2 silencing resulted in decrease in both AR mRNA and protein levels in androgen-free and R1881-treated cells [100], diminishing the expression of not only full-length AR but also ARV [98]. This effect on full-length AR and ARVs was recapitulated by the small molecule GATA-2 inhibitor K-7174 [98]. Simultaneously, GATA2 is necessary for optimal transcriptional activity of both full-length AR and ARV, as demonstrated by the inability of exogenously transfected ARs (both full-length AR and ARV) to drive transcription in the absence of GATA-2 [98]. As ARVs have emerged as a mechanism of CRPC resistance to androgen synthesis inhibitors, AR antagonists and even, potentially, chaperone inhibitors, the finding that inhibition of GATA-2 may not only target the full-length AR signaling axis but also decreases the expression and function of ARVs is critical. Interestingly, in studies looking at ETS factor reprogramming of the AR after deletion of PTEN (which is common in PCa), there is an even greater number of new AR binding sites on chromatin that contain the GATA-2 DNA-binding motif [101]. GATA-2 has been reported to regulate IGF-2 expression in PCa cells, resulting in the downstream activation of AR-independent genes vital for chemoresistance [102]. Taken together, these findings suggest a potential critical role for GATA-2 in both AR-dependent and AR-independent signaling and thus make it an attractive and promising candidate for PCa therapeutics.
AR Transcriptional Partners
It is important to note that AR itself has no intrinsic enzymatic activity; instead, it serves as a scaffold—recruiting various protein machineries that can modify chromatin and result in active transcription. AR-interacting partners on the chromatin represent an exciting pool of targets for innovative therapeutics either through inhibiting their activity or by preventing their recruitment to the AR. Although numerous proteins are involved in the initiation of transcription, three categories of proteins stand out as potentially offering some specificity for PCa over the ubiquitous and required transcription machinery in normal tissues and cells: the p160 family of steroid receptor coactivators (SRCs), lysine acetyltransferases (KATs), and lysine deacetylases (KDACs).
The p160 Steroid Receptor Coactivators
The p160 family of steroid receptor coactivators, including steroid receptor coactivator-1 (SRC-1, or NCOA1, RIP160), steroid receptor coactivator-2 (SRC-2, or NCOA2, GRIP1, TIF2), and steroid receptor coactivator-3 (SRC-3, or NCOA3, AIB1, RAC3), have been well established to play an important role in endocrine-related cancers, including ovarian [103], breast [104], endometrium [105], and prostate [98, 106–109]. The p160 SRCs have also been implicated in other diseases, as well as in lipid, carbohydrate, amino acid, and drug metabolism (reviewed in [110] and [111]). As the SRCs are involved in a number of diseases and cancers, targeting these proteins may have high therapeutic potential. One of the key challenges for successful inhibition, however, will be finding a therapeutic window such that the SRC levels are low enough to benefit the patient (Fig. 4c) but does not impact the important transcriptional roles played by the SRCs in normal tissues and cells.
The p160 SRCs are best known for their eponymous functions as coactivators for steroid receptors (including AR, estrogen receptor, progesterone receptor, mineralocorticoid receptor, glucocorticoid receptor (reviewed in [112]) but have also been shown to coactivate a variety of other transcription factors including NF-κB, TAT, and AP-1 among others [113–116]. Among the conserved features in the p160 family of SRCs are a basic helix-loop-helix Per/ARNT/Sim (bHLH-PAS) domain through which it can mediate various protein-protein interactions. They also contain three LxxLL motifs, which serve to recognize and bind activated steroid hormone receptors. The C terminus of the SRCs contains two activation domains which can recruit CREB-binding protein (CBP) and p300, as well as other coactivators and histone modifiers, such as CARM-1 and PRMT1 [117–119]. These features provide the backbone for the many interactions and activities attributed to the SRCs.
The role of the SRCs in PCa has been investigated for well over a decade, with one of the first studies implicating the SRCs arising in 2001. Patients with PCa that recurred after androgen deprivation therapy were shown to have elevated expression of SRC-1 and SRC-2, which is sufficient to potentiate AR signaling despite suboptimal levels of androgen [38]. Since then, overexpression of SRC-1, SRC-2, and SRC-3 mRNA has been observed in patients with primary PCa as well as patients treated with androgen deprivation therapy, although there is some heterogeneity among the findings [120]. This has been recapitulated recently in an expression dataset of patients with primary and metastatic PCas, which show increased levels of overexpression of the SRCs as disease progresses [121]. This is emphasized through the observation that the p160 SRCs have been shown to coactivate the AR not only with androgens but even in the presence of partial agonists [122].
SRC-1 has been shown to be important in both ligand-dependent PCas and CRPCs that retain expression of the AR [123]. SRC-1 has also been proposed to play a role in mediating resistance after androgen deprivation therapy [38]. Perhaps the most well-studied p160 SRC in PCa is SRC-2. SRC-2 has been shown to have a strong relationship with androgens and the AR itself; a study that profiled SRC-2 expression in cell lines and patient samples in normal and androgen deprivation conditions showed that SRC-2 mRNA and protein are increased in PCa cells in the absence of androgens and that SRC-2 overexpression is correlated with biochemical recurrence in patients [124]. Recent studies have shown that increased mRNA levels of SRC-2 (frequently associated with NCOA2 gene amplification) may occur in up to 20 % of primary and 63 % of metastatic tumors [121]. In mouse models, SRC-2 has been shown to be induced upon castration and, in this model, has been demonstrated to be critical for the development of CRPC [108].
SRC-3 has also been shown to play an important role in the progression of PCa. Although SRC-3 has been shown to be upregulated at the mRNA level in patients with PCa [121], the number of patients with increased mRNA is unimpressive when compared with SRC-1 or SRC-2. However, immunohistochemistry for SRC-3 in patient tissue samples has demonstrated frequent overexpression at the protein level and has associated it with higher grade prostate tumors as well as worse overall survival [125]. This finding suggests that SRC-3 may be extensively regulated at the post-translational level in PCa. For example, the E3 ubiquitin ligase adaptor speckle-type poxvirus and zinc finger (POZ) domain protein (SPOP) binds SRC-3 and promotes its ubiquitination and proteolysis. Mutations in the SPOP substrate-binding cleft are frequent in human PCas and interfere with SRC-3 ubiquitination and degradation, resulting in higher protein levels [126, 127]. In vitro, SRC-3 has been shown to be required for PCa cell proliferation and survival [107]. SRC-3 has also been implicated as positively regulating genes in the insulin-like growth factor/AKT signaling pathway in PCa cells, resulting in increased cell proliferation [128]. In addition to playing a role in cell proliferation, SRC-3 has also been shown to participate in driving a more metastatic-like program in PCa cells. In this study, SRC-3 was associated with increased cell migration and invasion by regulating matrix metalloproteinases [129]. The importance of SRC-3 (and SRCs in general) in PCa was highlighted in a study of the impact of SRC-1 knockout in a prostate tumorigenesis model in mice. The authors observed an increase in SRC-3 expression during prostate tumorigenesis, which they explained as a possible compensatory effect that allows cancer formation despite the loss of SRC-1 [130]. This is an important concept, as the SRCs (as well as other proteins that have structurally similar family members) may be able to compensate for each other, and thus, efforts to inhibit SRCs may need to be directed toward inhibiting all three rather than specifically targeting only one member.
Evidence for targeting the SRCs to inhibit the AR signaling axis does exist: Peptide-mediated targeted inhibition of the p160 coactivator interface on the AR itself results in a loss of AR activity in both androgen-sensitive, as well as castration-resistant PCa cells [115]. Most nuclear receptors, through their AF-2 domain (which is typically located on the carboxy-terminus), interact with the LxxLL motifs on the p160 SRCs. One study, investigating the relationship between SRC-1 and the AR, mutated the LxxLL domain on SRC-1, rendering that domain nonfunctional. Surprisingly, the mutant SRC-1 was still able to activate the AR and potentiate AR signaling. The authors discovered that SRC-1 can interact with the glutamate-rich region located in the amino-terminal AF-1 domain on the AR [131]. Importantly, targeting the SRCs may also serve to interfere with ARV signaling, as evidenced through the observation that overexpressing FOXO1—which binds to the Tau-5 domain on the AR (conserved on both full-length and most characterized ARVs)—can compete for SRC-1 binding and abrogate ARV transcriptional activity [132].
Attempts at targeting the SRCs for therapeutic benefit have begun with a number of compounds proposed as inhibitors for the p160 family. Gossypol, verrucarin A, and the cardiac glycoside bufalin have all been shown to inhibit SRC activity and to have activity against breast cancer cells expressing high levels of SRCs [133–135]. Bufalin was shown to decrease SRC-3 protein levels, although it may also have additional mechanisms for SRC inhibition [134]. Bufalin inhibits all three SRCs, which may help avoid compensatory effects. In addition, it has been shown that the microRNA miR137 is able to target SRC-1, SRC-2, and SRC-3, providing an alternative option for combined inhibition of the entire class of p160 SRCs [136]. Although there is concern that the SRCs are required for normal cell function and that their inhibition may thus prove too toxic, they remain—at least in theory—a potentially valuable therapeutic target. Many of the mechanisms of resistance in PCa arise through reestablishing at least part of the AR signaling axis: increased expression of the AR, mutations in the LBD, functional ARVs, or the use of other steroid receptors. All of these mechanisms of resistance signal through the p160 SRCs; therefore, effectively inhibiting the SRCs would provide a relatively comprehensive blockade of most of the currently known resistance mechanisms in PCa.
KATs
Another group of AR transcriptional partners are the KATs (or histone acetyltransferases (HATs)). KATs, such as p300 and CBP, are, in general, proteins that acetylate lysine residues on histones or other proteins by utilizing acetyl-CoA as a donor. KATs, and the acetylation they catalyze, are generally considered to play an activating role in transcription. As a result, the therapeutic potential from their inhibition in cancer—if specificity can be achieved—could be high. For PCa, research into inhibition of KATs has mixed results: PCa-specific inhibition of p300 and CBP—which play key roles in the regulation of transcription—has been difficult to achieve due to the ubiquitous expression of these proteins. However, another KAT, Tat-interacting protein 60 (Tip60), seems to have high potential as a target for novel therapies.
The association between CBP or p300 and nuclear receptors, and the importance of these KATs for efficient nuclear receptor signaling and transcription, was first demonstrated in 1996. In this study, endogenous CBP and p300 was required for signaling of the retinoic acid and glucocorticoid receptors [137]. The role of KATs in PCa became even more prominent when it was shown that, in the absence of androgen, interleukin-6 (IL-6) was able to transactivate AR. In this setting, p300 mediated the transcriptional activity of un-liganded AR, and silencing p300 by RNAi was sufficient to mitigate this IL-6-activated AR [138]. The role of CBP was also established when it was shown that CBP enhances AR transcriptional activity in the presence of androgen, as well as in the presence of first-generation anti-androgens hydroxyflutamide and bicalutamide [139].
While inhibitors of KDACs (the deacetylating counterparts to KATs) have been widely studied, less work has been done on developing small molecule inhibitors for KATs. This may be, in part, due to the complexity in teasing apart differences among several closely related KATs. Although CBP and p300 are commonly grouped together (and are often written collectively as CBP/p300), differences between their activities have been documented: One study noted that inhibition of p300, but not CBP, resulted in increased cell death, better inhibition of AR function, and a decrease in cell invasion in PCa cells (Fig. 4d). This study used C646, an inhibitor for both CBP and p300, but refined the specificity of their results through RNAi [140]. Interestingly, this same study noted that the ability of AR to bind to chromatin was not affected by the use of C646. Another well-studied agent for KAT inhibition is curcumin—which is notable as a component of the spice turmeric and has been implicated as an anticancer agent for decades [141]. In vitro use of curcumin in prostate lines has resulted in decreased AR activity in both androgen-dependent as well as castration-resistant PCa cells [142]. Despite this, the ubiquitous expression of KATs such as CBP and p300 and their importance in normal tissue as well as cancer cells severely limit the therapeutic window in which to treat patients.
Tip60, a less well-known (compared to CBP and p300) acetyltransferase, has also been studied for its role in PCa and AR signaling. Tip60 was first shown to interact with the AR in 1999. In this screen, Tip60—originally known as an interacting protein with the human immunodeficiency virus protein TAT—was discovered to interact with full-length AR, as well as estrogen and progesterone receptors, and to coactivate transcription of liganded AR to similar degrees as CBP, p300, and SRC-1 [143]. After the initial report, Tip60 was discovered to acetylate lysines in the AR hinge region, which was determined to be required for its transactivating capacity for the AR [144]. Interestingly, Tip60 has also been shown to play key roles in maintenance of DNA integrity, as deletion of its acetyltransferase activity has been shown to result in DNA damage, halting of cell cycle progression, and induction of apoptosis [145, 146]. Analysis of the cellular localization of Tip60 has revealed that Tip60 has a relatively diffuse expression pattern throughout the cell in benign prostate tissue and in localized PCa. However, there is an accumulation of Tip60 protein after androgen withdrawal, and significant nuclear localization has been observed in CRPC disease—potentially contributing to the castration resistance phenotype [147].
Tip60, therefore, remains a potential target. Recently, several publications have investigated the use of Tip60 inhibitors for antagonizing AR signaling and PCa cell growth. NU9056 was identified through a screen for acetyltransferase inhibitors and was determined to have specificity for Tip60 over other KATs (with over 29-fold selectivity over p300). The same study showed that NU9056 decreased cell viability in a number of PCa cell lines with IC50s in the low micromolar range, suggesting that Tip60 may be a promising candidate target for novel therapeutics [148]. More recently, another group has developed and tested another small molecule inhibitor for Tip60—TH1834—reporting similar inhibition of cell viability when used in breast cancer [149]. The dual roles of Tip60—coactivating the AR and maintaining the integrity of DNA—peg Tip60 as a very attractive candidate for therapeutic intervention in PCa.
KDACs
The counteracting enzymes to the KATs are the KDACs (or histone deacetylases (HDACs)). The KDACs consist of four classes of proteins, termed class I (including HDAC1, HDAC2, and HDAC3), II (including HDAC7), III (including the sirtuins), and IV. KDACs are primarily responsible for removing the acetylation modification from lysines on histones or other target proteins. As the counterpart to KATs, KDACs and deacetylation are generally considered to be more repressive with regard to transcription [150, 151], which may make them a less obvious target for inhibition in PCa therapy. However, KDACs have also been shown to play an important role in regulating post-translational modifications on a wide array of transcription factors and proteins, including the AR [152], GATA-2 [153], and many more general cancer-important proteins including c-Myc, E2F, Rb, and p53, among others (reviewed in [154]). Numerous studies have looked into KDAC inhibition, often with promising in vitro or preclinical data, although as will be explored in this section, little success so far has been observed for PCa in clinical trials.
For PCa, much of the focus on KDACs has been on class I KDACs, including HDAC1, HDAC2, and HDAC3. The dysregulation of class I KDACs has been implicated in cancer, as they have been established to play key roles in the regulation of cell cycle [155, 156]. For PCa, class I KDAC dysregulation has been observed even in early stages (including tumor initiation) [157]. Through IHC analysis of patient samples, it has been observed that HDAC1, HDAC2, and HDAC3 are all strongly expressed in tumor sections, and each has been associated with earlier biochemical recurrence and enhanced cellular proliferation, while HDAC1 and HDAC2 have also been associated with dedifferentiation of tumors [157].
Despite the correlation between increased HDAC1 expression levels and PCa cell growth, the relationship between HDAC1 and AR is more complex. It has been demonstrated that acetylation of AR enhances its ability to recruit coactivators and to drive transcription [158] and that HDAC1 may play a role in deacetylating and repressing AR transcription [144]. In contrast, another study has demonstrated that HDAC1 and HDAC3 are important for AR function in androgen-sensitive as well as CRPC cells and that inhibition can decrease not only AR target genes but also xenograft tumor size [159]. In this study, KDAC inhibition did not directly repress the AR itself—instead, KDAC inhibition blocked the assembly of the RNA polymerase II complex required for transcription. Importantly, the authors draw attention to this biphasic response to KDAC inhibition and noted that the level of KDAC inhibition needed to achieve an AR-inhibitory effect was unlikely to be achievable in patients [159]. In addition to HDAC1, sirtuin 1—a member of the class III KDACs—has also been shown to play an important role acting as a corepressor for ligand-dependent AR transcriptional activity. Perhaps more important for therapeutic approaches, the same study also identified sirtuin 1 as being required for the AR-inhibitory activity of the first-generation AR antagonist bicalutamide [160].
Despite the evidence suggesting that KDACs—by deacetylating the AR—impair coactivator recruitment to AR and thus may repress AR-mediated transcription, the importance of KDACs in driving cell cycle and cell proliferation have led to numerous studies investigating the use of KDAC inhibitors for inhibiting cancers (including PCa). KDAC inhibitors have been documented to result in cell death in a number of cancer types with a variety of proposed mechanisms [161]. With the large number of KDAC inhibitors available for the specific classes, or individual KDAC members, the focus here will remain on those used in clinical trials for PCa (descriptions of the KDAC classes and examples of inhibitors reviewed in [162]).
Although several KDAC inhibitors have obtained FDA approval for treatment of hematologic malignancies (both vorinostat in 2006 [163] and romidepsin in 2009 [164] were approved for cutaneous T cell lymphoma, and early in 2015, panobinostat was approved for patients progressing with multiple myeloma [165]), KDAC inhibitors have yet to be proven to be effective in human trials for PCa. Vorinostat—a KDAC inhibitor which blocks activity of both class I and class II KDACs [166]—failed to decrease circulating PSA levels in a clinical trial for PCa patients who had progressed on one previous chemotherapy, while also resulting in unacceptably toxicity for patients [167]. And earlier this year, a phase II study using SB939 (pracinostat) in patients with recurrent or metastatic PCa was completed. SB939 is an orally active, pan-KDAC inhibitor [168], but although SB939 was tolerated at the given dose, the study concluded that SB939 did not meet primary endpoints for PSA decline and should not be studied further for single-agent use in CRPC [169].
Although the need for novel therapeutics in PCa is great, the complexities of KDAC inhibition—especially in PCa—raise concern about the use of these inhibitors in patients. With the above mentioned evidence that KDACs deacetylate and thus repress AR activity [144] (and thus that inhibition of KDACs may increase or stabilize the AR signaling axis) and the antithetical evidence that inhibition of KDACs may decrease the ability of AR to recruit functional transcriptional complexes (albeit at very high concentrations) [159], and thus that potent inhibition of KDACs may decrease the AR signaling axis, it may not be easily feasible to utilize inhibitors that have activity across the different classes (or maybe even for multiple members) of KDAC enzymes. More research needs to be done to achieve inhibitors with higher specificity and to know which KDACs to inhibit and at what stage. Although, in general, KDAC inhibitors appear to have a low toxicity profile in patients [170], it is worth considering that several KDAC inhibitors have had difficulties with toxicity in clinical trials. Vorinostat—which has been demonstrated to have a fairly mild toxicity profile and is currently FDA approved for some hematologic cancers—failed in a CRPC trial, in part due to unacceptable toxicity [171]. Another KDAC inhibitor, romidepsin, resulted in withdrawal of 11/35 CRPC patients from a phase II trial due to toxicity issues [172]. This may suggest that patients with CRPC may be more susceptible than other oncologic patients to the broader effects of KDAC inhibition. Such a difference may be related to the systemic androgen deprivation instituted in PCa patients, as a phase II study using vorinostat combined with tamoxifen for patients with hormone therapy-resistant breast cancer showed that vorinostat was well tolerated [173]. Still, interest in KDAC inhibitors as a potential route for PCa therapeutics remains, and it is likely that more inhibitors will be brought to clinical trials.
Other AR-Interacting Proteins and miRNAs
Although chaperones, pioneer factors, steroid receptor coactivators, and chromatin remodelers (including KDACs and KATs) make up perhaps the more well-studied AR-interacting molecules, there are numerous other factors and molecules to consider targeting.
One of the earlier proteins shown to directly interact with the AR is AR-associated protein 70 (ARA70), which was identified in 1996, and shown to potentiate the transcriptional activity of the AR [174]. More work on ARA70 has indicated that there are actually two splice variants, ARA70-α and ARA70-β, and that these have distinct and opposing functions. ARA70-β has been shown to promote cell growth and invasion in Matrigel, and overexpression of ARA70-β was demonstrated to drive transcription of genes important for cell division and adhesion. In contrast, ARA70-α may act as an inhibitor for the AR [175].
Homeobox protein B-13 (HOXB13) is a member of the homeobox family, which contains a number of transcription factors which share the homeobox DNA-binding domain and are often associated with regulating development. HOXB13 was reported to have anti-proliferative effects on PCa cells in vitro [176] and to play a role in the localization of AR to chromatin [177]. Interestingly, a germline mutation in HOXB13, G84E, is associated with a significantly increased risk for hereditary PCa [178]. HOXB13 had been shown to be important in the posterior development of mice and that HOXB13 expression persisted in some adult organs, including the prostate [179]. This work was followed up with a HOXB13 knockout study that demonstrated that HOXB13 is required for normal development of the prostate [180]. In PCa, the role of HOXB13 is controversial. Overexpression of HOXB13 in LNCaP PCa cells has been shown to inhibit cell proliferation, and this effect was reversed upon androgen stimulation, suggesting that HOXB13 may be a repressor [176]. Other studies have revealed a more complex role. HOXB13 has been shown to shift the localization of AR away from AREs on chromatin (through interacting with the AR DNA-binding domain) and instead result in the recruitment of androgen-responsive complexes to promoters with a HOXB13-response element [177]. A more recent study also demonstrated reprogramming of the AR cistrome during PCa tumorigenesis and has implicated FoxA1 and HOXB13 as key mediators of this AR shifting to new sites on chromatin [181]. Treatment with siRNA against HOXB13 resulted in loss of cell proliferation in LNCaP cells [177, 181]. Although no specific inhibitor of HOXB13 has been reported, it is interesting to note that there is a report that a FoxA1 enhancer element regulates HOXB13 in the prostate, potentially linking an important pioneer factor for the AR (FoxA1) with an interesting coregulatory transcription factor (HOXB13) [182].
Other potentially interesting molecules that may impact and disrupt the AR signaling axis are microRNAs (miRNAs). Several miRNAs, including miRNAs that are suppressed in primary and, in particular, metastatic PCa, can target and regulate the expression of the AR itself and its p160 SRC coactivators [183, 184]. One of the most notable examples is miR-31. miR-31 expression levels have been shown to inversely correlate with those of AR, while expression of miR-31 is decreased in PCa patient samples; this decrease has been shown to occur through enhanced promoter methylation [185]. In another example, miR-137 has been shown to target and decrease the expression of all three p160 SRCs in PCa cells. Similar to miR-31, miR-137 is epigenetically silenced in many cancers [136]. The methyltransferases responsible for promoter methylation or hypermethylation of miRNA genes or other proteins that may inhibit or regulate the AR or interacting factors may also be considered as potential therapeutic targets to disrupt the AR signaling axis. Several well-studied demethylating agents, including 5-azacytidine and 5-aza-2′-deoxycytidine, have been brought to clinical trials in patients with liquid tumors. Additionally, 5-azacytidine has shown some promising results in a phase II clinical trial for metastatic PCa with demonstrated clinical resistance to docetaxel. In this study, a majority of patients demonstrated a decrease in PSA levels after 12 weeks of therapy [186].
Conclusions and Future Directions
Identifying new targets and developing new therapeutics for these targets is crucial to continue extending life span and improving quality of life for men suffering from PCa. Despite the historical benefit of directly targeting the AR or the synthesis of its ligands, it is becoming increasingly apparent that directly inhibiting the AR is not enough; PCa overcomes these therapies, often remaining reliant upon the AR signaling axis.
Nuclear receptors, including the AR, rely on a vast network of cooperating molecules to enact their signaling effects on the cell. Taking advantage of these interacting proteins for therapeutics may provide an additional avenue to combat the castration-resistant phenotype in PCa. Research into AR-associated proteins and development of inhibitors for these molecules has shown that there are opportunities for clinical benefit, with high potential for next-generation PCa treatment or combination therapies.
Significant advances have been made since the first orchiectomy was shown to have activity against PCa, yet over the past 70 years, the primary focus remains directly targeting the AR LBD or ligand. As technology advances, and our understanding of PCa biology moves forward, it is critical that PCa research moves beyond the “low-hanging fruit” of inhibiting the LBD of the AR. Given the continued resistance to AR-directed therapies, and the potential role that ARVs may play in developing resistance to therapies, the development of targeted therapeutics that disrupt protein-protein interactions presents an attractive direction. By focusing on disrupting interactions with the AR, especially in combination with inhibition of the AR itself, future therapeutic agents may provide more robust inhibition of PCa cells and ideally will pose a roadblock to the development of resistance. In addition to developing inhibitors of protein interfaces or of AR-interacting proteins, the use of targeting agents, such as PSMA-targeted nanoparticles, will likely be a key component of more effective drug development. Future research efforts should be directed toward moving beyond the AR and disrupting other elements of the AR signaling axis, likely with rational combinations of other targeted therapies, in order to make continued advances in our understanding of PCa and CRPC biology and to bolster our ability to effectively treat this disease.
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136(5):E359–86. doi:10.1002/ijc.29210
Society AC (2014) Cancer facts & figures. Am Cancer Soc 2014:1–72
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015 - Siegel - 2015 - CA: a Cancer Journal for Clinicians - Wiley Online Library. Cancer J Clin. doi:10.3322/caac.21254/pdf
Huggins C, Hodges CV (1941) Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941:9–12
Mitsiades N (2013) A road map to comprehensive androgen receptor axis targeting for castration-resistant prostate cancer. Cancer Res 73(15):4599–4605. doi:10.1158/0008-5472.CAN-12-4414
Grino PB, Griffin JE, Wilson JD (1990) Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endocrinology 126(2):1165–1172. doi:10.1210/endo-126-2-1165
Mitsiades N, Sung CC, Schultz N, Danila DC, He B, Eedunuri VK, Fleisher M, Sander C, Sawyers CL, Scher HI (2012) Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res 72(23):6142–6152. doi:10.1158/0008-5472.CAN-12-1335
Lacy JM, Kyprianou N (2014) A tale of two trials: the impact of 5α-reductase inhibition on prostate cancer (Review). Oncol Lett 8(4):1391–1396. doi:10.3892/ol.2014.2388
Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, Lieber MM, Cespedes RD, Atkins JN, Lippman SM, Carlin SM, Ryan A, Szczepanek CM, Crowley JJ, Coltman CA (2003) The influence of finasteride on the development of prostate cancer. N Engl J Med 349(3):215–224. doi:10.1056/NEJMoa030660
Andriole GL, Bostwick DG, Brawley OW, Gomella LG, Marberger M, Montorsi F, Pettaway CA, Tammela TL, Teloken C, Tindall DJ, Somerville MC, Wilson TH, Fowler IL, Rittmaster RS, REDUCE Study Group (2010) Effect of dutasteride on the risk of prostate cancer. N Engl J Med 362(13):1192–1202. doi:10.1056/NEJMoa0908127
Thompson IM Jr, Goodman PJ, Tangen CM, Parnes HL, Minasian LM, Godley PA, Lucia MS, Ford LG (2013) Long-term survival of participants in the prostate cancer prevention trial. N Engl J Med 369(7):603–610. doi:10.1056/NEJMoa1215932
Heinlein CA, Chang C (2004) Androgen receptor in prostate cancer. Endocr Rev 25(2):276–308. doi:10.1210/er.2002-0032
Wilson CM, McPhaul MJ (1996) A and B forms of the androgen receptor are expressed in a variety of human tissues. Mol Cell Endocrinol 120(1):51–57
Gregory CW, He B, Wilson EM (2001) The putative androgen receptor—a form results from in vitro proteolysis. J Mol Endocrinol 27(3):309–319
Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J (2009) Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 69(1):16–22. doi:10.1158/0008-5472.CAN-08-2764
Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ (2008) Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res 68(13):5469–5477. doi:10.1158/0008-5472.CAN-08-0594
Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, Dehm SM (2011) Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res 71(6):2108–2117. doi:10.1158/0008-5472.CAN-10-1998
Tepper CG, Boucher DL, Ryan PE, Ma A-H, Xia L, Lee L-F, Pretlow TG, Kung H-J (2002) Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res 62(22):6606–6614
Jagla M, Fève M, Kessler P, Lapouge G, Erdmann E, Serra S, Bergerat J-P, Céraline J (2007) A splicing variant of the androgen receptor detected in a metastatic prostate cancer exhibits exclusively cytoplasmic actions. Endocrinology 148(9):4334–4343. doi:10.1210/en.2007-0446
Ahrens-Fath I, Politz O, Geserick C, Haendler B (2005) Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J 272(1):74–84. doi:10.1111/j.1742-4658.2004.04395.x
Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL (2010) Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A 107(39):16759–16765. doi:10.1073/pnas.1012443107
Darshan MS, Loftus MS, Thadani-Mulero M, Levy BP, Escuin D, Zhou XK, Gjyrezi A, Chanel-Vos C, Shen R, Tagawa ST, Bander NH, Nanus DM, Giannakakou P (2011) Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res 71(18):6019–6029. doi:10.1158/0008-5472.CAN-11-1417
Zhu M-L, Horbinski CM, Garzotto M, Qian DZ, Beer TM, Kyprianou N (2010) Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res 70(20):7992–8002. doi:10.1158/0008-5472.CAN-10-0585
Zhang G, Liu X, Li J, Ledet E, Alvarez X, Qi Y, Fu X, Sartor O, Dong Y, Zhang H (2015) Androgen receptor splice variants circumvent AR blockade by microtubule-targeting agents. Oncotarget
Thadani-Mulero M, Portella L, Sun S, Sung M, Matov A, Vessella RL, Corey E, Nanus DM, Plymate SR, Giannakakou P (2014) Androgen receptor splice variants determine taxane sensitivity in prostate cancer. Cancer Res 74(8):2270–2282. doi:10.1158/0008-5472.CAN-13-2876
Schurko B, Oh WK (2008) Docetaxel chemotherapy remains the standard of care in castration-resistant prostate cancer. Nat Clin Pract Oncol 5(9):506–507. doi:10.1038/ncponc1201
de Bono JS, Oudard S, Ozguroglu M, Hansen S (2010) Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. doi:10.1016/S0140-6736(10)61389-X
Sweeney CJ, Chen Y-H, Carducci M, Liu G, Jarrard DF, Eisenberger M, Wong Y-N, Hahn N, Kohli M, Cooney MM, Dreicer R, Vogelzang NJ, Picus J, Shevrin D, Hussain M, Garcia JA, DiPaola RS (2015) Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med 373(8):737–746. doi:10.1056/NEJMoa1503747
Martin SK, Bañuelos CA, Sadar MD, Kyprianou N (2015) N-terminal targeting of androgen receptor variant enhances response of castration resistant prostate cancer to taxane chemotherapy. Mol Oncol 9(3):628–639. doi:10.1016/j.molonc.2014.10.014
Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS (2008) Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 68(11):4447–4454. doi:10.1158/0008-5472.CAN-08-0249
Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, Chen S, Balk SP (2011) Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res 71(20):6503–6513. doi:10.1158/0008-5472.CAN-11-0532
Ford OH III, Gregory CW, Kim D, Smitherman AB, Mohler JL (2003) Androgen receptor gene amplification and protein expression in recurrent prostate cancer. J Urol 170(5):1817–1821. doi:10.1097/01.ju.0000091873.09677.f4
Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP (1995) In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 9(4):401–406. doi:10.1038/ng0495-401
Sufrin G, Coffey DS (1976) Flutamide. Mechanism of action of a new nonsteroidal antiandrogen. Invest Urol 13(6):429–434
Furr BJA, Tucker H (1996) The preclinical development of bicalutamide: pharmacodynamics and mechanism of action. Urology 47(1):13–25. doi:10.1016/S0090-4295(96)80003-3
Scher HI, Kelly WK (1993) Flutamide withdrawal syndrome: its impact on clinical trials in hormone-refractory prostate cancer. J Clin Oncol 11(8):1566–1572
Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, Balk SP (1995) Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med 332(21):1393–1398. doi:10.1056/NEJM199505253322101
Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, Wilson EM (2001) A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res 61(11):4315–4319
Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT (2005) Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc Natl Acad Sci 102(17):6201–6206. doi:10.1073/pnas.0500381102
Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL (2004) Molecular determinants of resistance to antiandrogen therapy. Nat Med 10(1):33–39. doi:10.1038/nm972
O’Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, Mason M, Harland S, Robbins A, Halbert G, Nutley B, Jarman M (2004) Hormonal impact of the 17|[alpha]|-hydroxylase|[sol]|C17,20-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer 90(12):2317–2325. doi:10.1038/sj.bjc.6601879
Bedoya DJ, Mitsiades N (2012) Abiraterone acetate, a first-in-class CYP17 inhibitor, establishes a new treatment paradigm in castration-resistant prostate cancer. Expert Rev Anticancer Ther 12(1):1–3. doi:10.1586/era.11.196
Bedoya DJ, Mitsiades N (2013) Clinical appraisal of abiraterone in the treatment of metastatic prostatic cancer: patient considerations, novel opportunities, and future directions. Onco Targets Ther 6:9–18. doi:10.2147/OTT.S24941
Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL (2009) Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324(5928):787–790. doi:10.1126/science.1168175
Attard G, Reid AHM, Yap TA, Raynaud F, Dowsett M, Settatree S, Barrett M, Parker C, Martins V, Folkerd E, Clark J, Cooper CS, Kaye SB, Dearnaley D, Lee G, de Bono JS (2008) Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol 26(28):4563–4571. doi:10.1200/JCO.2007.15.9749
Ryan CJ, Peng W, Kheoh T, Welkowsky E, Haqq CM, Chandler DW, Scher HI, Molina A (2014) Androgen dynamics and serum PSA in patients treated with abiraterone acetate. Prostate Cancer Prostatic Dis 17(2):192–198. doi:10.1038/pcan.2014.8
Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, Monahan JE, Stegmeier F, Roberts TM, SELLERS WR, Zhou W, Zhu P (2013) An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov 3(9):1030–1043. doi:10.1158/2159-8290.CD-13-0142
Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, Bell RH, Anderson SA, McConeghy B, Shukin R, Bazov J, Youngren J, Paris P, Thomas G, Small EJ, Wang Y, Gleave ME, Collins CC, Chi KN (2015) Androgen receptor gene aberrations in circulating cell-free DNA: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res. doi:10.1158/1078-0432.CCR-14-2666
Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, Hager JH (2013) A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov 3(9):1020–1029. doi:10.1158/2159-8290.CD-13-0226
Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, Zheng D, Sawyers CL (2013) Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 155(6):1309–1322. doi:10.1016/j.cell.2013.11.012
Isikbay M, Otto K, Kregel S, Kach J, Cai Y, Vander Griend DJ, Conzen SD, Szmulewitz RZ (2014) Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm Cancer 5(2):72–89. doi:10.1007/s12672-014-0173-2
Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, Nelson PS, Montgomery RB (2011) Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res 17(18):5913–5925. doi:10.1158/1078-0432.CCR-11-0728
Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, Vakar-Lopez F, Vessella RL, Plymate SR (2011) Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS ONE 6(11), e27970. doi:10.1371/journal.pone.0027970
Sprenger CCT, Plymate SR (2014) The link between androgen receptor splice variants and castration-resistant prostate cancer. Horm Cancer 5(4):207–217. doi:10.1007/s12672-014-0177-y
Mudryj M, Tepper CG (2013) On the origins of the androgen receptor low molecular weight species. Horm Cancer 4(5):259–269. doi:10.1007/s12672-013-0152-z
Liu G, Sprenger C, Sun S, Epilepsia KS, Haugk K, Zhang X, Coleman I, Nelson PS, Plymate S (2013) AR variant ARv567es induces carcinogenesis in a novel transgenic mouse model of prostate cancer. Neoplasia 15(9):1009–1017
Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KAT, Dehm SM (2013) Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res 73(2):483–489. doi:10.1158/0008-5472.CAN-12-3630
Qu Y, Dai B, Ye D, Kong Y, Chang K, Jia Z, Yang X, Zhang H, Zhu Y, Shi G (2015) Constitutively active AR-V7 plays an essential role in the development and progression of castration-resistant prostate cancer. Sci Rep 5:7654. doi:10.1038/srep07654
Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, Isaacs JT, Isaacs WB, Nadal R, Paller CJ, Denmeade SR, Carducci MA, Eisenberger MA, Luo J (2014) AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 371(11):1028–1038. doi:10.1056/NEJMoa1315815
Yu Z, Chen S, Sowalsky AG, Voznesensky OS, Mostaghel EA, Nelson PS, Cai C, Balk SP (2014) Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin Cancer Res 20(6):1590–1600. doi:10.1158/1078-0432.CCR-13-1863
Vasaitis T, Belosay A, Schayowitz A, Khandelwal A, Chopra P, Gediya LK, Guo Z, Fang H-B, Njar VCO, Brodie AMH (2008) Androgen receptor inactivation contributes to antitumor efficacy of 17{alpha}-hydroxylase/17,20-lyase inhibitor 3beta-hydroxy-17-(1H-benzimidazole-1-yl)androsta-5,16-diene in prostate cancer. Mol Cancer Ther 7(8):2348–2357. doi:10.1158/1535-7163.MCT-08-0230
Penning TM, Byrns MC (2009) Steroid hormone transforming aldo-keto reductases and cancer. Ann N Y Acad Sci 1155(1):33–42. doi:10.1111/j.1749-6632.2009.03700.x
Adeniji AO, Chen M, Penning TM (2013) AKR1C3 as a target in castrate resistant prostate cancer. J Steroid Biochem Mol Biol 137:136–149. doi:10.1016/j.jsbmb.2013.05.012
Liedtke AJ, Adeniji AO, Chen M, Byrns MC, Jin Y, Christianson DW, Marnett LJ, Penning TM (2013) Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (Type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J Med Chem 56(6):2429–2446. doi:10.1021/jm3017656
Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung J-K, Watt K, Tam T, Yang YC, Bañuelos CA, Williams DE, McEwan IJ, Wang Y, Sadar MD (2010) Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 17(6):535–546. doi:10.1016/j.ccr.2010.04.027
Hoelder S, Clarke PA, Workman P (2012) Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol Oncol 6(2):155–176. doi:10.1016/j.molonc.2012.02.004
Jin L, Wang W, Fang G (2014) Targeting protein-protein interaction by small molecules. Annu Rev Pharmacol Toxicol 54(1):435–456. doi:10.1146/annurev-pharmtox-011613-140028
Mullard A (2012) Protein-protein interaction inhibitors get into the groove. Nature reviews. Drug discovery. 173–175
Kularatne SA, Wang K, Santhapuram H-KR, Low PS (2009) Prostate-specific membrane antigen targeted imaging and therapy of prostate cancer using a PSMA inhibitor as a homing ligand. Mol Pharm 6(3):780–789. doi:10.1021/mp900069d
Hrkach J, Hoff Von D, Mukkaram Ali M, Andrianova E, Auer J, Campbell T, De Witt D, Figa M, Figueiredo M, Horhota A, Low S, McDonnell K, Peeke E, Retnarajan B, Sabnis A, Schnipper E, Song JJ, Song YH, Summa J, Tompsett D, Troiano G, Van Geen Hoven T, Wright J, LoRusso P, Kantoff PW, Bander NH, Sweeney C, Farokhzad OC, Langer R, Zale S (2012) Preclinical development and clinical translation of a PSMA-targeted docetaxel nanoparticle with a differentiated pharmacological profile. Sci Transl Med 4(128):128ra39–128ra39. doi:10.1126/scitranslmed.3003651
Myung J-K, Bañuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, Yang YC, Tavakoli I, Haile S, Watt K, McEwan IJ, Plymate S, Andersen RJ, Sadar MD (2013) An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest 123(7):2948–2960. doi:10.1172/JCI66398
Fang Y, Fliss AE, Robins DM, Caplan AJ (1996) Hsp90 regulates androgen receptor hormone binding affinity in vivo. J Biol Chem 271(45):28697–28702
Joab I, Radanyi C, Renoir M, Buchou T, Catelli MG, Binart N, Mester J, Baulieu EE (1984) Common non-hormone binding component in non-transformed chick oviduct receptors of four steroid hormones. Nature 308(5962):850–853
Veldscholte J, Berrevoets CA, Zegers ND, van der Kwast TH, Grootegoed JA, Mulder E (1992) Hormone-induced dissociation of the androgen receptor-heat-shock protein complex: use of a new monoclonal antibody to distinguish transformed from nontransformed receptors. Biochemistry 31(32):7422–7430
Pratt WB, Toft DO (1997) Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev 18(3):306–360. doi:10.1210/edrv.18.3.0303
Vanaja DK, Mitchell SH, Toft DO, Young CYF (2002) Effect of geldanamycin on androgen receptor function and stability. Cell Stress Chaperones 7(1):55–64
Supko JG, Hickman RL, Grever MR, Malspeis L (1995) Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol 36(4):305–315. doi:10.1007/BF00689048
Heath EI, Hillman DW, Vaishampayan U, Sheng S, Sarkar F, Harper F, Gaskins M, Pitot HC, Tan W, Ivy SP, Pili R, Carducci MA, Erlichman C, Liu G (2008) A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin Cancer Res 14(23):7940–7946. doi:10.1158/1078-0432.CCR-08-0221
He S, Zhang C, Shafi AA, Sequeira M, Acquaviva J, Friedland JC, Sang J, Smith DL, Weigel NL, Wada Y, Proia DA (2013) Potent activity of the Hsp90 inhibitor ganetespib in prostate cancer cells irrespective of androgen receptor status or variant receptor expression. Int J Oncol 42(1):35–43. doi:10.3892/ijo.2012.1698
Heath EI, Stein MN, Vaishampayan U, Antonarakis ES, Liu G, Sheng S, Farrow K, Smith DW, Heilbrun LK (2013) Phase II trial of single-agent ganetespib (STA-9090), a heat shock protein 90 (Hsp90) inhibitor in heavily pretreated patients with metastatic castration-resistant prostate cancer (mCRPC) post docetaxel-based chemotherapy: results of a Prostate Cancer Clinical Trials Consortium (PCCTC) study. J Clin Oncol 31:(suppl–abstr 5085)
Shafi AA, Cox MB, Weigel NL (2013) Androgen receptor splice variants are resistant to inhibitors of Hsp90 and FKBP52, which alter androgen receptor activity and expression. Steroids 78(6):548–554. doi:10.1016/j.steroids.2012.12.013
Elbi C, Walker DA, Romero G, Sullivan WP, Toft DO, Hager GL, DeFranco DB (2004) Molecular chaperones function as steroid receptor nuclear mobility factors. Proc Natl Acad Sci 101(9):2876–2881. doi:10.1073/pnas.0400116101
Freeman BC, Yamamoto KR (2002) Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 296(5576):2232–2235. doi:10.1126/science.1073051
Reebye V, Querol Cano L, Lavery DN, Brooke GN, Powell SM, Chotai D, Walker MM, Whitaker HC, Wait R, Hurst HC, Bevan CL (2012) Role of the HSP90-associated cochaperone p23 in enhancing activity of the androgen receptor and significance for prostate cancer. Mol Endocrinol 26(10):1694–1706. doi:10.1210/me.2012-1056
Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N (2002) Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem 277(42):39858–39866. doi:10.1074/jbc.M206322200
Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS (2002) Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell 9(2):279–289
Clark KL, Halay ED, Lai E, Burley SK (1993) Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 364(6436):412–420. doi:10.1038/364412a0
Gao N, Zhang J, Rao MA, Case TC, Mirosevich J, Wang Y, Jin R, Gupta A, Rennie PS, Matusik RJ (2003) The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol Endocrinol 17(8):1484–1507. doi:10.1210/me.2003-0020
Wang Q, Li W, Liu XS, Carroll JS, JAnne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M (2007) A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell 27(3):380–392. doi:10.1016/j.molcel.2007.05.041
Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, Wu T, Regan MM, Meyer CA, Carroll JS, Manrai AK, JAnne OA, Balk SP, Mehra R, Han B, Chinnaiyan AM, Rubin MA, True L, Fiorentino M, Fiore C, Loda M, Kantoff PW, Liu XS, Brown M (2009) Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 138(2):245–256. doi:10.1016/j.cell.2009.04.056
Gerhardt J, Montani M, Wild P, Beer M, Huber F, Hermanns T, Müntener M, Kristiansen G (2012) FOXA1 promotes tumor progression in prostate cancer and represents a novel hallmark of castration-resistant prostate cancer. Am J Pathol 180(2):848–861. doi:10.1016/j.ajpath.2011.10.021
Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A, Sankila A, Turunen J-P, Lundin M, Konsti J, Vesterinen T, Nordling S, Kallioniemi O, Hautaniemi S, JAnne OA (2011) Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J 30(19):3962–3976. doi:10.1038/emboj.2011.328
Robinson JLL, Hickey TE, Warren AY, Vowler SL, Carroll T, Lamb AD, Papoutsoglou N, Neal DE, Tilley WD, Carroll JS (2014) Elevated levels of FOXA1 facilitate androgen receptor chromatin binding resulting in a CRPC-like phenotype. Oncogene 33(50):5666–5674. doi:10.1038/onc.2013.508
Jin H-J, Zhao JC, Wu L, Kim J, Yu J (2014) Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat Commun 5:1–14. doi:10.1038/ncomms4972
Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, Glass CK, Rosenfeld MG, Fu X-D (2011) Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474(7351):390–394. doi:10.1038/nature10006
Krause WC, Shafi AA, Nakka M, Weigel NL (2014) Androgen receptor and its splice variant, AR-V7, differentially regulate FOXA1 sensitive genes in LNCaP prostate cancer cells. Int J Biochem Cell Biol 54:49–59. doi:10.1016/j.biocel.2014.06.013
Böhm M, Locke WJ, Sutherland RL, Kench JG, Henshall SM (2009) A role for GATA-2 in transition to an aggressive phenotype in prostate cancer through modulation of key androgen-regulated genes. Oncogene 28(43):3847–3856. doi:10.1038/onc.2009.243
He B, Lanz RB, Fiskus W, Geng C, Yi P, Hartig SM, Rajapakshe K, Shou J, Wei L, Shah SS, Foley C, Chew SA, Eedunuri VK, Bedoya DJ, Feng Q, Minami T, Mitsiades CS, Frolov A, Weigel NL, Hilsenbeck SG, Rosen DG, Palzkill T, Ittmann MM, Song Y, Coarfa C, O’Malley BW, Mitsiades N (2014) GATA2 facilitates steroid receptor coactivator recruitment to the androgen receptor complex. Proc Natl Acad Sci U.S.A. 201421415. doi:10.1073/pnas.1421415111
Chiang YT, Wang K, Fazli L, Qi RZ, Gleave ME, Collins CC, Gout PW, Wang Y (2014) GATA2 as a potential metastasis-driving gene in prostate cancer. Oncotarget 5(2):451–461
Wu D, Sunkel B, Chen Z, Liu X, Ye Z, Li Q, Grenade C, Ke J, Zhang C, Chen H, Nephew KP, Huang TH-M, Liu Z, Jin VX, Wang Q (2014) Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res 42(6):3607–3622. doi:10.1093/nar/gkt1382
Chi P, Rockowitz S, Iaquinta PJ, Shamu T, Shukla S, Gao D, Sirota I, Carver BS, Wongvipat J, Scher HI, Zheng D, Chen Y, Sawyers CL (2013) ETS factors reprogram the androgen receptor cistrome and prime prostate tumorigenesis in response to PTEN loss. Nat Med 1–9. doi:10.1038/nm.3216
Vidal SJ, Rodriguez-Bravo V, Quinn SA, Rodriguez-Barrueco R, Lujambio A, Williams E, Sun X, de la Iglesia-Vicente J, Lee A, Readhead B, Chen X, Galsky M, Esteve B, Petrylak DP, Dudley JT, Rabadan R, Silva JM, Hoshida Y, Lowe SW, Cordon-Cardo C, Domingo-Domenech J (2015) A targetable GATA2-IGF2 axis confers aggressiveness in lethal prostate cancer. Cancer Cell 27(2):223–239. doi:10.1016/j.ccell.2014.11.013
Tanner MM, Grenman S, Koul A, Johannsson O, Meltzer P, Pejovic T, Borg A, Isola JJ (2000) Frequent amplification of chromosomal region 20q12-q13 in ovarian cancer. Clin Cancer Res 6(5):1833–1839
Wang Y, Wu M-C, Sham JST, Zhang W, Wu W-Q, Guan X-Y (2002) Prognostic significance ofc-myc andAIB1 amplification in hepatocellular carcinoma. Cancer 95(11):2346–2352. doi:10.1002/cncr.10963
Kershah SM, Desouki MM, Koterba KL, Rowan BG (2004) Expression of estrogen receptor coregulators in normal and malignant human endometrium. Gynecol Oncol 92(1):304–313. doi:10.1016/j.ygyno.2003.10.007
Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C, Fiskus W, Rajendran M, Chew SA, Zimmermann M, Bond R, He B, Coarfa C, Mitsiades N (2014) Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res 74(19):5631–5643. doi:10.1158/0008-5472.CAN-14-0476
Zhou H-J, YAN J, Luo W, Ayala G, Lin S-H, Erdem H, Ittmann M, Tsai SY, Tsai M-J (2005) SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res 65(17):7976–7983. doi:10.1158/0008-5472.CAN-04-4076
Qin J, Lee H-J, Wu S-P, Lin S-C, Lanz RB, Creighton CJ, DeMayo FJ, Tsai SY, Tsai M-J (2014) Androgen deprivation-induced NCoA2 promotes metastatic and castration-resistant prostate cancer. J Clin Invest 124(11):5013–5026. doi:10.1172/JCI76412
Xu J, Wu R-C, O’Malley BW (2009) Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. 1–16. doi:10.1038/nrc2695
Stashi E, York B, O’Malley BW (2014) Steroid receptor coactivators: servants and masters for control of systems metabolism. Trends Endocrinol Metab 25(7):337–347. doi:10.1016/j.tem.2014.05.004
Dasgupta S, Lonard DM, O’Malley BW (2014) Nuclear receptor coactivators: master regulators of human health and disease. Annu Rev Med 65:279–292. doi:10.1146/annurev-med-051812-145316
Leo C, Chen JD (2000) The SRC family of nuclear receptor coactivators. Gene 245(1):1–11
Kino T, Slobodskaya O, Pavlakis GN, Chrousos GP (2002) Nuclear receptor coactivator p160 proteins enhance the HIV-1 long terminal repeat promoter by bridging promoter-bound factors and the Tat-P-TEFb complex. J Biol Chem 277(4):2396–2405. doi:10.1074/jbc.M106312200
Gao Z, Chiao P, Zhang X, Lazar M, Seto E, Young HA, Ye J (2005) Coactivators and corepressors of NF-KB in IkB alpha gene promoter. J Biol Chem 13(4), e97. doi:10.2196/jmir.1827
Nakka M, Agoulnik IU, Weigel NL (2013) Targeted disruption of the p160 coactivator interface of androgen receptor (AR) selectively inhibits AR activity in both androgen-dependent and castration-resistant AR-expressing prostate cancer cells. Int J Biochem Cell Biol 45(4):763–772. doi:10.1016/j.biocel.2012.12.012
Lee SK, Kim HJ, Na SY, Kim TS, Choi HS, Im SY, Lee JW (1998) Steroid receptor coactivator-1 coactivates activating protein-1-mediated transactivations through interaction with the c-Jun and c-Fos subunits. J Biol Chem 273(27):16651–16654
Koh SS, Li H, Lee Y-H, Widelitz RB, Chuong C-M, Stallcup MR (2002) Synergistic coactivator function by coactivator-associated arginine methyltransferase (CARM) 1 and beta-catenin with two different classes of DNA-binding transcriptional activators. J Biol Chem 277(29):26031–26035. doi:10.1074/jbc.M110865200
He B, Kemppainen JA, Wilson EM (2000) FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J Biol Chem 275(30):22986–22994. doi:10.1074/jbc.M002807200
Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR (1998) Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev 12(21):3343–3356. doi:10.1101/gad.12.21.3343
Linja MJ, Porkka KP, Kang Z, Savinainen KJ, JAnne OA, Tammela TLJ, Vessella RL, Palvimo JJ, Visakorpi T (2004) Expression of androgen receptor coregulators in prostate cancer. Clin Cancer Res 10(3):1032–1040
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18(1):11–22. doi:10.1016/j.ccr.2010.05.026
Agoulnik IU, Weigel NL (2009) Coactivator selective regulation of androgen receptor activity. Steroids 74(8):669–674. doi:10.1016/j.steroids.2009.02.007
Agoulnik IU, Vaid A, Bingman WE, Erdeme H, Frolov A, Smith CL, Ayala G, Ittmann MM, Weigel NL (2005) Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res 65(17):7959–7967. doi:10.1158/0008-5472.CAN-04-3541
Agoulnik IU, Vaid A, Nakka M, Alvarado M, Bingman WE, Erdem H, Frolov A, Smith CL, Ayala GE, Ittmann MM, Weigel NL (2006) Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res 66(21):10594–10602. doi:10.1158/0008-5472.CAN-06-1023
Gnanapragasam VJ, Leung HY, Pulimood AS, Neal DE, Robson CN (2001) Expression of RAC 3, a steroid hormone receptor co-activator in prostate cancer. Br J Cancer 85(12):1928–1936. doi:10.1054/bjoc.2001.2179
Geng C, He B, Xu L, Barbieri CE (2013) Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. In. doi:10.1073/pnas.1304502110/-/DCSupplemental
Theurillat J-PP, Udeshi ND, Errington WJ, Svinkina T, Baca SC, Pop M, Wild PJ, Blattner M, Groner AC, Rubin MA, Moch H, Privé GG, Carr SA, Garraway LA (2014) Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 346(6205):85–89. doi:10.1126/science.1250255
Yan J, YU C-T, Ozen M, Ittmann M, Tsai SY, Tsai M-J (2006) Steroid receptor coactivator-3 and activator protein-1 coordinately regulate the transcription of components of the insulin-like growth factor/AKT signaling pathway. Cancer Research 66(22):11039–11046. doi:10.1158/0008-5472.CAN-06-2442
YAN J, Erdem H, Li R, Cai Y, Ayala G, Ittmann M, Yu-Lee L-Y, Tsai SY, Tsai M-J (2008) Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res 68(13):5460–5468. doi:10.1158/0008-5472.CAN-08-0955
Tien JC-Y, Zhou S, Xu J (2009) The role of SRC-1 in murine prostate cancinogenesis is nonessential due to a possible compensation of SRC-3/AIB1 overexpression. Int J Biol Sci 5(3):256–264
Powell SM, Christiaens V, Voulgaraki D, Waxman J, Claessens F, Bevan CL (2004) Mechanisms of androgen receptor signalling via steroid receptor coactivator-1 in prostate. Endocr Relat Cancer 11(1):117–130
Bohrer LR, Liu P, Zhong J, Pan Y, Angstman J, Brand LJ, Dehm SM, Huang H (2013) FOXO1 binds to the TAU5 motif and inhibits constitutively active androgen receptor splice variants. Prostate 73(10):1017–1027. doi:10.1002/pros.22649
Wang Y, Lonard DM, Yu Y, Chow D-C, Palzkill TG, O’Malley BW (2011) Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol Endocrinol 25(12):2041–2053. doi:10.1210/me.2011-1222
Wang Y, Lonard DM, Yu Y, Chow D-C, Palzkill TG, Wang J, Qi R, Matzuk AJ, Song X, Madoux F, Hodder P, Chase P, Griffin PR, Zhou S, Liao L, Xu J, O’Malley BW (2014) Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res 74(5):1506–1517. doi:10.1158/0008-5472.CAN-13-2939
Yan F, Yu Y, Chow D-C, Palzkill T, Madoux F, Hodder P, Chase P, Griffin PR, O’Malley BW, Lonard DM (2014) Identification of verrucarin A as a potent and selective steroid receptor coactivator-3 small molecule inhibitor. Agoulnik IU, ed. PLoS ONE 9(4), e95243. doi:10.1371/journal.pone.0095243
Kumar Eedunuri V, Rajapakshe K, Fiskus W, Geng C, Anne Chew S, Foley C, Shah SS, Shou J, Mohamed JS, Coarfa C, O’Malley BW, Mitsiades N (2015) MiR137 targets p160 Steroid Receptor Coactivators SRC1, SRC2 and SRC3 and inhibits cell proliferation. Mol Endocrinol. me20151080. doi:10.1210/me.2015-1080
Chakravarti D, LaMorte VJ, Nelson MC, Nakajima T (1996) Role of CBP/P300 in nuclear receptor signalling. Nature 383(6595):99–103. doi:10.1038/383099a0
Debes JD, Schmidt LJ, Huang H, Tindall DJ (2002) p300 mediates androgen-independent transactivation of the androgen receptor by interleukin 6. Cancer Res 62(20):5632–5636
Comuzzi B, Lambrinidis L, Rogatsch H, Godoy-Tundidor S, Knezevic N, Krhen I, Marekovic Z, Bartsch G, Klocker H, Hobisch A, Culig Z (2003) The transcriptional co-activator cAMP response element-binding protein-binding protein is expressed in prostate cancer and enhances androgen- and anti-androgen-induced androgen receptor function. Am J Pathol 162(1):233–241. doi:10.1016/S0002-9440(10)63814-X
Santer FR, Höschele PPS, Oh SJ, Erb HHH, Bouchal J, Cavarretta IT, Parson W, Meyers DJ, Cole PA, Culig Z (2011) Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol Cancer Ther 10(9):1644–1655. doi:10.1158/1535-7163.MCT-11-0182
Kuttan R, Bhanumathy P, Nirmala K, George MC (1985) Potential anticancer activity of turmeric (Curcuma longa). Cancer Lett 29(2):197–202
Shah S, Prasad S, Knudsen KE (2012) Targeting pioneering factor and hormone receptor cooperative pathways to suppress tumor progression. Cancer Res 72(5):1248–1259. doi:10.1158/0008-5472.CAN-11-0943
Brady ME, Ozanne DM, Gaughan L, Waite I, Cook S, Neal DE, Robson CN (1999) Tip60 is a nuclear hormone receptor coactivator. J Biol Chem 274(25):17599–17604
Gaughan L, Logan IR, Cook S, Neal DE, Robson CN (2002) Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J Biol Chem 277(29):25904–25913. doi:10.1074/jbc.M203423200
Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y (2000) Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102(4):463–473
Sun Y, Jiang X, Chen S, Fernandes N, Price BD (2005) A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci 102(37):13182–13187. doi:10.1073/pnas.0504211102
Halkidou K, Gnanapragasam VJ, Mehta PB, Logan IR, Brady ME, Cook S, Leung HY, Neal DE, Robson CN (2003) Expression of Tip60, an androgen receptor coactivator, and its role in prostate cancer development. Oncogene 22(16):2466–2477. doi:10.1038/sj.onc.1206342
Coffey K, Blackburn TJ, Cook S, Golding BT, Griffin RJ, Hardcastle IR, Hewitt L, Huberman K, McNeill HV, Newell DR, Roche C, Ryan-Munden CA, Watson A, Robson CN (2012) Characterisation of a Tip60 specific inhibitor, NU9056, in prostate cancer. PLoS ONE 7(10), e45539. doi:10.1371/journal.pone.0045539
Gao C, Bourke E, Scobie M, Famme MA, Koolmeister T, Helleday T, Eriksson LA, Lowndes NF, Brown JAL (2014) Rational design and validation of a Tip60 histone acetyltransferase inhibitor. Sci Rep 4:5372. doi:10.1038/srep05372
Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393(6683):386–389. doi:10.1038/30764
Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19(2):187–191. doi:10.1038/561
Fu M, Wang C, Wang J, Zhang X, Sakamaki T, Yeung YG, Chang C, Hopp T, Fuqua SAW, Jaffray E, Hay RT, Palvimo JJ, JAnne OA, Pestell RG (2002) Androgen receptor acetylation governs trans activation and MEKK1-induced apoptosis without affecting in vitro sumoylation and trans-repression function. Mol Cell Biol 22(10):3373–3388
Hayakawa F, Towatari M, Ozawa Y, Tomita A, Privalsky ML, Saito H (2004) Functional regulation of GATA-2 by acetylation. J Leukoc Biol 75(3):529–540. doi:10.1189/jlb.0603389
Glozak MA, Sengupta N, Zhang X, Seto E (2005) Acetylation and deacetylation of non-histone proteins. Gene 363:15–23. doi:10.1016/j.gene.2005.09.010
Gui C-Y, Ngo L, Xu WS, Richon VM, Marks PA (2004) Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci 101(5):1241–1246. doi:10.1073/pnas.0307708100
Wilson AJ, Byun D-S, Popova N, Murray LB, L’Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM (2006) Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem 281(19):13548–13558. doi:10.1074/jbc.M510023200
Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M, Kristiansen G (2008) Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer 98(3):604–610. doi:10.1038/sj.bjc.6604199
Fu M, Rao M, Wang C, Sakamaki T, Wang J, Di Vizio D, Zhang X, Albanese C, Balk S, Chang C, Fan S, Rosen E, Palvimo JJ, JAnne OA, Muratoglu S, Avantaggiati ML, Pestell RG (2003) Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol Cell Biol 23(23):8563–8575
Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N, Sawyers CL (2009) Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res 69(3):958–966. doi:10.1158/0008-5472.CAN-08-2216
Dai Y, Ngo D, Forman LW, Qin DC, Jacob J, Faller DV (2007) Sirtuin 1 is required for antagonist-induced transcriptional repression of androgen-responsive genes by the androgen receptor. Mol Endocrinol 21(8):1807–1821. doi:10.1210/me.2006-0467
Bolden JE, Shi W, Jankowski K, Kan C-Y, Cluse L, Martin BP, MacKenzie KL, Smyth GK, Johnstone RW (2013) HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis 4, e519. doi:10.1038/cddis.2013.9
Abbas A, Gupta S (2008) The role of histone deacetylases in prostate cancer. Epigenetics 3(6):300–309
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12(10):1247–1252. doi:10.1634/theoncologist.12-10-1247
Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, Zain J, Prince HM, Leonard JP, Geskin LJ, Reeder C, Joske D, Figg WD, Gardner ER, Steinberg SM, Jaffe ES, Stetler-Stevenson M, Lade S, Fojo AT, Bates SE (2009) Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol 27(32):5410–5417. doi:10.1200/JCO.2008.21.6150
Fenichel MP (2015) FDA approves new agent for multiple myeloma. J Natl Cancer Inst. djv165
Richon VM (2006) Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br J Cancer 95:S2–S6. doi:10.1038/sj.bjc.6603463
Bradley D, Rathkopf D, Dunn R, Stadler WM, Liu G, Smith DC, Pili R, Zwiebel J, Scher H, Hussain M (2009) Vorinostat in advanced prostate cancer patients progressing on prior chemotherapy (National Cancer Institute Trial 6862): trial results and interleukin-6 analysis: a study by the Department of Defense Prostate Cancer Clinical Trial Consortium and University of Chicago Phase 2 Consortium. Cancer 115(23):5541–5549. doi:10.1002/cncr.24597
Wang H, Yu N, Chen D, Lee KCL, Lye PL, Chang JWW, Deng W, Ng MCY, Lu T, Khoo ML, Poulsen A, Sangthongpitag K, Wu X, Hu C, Goh KC, Wang X, Fang L, Goh KL, Khng HH, Goh SK, Yeo P, Liu X, Bonday Z, Wood JM, Dymock BW, Kantharaj E, Sun ET (2011) Discovery of (2E)-3-{2-butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl}-N-hydroxyacrylamide (SB939), an orally active histone deacetylase inhibitor with a superior preclinical profile. J Med Chem 54(13):4694–4720. doi:10.1021/jm2003552
Eigl BJ, North S, Winquist E, Finch D, Wood L, Sridhar SS, Powers J, Good J, Sharma M, Squire JA, Bazov J, Jamaspishvili T, Cox ME, Bradbury PA, Eisenhauer EA, Chi KN (2015) A phase II study of the HDAC inhibitor SB939 in patients with castration resistant prostate cancer: NCIC clinical trials group study IND195. Invest New Drugs 33(4):969–976. doi:10.1007/s10637-015-0252-4
Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL (2010) Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals 3(9):2751–2767. doi:10.3390/ph3092751
Schneider BJ, Kalemkerian GP, Bradley D, Smith DC, Egorin MJ, Daignault S, Dunn R, Hussain M (2012) Phase I study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Invest New Drugs 30(1):249–257. doi:10.1007/s10637-010-9503-6
Molife LR, Attard G, Fong PC, Karavasilis V, Reid AHM, Patterson S, Riggs CE, Higano C, Stadler WM, McCulloch W, Dearnaley D, Parker C, de Bono JS (2010) Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann Oncol 21(1):109–113. doi:10.1093/annonc/mdp270
Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, Melisko M, Ismail-Khan R, Rugo H, Moasser M, Minton SE (2011) A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer 104(12):1828–1835. doi:10.1038/bjc.2011.156
Yeh S, Chang C (1996) Cloning and characterization of a specific coactivator, ARA70, for the androgen receptor in human prostate cells. Proc Natl Acad Sci 93(11):5517–5521
Peng Y, Li CX, Chen F, Wang Z, Ligr M, Melamed J, Wei J, Gerald W, Pagano M, Garabedian MJ, Lee P (2008) Stimulation of prostate cancer cellular proliferation and invasion by the androgen receptor co-activator ARA70. Am J Pathol 172(1):225–235. doi:10.2353/ajpath.2008.070065
Jung C, Kim R-S, Lee S-J, Wang C, Jeng M-H (2004) HOXB13 homeodomain protein suppresses the growth of prostate cancer cells by the negative regulation of T-cell factor 4. Cancer Res 64(9):3046–3051
Norris JD, Chang C-Y, Wittmann BM, Kunder RS, Cui H, Fan D, Joseph JD, McDonnell DP (2009) The homeodomain protein HOXB13 regulates the cellular response to androgens. Mol Cell 36(3):405–416. doi:10.1016/j.molcel.2009.10.020
Ewing CM, Ray AM, Lange EM, Zuhlke KA, Robbins CM, Tembe WD, Wiley KE, Isaacs SD, Johng D, Wang Y, Bizon C, Yan G, Gielzak M, Partin AW, Shanmugam V, Izatt T, Sinari S, Craig DW, Zheng SL, Walsh PC, Montie JE, Xu J, Carpten JD, Isaacs WB, Cooney KA (2012) Germline mutations in HOXB13 and prostate-cancer risk. N Engl J Med 366(2):141–149. doi:10.1056/NEJMoa1110000
Sreenath T, Orosz A, Fujita K, Bieberich CJ (1999) Androgen-independent expression of hoxb-13 in the mouse prostate. Prostate 41(3):203–207
Economides KD, Capecchi MR (2003) Hoxb13 is required for normal differentiation and secretory function of the ventral prostate. Development 130(10):2061–2069
Pomerantz MM, Li F, Takeda DY, Lenci R, Chonkar A, Chabot M, Cejas P, Vazquez F, Cook J, Shivdasani RA, Bowden M, Lis R, Hahn WC, Kantoff PW, Brown M, Loda M, Long HW, Freedman ML (2015) The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Publ Group. doi:10.1038/ng.3419
McMullin RP, Dobi A, Mutton LN, Orosz A, Maheshwari S, Shashikant CS, Bieberich CJ (2010) A FOXA1-binding enhancer regulates Hoxb13 expression in the prostate gland. Proc Natl Acad Sci U S A 107(1):98–103. doi:10.1073/pnas.0902001107
Östling P, Leivonen S-K, Aakula A, Kohonen P, Mäkelä R, Hagman Z, Edsjö A, Kangaspeska S, Edgren H, Nicorici D, Bjartell A, Ceder Y, Perälä M, Kallioniemi O (2011) Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res 71(5):1956–1967. doi:10.1158/0008-5472.CAN-10-2421
Coarfa C, Fiskus W, Eedunuri VK, Rajapakshe K, Foley C, Chew SA, Shah SS, Geng C, Shou J, Mohamed JS, O’Malley BW, Mitsiades N (2015) Comprehensive proteomic profiling identifies the androgen receptor axis and other signaling pathways as targets of microRNAs suppressed in metastatic prostate cancer. Oncogene. doi:10.1038/onc.2015.295
Lin P-C, Chiu Y-L, Banerjee S, Park K, Mosquera J-M, Giannopoulou E, Alves P, Tewari AK, Gerstein MB, Beltran H, Melnick AM, Elemento O, Demichelis F, Rubin MA (2013) Epigenetic repression of miR-31 disrupts androgen receptor homeostasis and contributes to prostate cancer progression. Cancer Res 73(3):1232–1244. doi:10.1158/0008-5472.CAN-12-2968
Singal R, Ramachandran K, Gordian E, Quintero C, Zhao W, Reis IM (2015) Phase I/II study of azacitidine, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel-based therapy. Clin Genitourin Cancer 13(1):22–31. doi:10.1016/j.clgc.2014.07.008
Acknowledgments
The authors acknowledge the joint participation by Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors state that they have no relevant financial interests to disclose.
Funding/Support
This work was also supported by the American Cancer Society RSG-14-218-01-TBG (to N.M.), the Prostate Cancer Foundation (N.M.), the Conquer Cancer Foundation of the American Society of Clinical Oncology Young Investigator and Career Development Awards (both to N.M.), and the Pilot/Feasibility Program of the Diabetes & Endocrinology Research Center (P30-DK079638) at Baylor College of Medicine (N.M.). N.M. is supported by NCI grant 1R01CA193321, is a Dan L. Duncan Scholar, a Caroline Wiess Law Scholar, and member of the Dan L. Duncan Cancer Center (supported by the NCI Cancer Center Support Grant P30CA125123).
Rights and permissions
About this article

Cite this article
Foley, C., Mitsiades, N. Moving Beyond the Androgen Receptor (AR): Targeting AR-Interacting Proteins to Treat Prostate Cancer. HORM CANC 7, 84–103 (2016). https://doi.org/10.1007/s12672-015-0239-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-015-0239-9