
Abstract
Despite new treatments for castrate-resistant prostate cancer (CRPC), the prognosis of patients with CRPC remains bleak due to acquired resistance to androgen receptor (AR)-directed therapy. The glucocorticoid receptor (GR) and AR share several transcriptional targets, including the anti-apoptotic genes serum and g lucocorticoid-regulated kinase 1 (SGK1) and Map kinase phosphatase 1 (MKP1)/dual specificity phosphatase 1 (DUSP1). Because GR expression increases in a subset of primary prostate cancer (PC) following androgen deprivation therapy, we sought to determine whether GR activation can contribute to resistance to AR-directed therapy. We studied CWR-22Rv1 and LAPC4 AR/GR-expressing PC cell lines following treatment with combinations of the androgen R1881, AR antagonist MDV3100, GR agonist dexamethasone, GR antagonists mifepristone and CORT 122928, or the SGK1 inhibitor GSK650394. Cell lines stably expressing GR (NR3C1)-targeted shRNA or ectopic SGK1-Flag were also studied in vivo. GR activation diminished the effects of the AR antagonist MDV3100 on tumor cell viability. In addition, GR activation increased prostate-specific antigen (PSA) secretion and induced SGKI and MKP1/DUSP gene expression. Glucocorticoid-mediated cell viability was diminished by a GR antagonist or by co-treatment with the SGK1 inhibitor GSK650394. In vivo, GR depletion delayed castrate-resistant tumor formation, while SGK1-Flag-overexpressing PC xenografts displayed accelerated castrate-resistant tumor initiation, supporting a role for SGK1 in GR-mediated CRPC progression. We studied several PC models before and following treatment with androgen blockade and found that increased GR expression and activity contributed to tumor-promoting PC cell viability. Increased GR-regulated SGK1 expression appears, at least in part, to mediate enhanced PC cell survival. Therefore, GR and/or SGK1 inhibition may be useful adjuncts to AR blockade for treating CRPC.
Similar content being viewed by others

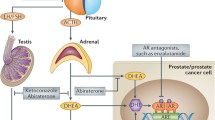

Introduction
Prostate cancer (PC) is the most common cancer diagnosis among men in developed countries [20]. In addition, PC remains the second leading cause of cancer death in men in the USA and is a significant cause of pain and suffering. Seminal work by Huggins and colleagues showed that PC growth is invariably driven by androgens, and as a result, metastatic PC is initially treated with castration-based therapies to eliminate testicular androgen production [18]. This approach, while initially effective in controlling metastatic PC and palliating many of the symptoms associated with metastasis, eventually fails. When metastatic PC progresses despite primary testosterone-lowering hormonal therapy, the disease is termed “castration-resistant prostate cancer” (CRPC) [46].
Surprisingly, even after a significant reduction in circulating androgens following pharmacological or surgical castration, AR signaling appears to drive CRPC tumorigenesis [7, 35]. Mechanisms include AR gene amplification, acquired somatic mutations, and alternative splicing; all of which can contribute to AR signaling to persist despite low concentrations of circulating androgens. AR activity can be successfully reduced with new hormone-based systemic therapies that are used to treat metastatic CRPC. Specifically, abiraterone, TOK-001 (galeterone), and TAK-700 (orteronel) can reduce extra-gonadal androgen synthesis thereby decreasing systemic levels of dihydroepiandrosterone (DHEA), androstenedione, and other androgens. Abiraterone is already approved by the Food and Drug Administration (FDA) for use in men with progressive, metastatic CRPC. Highly selective AR antagonists that effectively prevent AR nuclear localization and transcriptional activity have also been developed recently; these newer agents do not have the partial agonistic properties of previous AR modulators [9, 45]. Treatment with one such AR antagonist, enzalutamide (MDV3100), results in a significant improvement in overall survival of patients with progressive CRPC and is also FDA-approved. Unfortunately, the absolute magnitude of benefit is modest—compared to placebo, treatment with enzalutamide in patients with CRPC that has clinically progressed despite docetaxel chemotherapy improves improves the median overall survival by 4.8 months, and survival following progression remains less than a year [34].
Tumor progression following anti-AR treatment appears to be mediated by several mechanisms that can bypass AR inhibition. For example, AR-targeted therapy may select for cells expressing constitutively active AR mutations or may lead to selection of cells with oncogenic intracellular signaling pathways, such as phosphoinositide 3-kinase (PI3K) activation [6, 35]. Here, we explore the hypothesis that increased glucocorticoid receptor (GR) activity following exposure to androgen blockade can bypass AR antagonism and cause tumor progression.
GR activation has pleiotropic physiological effects and regulates metabolic, inflammatory, and cell survival pathways in various ways depending on tissue type. In hematological malignancies, GR activation is associated with increased apoptosis; however, in sarcomas, GR activation is mitogenic and in most epithelial cells GR activity has potent anti-apoptotic effects [15, 43]. GR and AR are similar not only in structure but also share target gene response elements and regulate a subset of common genes [5, 10]. Furthermore, similar to AR signaling in prostate cancer, GR has an established role in regulating cell survival and anti-apoptotic genes in breast cancer [47, 48]. Interestingly, the poor prognosis associated with increased GR expression in primary breast cancers is dependent on estrogen receptor (ER) expression; high GR expression is associated with a poor prognosis in patients with early-stage ER-negative breast cancer; conversely, high GR expression is associated with an improved prognosis in patients with ER-positive disease [30].
GR’s role in PC is not well understood. Clinically, high-dose glucocorticoids can have palliative benefits in metastatic prostate cancer patients by reducing bone pain and increasing appetite [13]. Furthermore, there have been reports that GR activation, in preclinical models of PC with intact AR signaling, can be growth inhibitory [29, 38, 51]. In contrast, in PC cell lines that lack AR expression, but have high levels of GR expression, GR signaling promotes cell proliferation [14, 25, 44, 49]. Previous results from our laboratory [41] and others [50] have shown that immunohistochemically detected GR expression is significantly increased in primary PC samples from patients exposed to androgen deprivation when compared to PC specimens from previously untreated patients [41, 50]. This observation suggested that increased GR expression might follow exposure of PC to anti-androgens or androgen-lowering therapies and the potential for GR to bypass inhibited AR pathways.
Here, we test the hypothesis that following AR pathway inhibition, increased GR expression and subsequent GR activation mediate PC resistance to anti-AR therapy, potentially through inducing expression of genes encoding pro-cell survival proteins. Using both in vitro and in vivo models of human PC, we found that GR expression and activation mitigate the growth inhibitory effects of AR blockade, leading to CRPC progression. Furthermore, GR activation appears to bypass the effects of reducing AR signaling through increasing the expression of established anti-apoptotic genes such as serum/glucocorticoid-regulated kinase 1 (SGK1). Moreover, increased expression of SGK1 alone is sufficient to accelerate CRPC xenograft tumor growth.
Materials and Methods
Cell Lines and Culture Materials
All cell lines were routinely screened for mycoplasma contamination using the American Type Tissue Culture (ATCC) Universal Mycoplasma Detection Kit (Manassas, VA). The PC3 human prostate carcinoma cell line was purchased from ATCC. The DU145, LNCaP, CWR-22Rv1, VCaP, and LAPC4 cells were a generous gift of Dr. John Isaacs (Johns Hopkins University) and have been previously characterized [26]. All cell lines, except the LAPC4 and VCaP cell lines, were cultured in RPMI 1640 (Mediatech) supplemented with 10 % fetal calf serum (FCS) (Atlanta Biologicals) and 1 % penicillin (100 units/ml)/streptomycin (100 Ag/ml; BioWhittaker/Cambrex). The LAPC4 human PC cell line was cultured in IMDM (ATCC) supplemented with 10 % FCS, 1 % penicillin/streptomycin, and 1 nM R1881 (Sigma-Aldrich), and VCaP cells lines were cultured in DMEM (Mediatech) supplemented with 10 % FCS and 1 % penicillin/streptomycin. Charcoal stripped serum was utilized when specified (Atlanta Biologicals). Secreted total prostate-specific antigen (PSA) was measured from collected media using the Elecsys Total PSA Assay (Roche).
Cell Treatments
LAPC4 and CWR-22Rv1 cells lines were plated at 5 × 104 in 6-cm cell culture dishes using standard media listed above, supplemented with either 10 or 1 % charcoal stripped serum as specified. Cells were treated for 2 h, 3 h, 7 h, or 14 days with combinations of the following chemical treatments (or vehicle control) as described below: 1 nM R1881 (Sigma-Aldrich), 10 μM MDV (Selleck Chemicals and Medivation), 100 nM dexamethasone (Sigma-Aldrich), 10 μM Casodex (Sigma-Aldrich), 100 nM mifepristone (Sigma-Aldrich), 1 μM CORT 122928 (Corcept Therapeutics), and 1 μM GSK 650394 (Tocris Bioscience). For doxycycline treatment, cells were grown in 1 μg/ml (in water) doxycycline (Sigma-Aldrich).
Cell Line Generation
For generation of SGK1-overexpressing cell lines, LAPC4 and CWR-22Rv1 cell lines were grown in the standard media conditions and plated at 5 × 105 per 6-cm dish. pLPCX-Flag-SGK1 and pLPCX empty vector plasmids, described previously [16], were transfected using 5 μg plasmid DNA with the Lipofectamine LTX kit according to manufacturer’s instructions (Invitrogen). Cells were then selected using puromycin (1 μg/ml, Invitrogen); stable cell line pools were generated and screened via Western blot for overexpression of SGK1. For GR knockdown, two separate systems were employed. For the LAPC4 cells, the RNAi-Ready pSIREN-RetroQ plasmid (6.4 Kb, Invitrogen) was cut with BamH1 and EcoR1 restriction endonuclease enzymes to clone in a GR-targeted short hairpin RNA modified from published GR-targeted short hairpin RNA (shRNA) studies [19]: upper, 5′-GAT CCG CGG GAG AAG ACG ATT CAT TCC TTT TTC AAG AGA AAA GGA ATG AAT CGT CTT CTC CCG TTT TTT ACG CGT G-3′; lower, 5′-AAT TCA CGC GTA AAA AAC GGG AGA AGA CGA TTC ATT CCT TTT CTC TTG AAA AAG GAA TGA ATC GTC TTC TCC CGC G-3′. The scrambled shRNA purchased from the manufacturer (Invitrogen) was used as a control. The cloned sequences were confirmed with U6 primer (5′-GGG CAG GAA GAG GGC CTA T-3′)-initiated sequencing. The PC cells were infected with high titer virus of either RNA1-Ready pSIREN-RetroQ-GRshRNA (GR shRNA) or RNA1-Ready pSIREN-RetroQ-scramble shRNA (ss shRNA) and selected with puromycin (1 μg/ml). GR depletion was assessed with Western blot analysis. A second GR knockdown method was utilized for the CWR-22Rv1 cell line. GR knockdown was achieved using the pTRIPZ doxycycline-inducible lentiviral shRNA system (Open Biosystems). HEK293T cells were plated in duplicate at 6 × 106 in 10-cm dishes. pTRIPZ shRNA GR plasmids (Open Biosystems, Lafayette, CO) were transfected using reagents from the Trans-Lentiviral Packaging System (Open Biosystems) according to manufacturer’s instructions. Virus was collected using a 0.45-μM filter and frozen down in 1.5-ml aliquots. Viral aliquots were added to 8.5 ml complete media and placed on target cells with Polybrene at 8 μg/ml overnight. Complete media were replaced for 24 h and then selected with puromycin (1 μg/ml, Invitrogen). After selection, stable pools were treated with doxycycline 1 μg/ml for 8 h, and GR depletion was confirmed with Western blot analysis.
Protein Lysate Preparation and Immunoblotting
Cell cultures were washed in ice-cold phosphate-buffered saline (PBS) and protein lysates prepared using radioimmunoprecipitation assay (RIPA) buffer as previously described [41]. Tumor lysates were prepared similarly using RIPA buffer and a glass dounce homogenizer. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Scientific). Total protein was resolved by SDS-PAGE on 4–20 % Precise Protein Gels (Pierce) and transferred to a nitrocellulose or PVDF membrane (Millipore). Membranes were blocked at 4 °C overnight or for 2 h at room temperature in either Tris-buffered saline (TBS)-Tween 20 plus 5 % nonfat dry milk or BSA (wt/vol). Primary antibodies were diluted in blocking solution and incubated with the membrane for 1 h or overnight. Following six washes for 5 min each in TBS-Tween 20, the membrane was incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h. After an identical series of washes, the HRP-conjugated secondary antibody was detected using the SuperSignal West Femto Maximum Sensitivity Chemiluminescence Substrate (Pierce). Alternatively, the LI-COR Odyssey imaging system was used to analyze immunoblots. Antibodies and concentrations were as follows: anti-SGK1 C-Term (1:500, Enzo Life Sciences [47]); anti-GR (1:1,000, Santa Cruz Biotechnology Cell Signaling); anti-AR (N20, 1:2,000, Santa Cruz); anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:10,000, Cell Signaling); and anti-beta-actin (1:10,000, Sigma Aldrich). The secondary antibody used was an anti-rabbit (Cell Signaling) at 1:10,000. For LI-COR Western blots, the secondary antibodies were anti-rabbit IRDye 800CW and anti-mouse IRDye 680RD (1:10,000, LI-COR).
Chromatin Immunoprecipitation (ChIP)
For ChIP experiments, 22Rv1 and LAPC4 cells grown in various hormonal conditions were harvested for DNA as previously described [22]. Cellular DNA and proteins were cross-linked using 1 % formalin for 15 min followed by treatment with glycine (final concentration 125 mmol/L) for 5 min. Harvested chromatin was sheared using sonication and micrococcal nuclease (Thermo Scientific) digestion. Ten percent of each lysate was set aside to analyze input protein and DNA. Appropriate DNA digestion was verified using a 3 % agarose gel for size detection. Immunoprecipiation (IP) was performed using rabbit ChIP grade antibodies for AR (N-terminal antibody raised against AR amino acids 1–50 of human origin, Santa Cruz) and GR (N-terminal antibody raised against amino acids 1–50 of human origin, Santa Cruz). A normal rabbit IgG antibody (Santa Cruz) was also used as a negative control. IP was confirmed with Western blot analysis (methods and antibodies described above) for GR and AR (Fig. S1).
Immunofluorescence
Immunofluorescence staining of xenograft tumors was performed on 6-μm paraffin tissue sections. After deparaffinization and rehydration, sections were treated with antigen retrieval buffer (DAKO) in a steamer for 20 min. Slides were incubated with 10 % normal goat serum (Cell Signaling) and with primary antibody/rabbit anti-GR H-300 (Santa Cruz, 1:100) and mouse anti-Ki67 (Dako, 1:50) diluted in PBS overnight at 4 °C. Slides were then incubated with secondary antibody/anti-mouse conjugated to Alexa 488 and anti-rabbit conjugated to Alexa Fluor 647 (Cell Signaling, 1:1,000). Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, diluted 1:1,000 in PBS) (Invitrogen) to visualize nuclei and mounted with Fluoromount mounting medium (Sigma). Immunofluorescence staining was imaged using the AMG EVOS-FL microscope or Leica-Axiovert 200. Arbitrary false coloring of images was implemented using ImageJ software. Images were captured at same intensity and exposure for all conditions within each cell line. Any contrast/brightness changes were made consistently across images. For GR IF enumeration (VCaP xenografts), five representative fields were captured at ×40 magnification, and the numbers of GR-expressing and nuclear (overlapping with DAPI) cells were counted.
Quantitative Real-Time PCR (qRT-PCR)
RNA was purified using the Qiagen RNeasy Mini Kit with the optional DNAse digestion kit (Qiagen, Valencia, CA) and quality tested using an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). For standard qRT-PCR, extracted RNA was converted to cDNA by reverse transcription using SuperScript® III Reverse Transcriptase (Invitrogen). mRNA transcript levels of SGK1, GR, PSA, Map kinase phosphatase 1 (MKP1), and GAPDH were quantified using Power SYBR® Green Master Mix (Invitrogen) using custom-designed primers (Table S1). Similarly, for targeted quantitative PCR of ChIP samples, primers were designed and implemented to amplify known AR/GR-binding regions within the promoters of PSA and SGK1 (Table S1) [1, 30]. For mRNA expression analysis, average change in threshold cycle (ΔCT) values was determined for each of the samples relative to endogenous GAPDH levels and compared to vehicle control or serum-starved control as indicated (ΔΔCT). Experiments were performed in triplicate to determine standard error of the mean (SEM) and Student’s t tests performed using SigmaPlot (version 11, Systat). For graphical representation of mRNA expression data, the fold change from control (2−ΔΔCT) was plotted on the y-axis for each condition with the error bar representing the standard error of the fold change [SE 2−ΔΔCT ≈ (ln2) × 2−ΔΔCT × (standard error of ΔCT)]. For ChIP qPCR samples, cycle threshold values were normalized to respective IgG values, and chromatin enrichment was plotted on the y-axis. Error bars for each ChIP condition represent the standard deviation of normalized CT values, and Student’s t tests were performed on these values. Real-time PCR experiments were repeated at least three independent times with representative experiment shown.
In Vitro Determination of Tumor Cell Survival
Cell lines were plated at 5 × 104 in 6-cm cell culture dishes in standard growth media. After 24 h, the media was removed, plates washed with room temperature PBS ×2, and fresh media with charcoal stripped serum with specified treatment condition. At specified days of treatment [45, 48], cell cultures were washed, trypsinized, spun to pellet, and resuspended in medium containing serum to neutralize the trypsin. Trypan blue exclusion assay was performed by adding 1:1 solution of trypan blue (Hyclone) to the cell suspension and then by counting viable cells. Counts were performed in a blinded fashion. Three biological replicates were assayed per condition, per time point, and the mean and standard error of the mean reported. A two-sided Student’s t test was performed to compare two conditions.
In Vivo Tumor Formation
All animal studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. In vivo tumor formation of LAPC4 and CWR-22Rv1 cells was conducted via a subcutaneous inoculation of 1 × 106 (LAPC4) or 2 × 105 (CWR-22Rv1) cells in 4–6-week-old male athymic nude mice (Harlan) using 100 μL of 75 % Matrigel and 25 % HBSS solution (BD Biosciences). To measure tumor take in a castrated host, host mice were surgically castrated 1 week prior to cell inoculation. Time to castrate resistant tumor formation was defined as the time from inoculation until tumors measured 100 mm3. To measure tumor progression to castration resistance, animal hosts were castrated when tumors reached 150 mm3 and followed until tumors doubled in volume, at which point progression endpoint was met and documented as such. For doxycycline treatment, animals were fed doxycycline-containing diet (200 mg/kg, Bio-Serve) ad libitum. Animals treated with mifepristone were dosed at 12 mg/kg/day using intraperitoneal injection. As in doxycycline treatment, mifepristone or vehicle treatment was started when tumors reached 150 mm3 and followed until tumors doubled in volume, at which point progression endpoint was met and documented as such. The pharmaceutical grade mifepristone (Corcept Therapeutics) used for in vivo work was first dissolved in ethanol (0.066 % w/v) and then diluted in sesame oil (1:10, Fisher Scientific) as a drug vehicle. Time-to-event (progression or tumor formation) Kaplan–Meier curves were generated using SigmaPlot (V11, Systat), and the curves were compared using a log-rank analysis. At the endpoint of all experiments, tumors were harvested for protein (snap frozen in liquid nitrogen and stored at −80) or fixed in formalin for subsequent tissue embedding.
Statistical methods and analyses were performed in collaboration with biostatisticians within the Biostatistics Core at the University of Chicago.
Results
GR Expression Increases in Prostate Cancer Following AR Inhibition
In primary human PC, GR protein expression frequently increases following anti-AR therapy [41, 50]. To investigate a possible role for increased GR signaling in CRPC progression, we first investigated GR expression in several commonly studied PC cell lines at baseline and following AR antagonism using Western blot analysis [31]. As shown in Fig. 1A, baseline GR expression was high in the AR-negative cell lines DU145 and PC3, while steady-state levels were very low in the AR-positive, AR-dependent cell lines LNCaP (mutated AR), LAPC4 (wild-type AR), and VCaP (wild-type AR). The CWR-22Rv1 (22Rv1) cell line is castration-resistant (i.e., it reliably forms xenografted tumors in a castrated host) and demonstrated high baseline GR expression and relatively low AR expression. Of note, the 22Rv1 cells express both a full length AR with a mutation in the ligand binding domain (LBD, H874Y) that is able to use other non-androgen hormones, such as the GR agonist dexamethasone as a ligand, as well as common AR splice variants found within human PC, that are active in the absence of ligand [3, 11].

GR expression in prostate cancer following androgen signaling inhibition. A Western blot of GR and AR expression for PC cell lines PC3, DU145, LNCaP, LAPC4, CWR-22Rv1, and VCaP. B NRC31 (GR) mRNA expression (above) and protein expression (below) in PC cell lines after long-term (>30 days) treatment with AR antagonist MDV3100. Error bars represent standard error of the mean, and asterisk indicates p < 0.05 paired t test. For Western blot, parental cell lines (P) are compared to the MDV3100 (MDV) treated. The GRα isoforms are labeled to the left of the immunoblot. C Xenografted PC tumors from host mice with intact androgen production versus 30-day-status post-castration were analyzed using an immunofluorescent anti-GR antibody and were also stained with DAPI. D Protein immunoblots from prostate cancer xenografts analyzed grown in intact (In) non-castrated mice, 48 h (48h), 7 days (7D), and 30 days (30D) post-castration
To determine whether PC cell lines cultured in the presence of the AR inhibition developed increased GR expression, we examined GR (NR3C1) mRNA and protein expression in cells treated with the potent AR antagonist MDV3100. Notably, when LNCaP, LAPC4, and VCaP PC cell lines were grown in 10 μM MDV3100 for >30 days, qRT-PCR revealed a significant increase in GR mRNA expression compared to the cell lines grown in control medium (Fig. 1B). LNCaP, which has no detectable GR expression under steady-state conditions, displayed the most robust upregulation of GR mRNA. These findings at the mRNA level were consistent with Western blot analysis, which also showed increased GR protein expression (Fig. 1B). The 22Rv1 line had no significant relative induction of GR mRNA or protein, although the basal level of 22Rv1 GR expression is the highest among the cell lines that express AR and may account for the lack of further induction of GR expression. In sum, these cell line data are consistent with our previous report showing increased GR expression occurring in a subset of patient tumors following AR inhibition [41].
PC cell line xenografts were then established in athymic male nude mice, and a subset of those animals was surgically castrated. The xenografts were removed from both intact (non-castrated) and castrated hosts after 30 days and examined for GR expression using immunofluorescence (Fig. 1C). We observed low tumor GR expression for LNCaP and LAPC4 cell lines and modestly elevated GR expression in the 22Rv1 xenograft grown in non-castrated mice. For both the LAPC4 and LNCaP cell lines, nuclear (staining overlays with DAPI nuclear staining) GR expression in tumor cells was significantly increased in castrated hosts compared to intact animals, suggesting the possibility of increased GR expression and activity. We also examined the proliferation marker Ki-67 within these xenografts to determine if the higher GR-expressing regions were more proliferative compared to the lower GR-expressing regions. For both LAPC4 and LNCaP xenografts, proliferation rates (percentage Ki-67 positivity) were decreased within the low-GR-expressing regions following castration. For the LAPC4 xenograft, we observed sustained Ki-67 staining in the high-GR region compared to low-GR region of the same tumor, suggesting a role for the GR in compensating for AR inhibition (Fig. S2). In the LNCaP xenograft from the non-castrated host, Ki-67 cell positivity was low and in the castrated host was diminished throughout (data not show).
We next examined GR protein levels by Western analysis within parallel tumor xenografts at various time points after castration (Fig. 1D). We found that total GR protein steadily increased within 30 days of post-castration. Of note, within xenograft lysates, there appeared to be a large fraction of GR isoform D [12], which alternatively could represent degraded GR. Compared to other AR-expressing PC xenografts within a castrated host, 22Rv1 xenografts did not exhibit a substantial increase in GR expression, similar to the in vitro findings above. However, within 22Rv1xenografts, a distinctly nuclear GR pattern was seen in the 30-day post-castration tumors (Fig. 1C). Of note, VCaP xenografts completely regressed 30 days after castration, making analysis of GR expression impossible at this time point. However, at 7 days post-castration, there was a noticeable increase in GR-expressing cells compared to the intact animal’s xenograft (Fig. 1C). Unlike the LAPC4 and LNCaP post-castration xenografts that showed distinct regions within the tumor with increased GR expression, for the VCaP cell line, the GR-expressing cells were distributed throughout the tumor and less focal. Compared to the non-castrated xenograft, there was a statistically significant increase in GR-expressing cells per imaged field (mean ± standard error of the mean number of GR+ cells 43.2 ± 3.3 versus 21.6 ± 3.1, p < 0.01). In sum, increased PC GR expression following AR antagonism or castration suggests a potential role for GR signaling in PC resistance to AR inhibition. Given these results and the increase in GR expression within a significant subset of prostate cancers we observed previously, castration-resistant prostate cancer appears to be associated with the acquisition of increased nuclear GR expression and potentially activity.
To further examine GR expression in the presence and absence of defined ligands, we studied these cell lines in media supplemented with 10 % charcoal stripped serum and various combinations of the synthetic AR agonist R1881 (1 nM) or GR agonist dexamethasone (Dex, 100 nM) and the AR antagonist MDV3100 (10 μM). Consistent with Fig. 1, 22Rv1 cells had no significant increase in GR (NR3C1) mRNA expression following 3 days of MDV3100-mediated AR antagonism (Fig. 2A). However, this short 3-day treatment demonstrated that in LAPC4 cells, there was a clear increase in GR mRNA with MDV3100. A similar pattern with GR protein at 3 days was detected by Western blotting (data not shown). Consistent changes in both mRNA and protein levels suggest that regulation of GR, following AR inhibition, occurs transcriptionally and not translationally. Longer AR antagonist exposure led to even further increases in GR protein levels within the LAPC4 cells (Fig. 2B). Consistent with previous reports, we observed that AR is stabilized (more AR protein visualized) in LAPC4 cells following androgen-mediated activation [51]. Concomitant with increased GR expression, MDV3100 treatment resulted in decreased AR protein levels (Fig. 2B). To ensure these changes were not specific to MDV3100, a second AR antagonist, Casodex (CDX, bicalutamide), was used. After 3 days of treatment with CDX, GR expression increased similarly (Fig. S2B). We also explored the expression of AR and GR for the LAPC4 and 22Rv1 cells after 14 days of various hormonal manipulations and found that the changes seen after 3 days persisted at this time point (Fig. 2C). Furthermore, within the 22Rv1 cell line, we noted a slight increase in the expression of GR with the addition of MDV3100 to R1881 and Dex. Taken together, these data further suggest an association between AR inhibition and subsequently increased GR expression. Moreover, these data support a model in which GR expression in PC increases independent of how AR signaling is blocked. Surgical castration (removing AR ligand) or treatment with a specific AR antagonist (MDV3100, CDX) both resulted in increased GR expression.

GR expression/activation levels following AR inhibitor therapy. Hormonal therapies utilized included: R1881 (1 nM), a synthetic androgen; dexamethasone (Dex, 100 nM), a GR agonist; and MDV3100 (MDV, 10 μM), an AR antagonist. A 22Rv1 and LAPC4 GR mRNA expression analysis by qRT-PCR after 3 days of treatment; normalized to GAPDH and depicted as fold change relative to vehicle. B Western blot analysis of 22Rv1 and LAPC4 GR and AR protein expression after 3 and 7 days of treatment. All conditions were treated with Dex and R1881. Asterisk denotes AR splice variants within 22Rv1 cells. C 22Rv1 and LAPC4 cells were treated for 14 days under various conditions
GR Signaling Reduces the Efficacy of AR Inhibition
To begin exploring the functional relationship between AR and GR signaling in PC, we next asked whether GR activation could alter the antiproliferative effects of blocking AR activity. The LAPC4 and 22Rv1 PC cell lines were grown in media supplemented with charcoal stripped serum, exogenous androgen, and various combinations of MDV3100 and +/− dexamethasone (100 nM) for 14 days. Trypan blue exclusion assay was then used to determine the number of live cancer cells. Dex-induced GR activation led to increased relative cell viability of PC cells in the presence of R1881 and MDV3100 (Fig. 3A, B). MDV3100 significantly decreased cell viability for the LAPC4 cells (p < 0.05), whereas total viable cell numbers were not significantly changed for the castration-resistant 22Rv1 cells (Fig. 3A). Paradoxically, when 22Rv1 cells were grown with R1881 and Dex, the addition of enzalutamide increased cell viability (Fig. 3A). Although the total number of viable cells in the 22Rv1 cell line did not decrease upon enzalutamide treatment, in both cell lines, GR activation with Dex increased relative cell viability following AR antagonism with enzalutamide (Fig. 3B). Notably, Dex at 100 nM had no impact on LAPC4 cell viability; however, it did decrease cell proliferation in the 22Rv1 cells when AR signaling was intact (with R1881, Fig. 3A).

GR expression/activation and PC cell viability. Cells were cultured for 14 days under the same conditions described in Fig. 2 that also included mifepristone (Mif, 100 nM), a GR antagonist. Total viable cells (A) and relative viable cells (B) are shown. For relative viability (b), the control denominator is listed within the figure for each data set. R = R1881, D = Dex, MDV = MDV3100, and Mif = Mifepristone. Error bars represent standard error of the mean (SEM), and asterisk indicates p < 0.05 paired t test. C Kaplan–Meier analysis of time to initiation of castration-resistant tumors for LAPC4 cells stably expressing a GR-specific shRNA versus SS shRNA (p = 0.038, log-rank). Western blot analysis of GR protein expression [inset]. D Progression-free survival for 22Rv1 cells stably expressing doxycycline (Dox)-inducible shRNA construct with and without Dox treatment in the setting of castration (p = 0.009, log-rank). Western blot of GR showing GR depletion with Dox [inset]
Although testing cell viability in the presence of androgen (R1881) is most relevant given that all patients with prostate cancer have detectable androgen levels, we also performed a tumor cell viability experiment with androgen and glucocorticoid alone (Fig. S3B). Compared to vehicle control, R1881 led to increased number of viable cells for LAPC4 cells, while Dex alone had no substantial effect on viability. For the 22Rv1 cells, we saw a similar increase in viable cells with R1881. However, Dex alone also increased the number of viable cells. This may be due to the mutation in the full length AR for the 22Rv1 cells, which can bind to Dex as a ligand [3]. Of note, in both cell lines, higher dose Dex (1 μM) led to decreased total cell numbers in the context of intact AR signaling (R1881 + Dex condition, Fig. S3C), consistent with published data [51]. However, following AR antagonism, the corticosteroid Dex at both doses tested led to increased relative PC cell viability (Figs. 3A, B and S3C). Treatment of a third PC cell line, VCaP, yielded results similar to the LAPC4 and 22Rv1 cell lines; the addition of Dex led to increased cell viability compared to MDV3100 treatment alone (Fig. S3A).
To determine whether the pro-survival effects seen with Dex were mediated through GR signaling [32], mifepristone (Mif) was used to antagonize the GR. The addition of Mif reversed the cell survival effects of Dex in the setting of MDV3100 treatment (Fig. 3A, B). Because Mif has reported mixed agonist/antagonist activity and could act as ligand to mutant AR, a highly selective GR antagonist (CORT 122928) without detectable AR ligand binding (Dr. Robert Roe, Corcept Therapeutics, personal communication) was also examined. Similar to Mif, the protective effects of GR activation were reduced with CORT 122928 (Fig. S3D). In aggregate, these data support the hypothesis that GR activation blunts the effectiveness of AR blockade.
We next sought to test the hypothesis that the GR activity contributes to castration-resistant PC progression in vivo. To ensure clearance of systemic testicular androgens, the animals underwent surgical castration 1 week prior to tumor cell injections. Stably transfected pools of LAPC4 cells expressing an shRNA construct with validated target specificity [16] to GR (NR3C1) (~75 % depletion by densitometry, Fig. 3C inset), or a scrambled sequence (SS) shRNA control, were injected subcutaneously (SC) into immunocompromised mice. The animals were monitored for the development of castration-resistant PC tumors with serial measurements. As shown in Fig. 3C, LAPC4-GR shRNA cells demonstrated a significant decrease in the development of castration-resistant tumors, corresponding to an increase in castration-resistant tumor-free survival (p = 0.038). With this same shRNA construct, stably expressing 22Rv1 cell line pools showed no decrease in GR expression; therefore, a doxycycline (Dox)-inducible GR-specific knockdown (KD) construct (TRIPZ shRNAmir, Open Biosystems) was used. Several constructs were tested for the efficacy of GR knockdown, and the H12 TRIPZ plasmid was utilized for further experimentation as it had the most significant degree of knockdown in our cell line. With the TRIPZ H12 plasmid, there is a ~65 % knockdown (by densitometry) of the GR protein level within a stably selected pool of GR-KD cells with Dox treatment by Western blot (Fig. 3D, inset). Doxycycline had no effect on GR expression in the non-silencing control (NSC)-transfected cells. In addition, there was no effect on cell viability when the NSC cells were treated with Dox in vitro (data not shown). When 22Rv1 GR-KD cells were established as SC xenografts, the animals were surgically castrated and half were fed Dox-impregnated food. The animals were followed until CRPC tumor progression using serial tumor measurements. Given the aggressive nature of the 22Rv1 cell line, large castration-resistant tumors developed within days of castration. However, similar to the LAPC4 xenografts, the 22Rv1 xenografts had a statistically significant delay in castration-resistant progression when GR was depleted using GR shRNA expression (p = 0.009; Fig. 3D). In addition to delaying time to castration-resistant progression, the xenograft tumors following doxycycline-induced GR depletion were smaller then the non-depleted control tumors over time (two-way ANOVA, p < 0.01) (Fig. S4A). For the LAPC4 xenografts, as many xenografts did not initiate growth, comparison of tumor sizes between the two groups was not feasible. In both xenograft experiments, tumors were harvested and GR protein levels were examined by immunofluorescence, confirming continued shRNA-induced GR knockdown (Fig. S4B). Given the highly castration-resistant nature of the 22Rv1 line and their relatively small (but still statistically significant) delay in castration-resistant tumor development with GR depletion, a second in vivo model was utilized for this cell line. Upon castration-resistant tumor initiation, mice were treated with mifepristone (12 mg/kg/day) or vehicle daily. Mifepristone treatment was associated with an approximately 33 % prolongation of time to tumor progression compared to vehicle treatment (Fig. S4C). Collectively, these data support a role for GR signaling in CRPC progression and imply that blocking GR activity may delay tumor progression.
Increased GR Activation Can Induce Expression of AR-Regulated Pro-Cell Survival Genes in PC
Our results suggest that GR expression and activation can increase tumor cell survival and are pro-tumorigenic despite AR signaling inhibition. One possible mechanism for these observations is that GR could regulate the expression of shared AR and GR target genes in PC. The gene encoding PSA, KLK3, is an AR-regulated gene whose protein product is a serum biomarker used clinically to monitor PC disease burden. To explore the potential for GR to regulate canonical AR target genes following AR inhibition, KLK3 (PSA)was the initial gene interrogated. We first examined PSA production in PC cells under controlled conditions of AR and GR activation. As anticipated, following AR antagonism with MDV3100, there was a significant decrease in PSA secretion in conditioned media in both LAPC4 and 22Rv1 cell lines (Fig. 4A). Interestingly, in both cell lines, GR activation with Dex alone induced PSA production and when combined with R1881 led to PSA levels greater than those seen with R1881 alone. Furthermore, GR activation resulted in sustained PSA expression despite MDV3100 treatment. PSA secretion did not decrease significantly with the addition of MDV3100 to Dex treatment; however, as expected, PSA secretion decreased with concomitant mifepristone treatment (Fig. 4A).

Ligand-bound GR regulation of AR target genes in the absence of AR signaling. A Conditioned media were collected after 3 days of agonist and/or antagonist treatment, and secreted PSA was measured by ELISA. B SGK1 protein expression by Western blot analysis after 7 days of treatment. C Protein immunoblots from PC xenografts at various time points post-castration as in Fig. 1d. D SGK1 immunoblots of GR-depleted shRNA expressing cell lines shown in Fig. 2 showing decreased SGK1 expression (normalized densitometry shown) in the setting of GR depletion. E mRNA was collected from LAPC4 and 22Rv1 cells treated for 7 days under various hormonal conditions, and MKP1 gene expression was assessed by qRT-PCR. The conditions used included: R1881 (1 nM), a synthetic androgen; dexamethasone (Dex, 100 nM), a GR agonist; MDV3100 (MDV, 10 μM), an AR antagonist; and mifepristone (Mif, 100 nM), a GR antagonist
We next tested the hypothesis that in the context of AR antagonism, GR activation regulates AR target genes encoding proteins promoting cell viability. Prior to a global study of GR-regulated genes contributing to CRPC progression, we started with the known AR- and GR-regulated genes SGKI and MKP1. These genes encode proteins which are critical mediators of GR-regulated survival in the context of breast cancer and chemotherapy sensitivity [27, 48]. SGK1 encodes an AGC-family kinase that is known to be transcriptionally upregulated by both the AR and GR [36, 47]. In models of human PC, increased SGK1 expression facilitates AR-regulated cell survival, and in breast cancer, SGK1 expression confers relative resistance to chemotherapy-induced cell death [36, 37, 47]. Therefore, we examined SGK1 expression induced by GR activation during treatment with AR agonists and antagonists. Using Western blot analysis, we found that similar to PSA secretion, SGK1 expression in LAPC4 cells increased after AR and GR activation and was highest when both receptors were activated concomitantly (Fig. 4B). MDV3100 substantially decreased SGK1 expression despite the presence of R1881. Within this context of AR blockade, GR activation again increased SGK1 expression and this was reversed with the GR antagonist Mif. As was the case with PSA, for the 22Rv1 cells, SGK1 appeared to be most strongly regulated by the GR. In fact, SGK1 expression is decreased when Dex and R1881 are combined, compared to Dex alone, and again increased with AR antagonism by MDV3100. SGK1 expression in this line also decreased down to levels observed in the vehicle condition when treated with Mif. Interestingly, although SGK1 seems to be minimally regulated by AR in vitro, upon surgical castration in vivo, the 22Rv1 xenografts expressed much less SGK1 after 48 h (Fig. 4C). At 30 days post-castration, SGK1 levels again increased which may be due to the activation of the GR over time. In the LAPC4 xenografts, where GR expression increased post-castration, we saw sustained SGK1 levels throughout the time course of castration (Fig. 4C). Finally, we explored SGK1 expression in our GR-KD cell lines. In both cell lines, with the addition of R1881, MDV3100, and dexamethasone, GR knockdown decreased SGK1 expression (Fig. 4D).
MKP1 is another GR target gene whose induction is associated with increased cell survival [47, 48]. The role of MKP1 in PC is unknown. MKP1 protein was not detectable using available antibodies (data not shown), and evaluation via Western blotting was not possible in these cell lines. However, using quantitative RT-PCR, MKP-1 (dual specificity phosphatase 1 (DUSP1) mRNA expression increased in both cell lines following treatment with R1881 and dexamethasone, while steady-state mRNA levels decreased with MDV3100 and decreased further with GR antagonism (Fig. 4C). The pattern of anti-apoptotic gene regulation suggests that increased GR signaling may counteract diminished AR activity via the upregulation of pro-survival genes to facilitate cancer cell survival.
To determine whether or not GR directly regulates our AR target genes of interest, we first analyzed mRNA expression immediately following AR and/or GR modulation with various ligands and antagonists. PSA (KLK3) expression was lowest in the R1881 + MDV3100 condition, increased significantly with the addition of Dex despite AR antagonism, and decreased with Mif (Fig. 5A). These trends were even more apparent when looking at SGK1 expression (Fig. 6A). Similar results were seen with the pro-survival gene MKP1 (data not shown).

Increased regulation of PSA by the GR following AR antagonism. A mRNA was collected from LAPC4 and 22Rv1 cells treated for 2 h under various hormonal conditions, and PSA gene expression was assessed by qRT-PCR. B Both 22Rv1 and LAPC4 cells were treated for 3 days in either vehicle, R1881 (1 nM), or R1881 + MDV3100 (10 μM) containing media. Conditions specifying dexamethasone treatment were stimulated with Dex (100 nM) for 1 h prior to chromatin harvest. Targeted qPCR was performed for the PSA promoter after ChIP, and y-values represent fold chromatin enrichment (relative to IgG controls for each condition). Error bars represent standard deviation of mean cycle threshold values, and asterisk indicates p < 0.05 of performed paired t tests (only included for key statistical analysis)

Increased regulation of SGK1 by the GR following AR antagonism. A mRNA was collected from LAPC4 and 22Rv1 cells treated for 2 h under various hormonal conditions, and SGK1 gene expression was assessed by qRT-PCR. B Both 22Rv1 and LAPC4 cells were treated for 3 days in either vehicle, R1881 (1 nM), or R1881 + MDV3100 (10 μM)-containing media. Conditions specifying dexamethasone treatment were stimulated with Dex (100 nM) for 1 h prior to chromatin harvest. Targeted qPCR was performed for the region within the SGK1 promoter after ChIP, and y-values represent fold chromatin enrichment (relative to IgG controls for each condition). Error bars represent standard deviation of mean cycle threshold values, and asterisk indicates p < 0.05 of performed paired t tests (only included for key statistical analysis)
To further understand the mechanism of increased expression of these specific AR target genes following GR activation, we examined GR occupancy within their respective promoter regions. ChIP experiments were conducted in both 22Rv1 and LAPC4 lines under various hormonal conditions. Of note, to parallel the clinical condition where patients are treated with AR antagonists chronically, we performed these assays with AR activation at steady state (3 days of treatment with R1881 ± MDV3100). Specifically, we sought to examine the difference in GR chromatin binding between conditions where AR is chronically activated with androgen compared to its inhibition with a second-generation AR antagonist such as MDV3100. Targeted qPCR was performed following either AR or GR ChIP for PSA and SGK1 known AR binding regions within their respective promoters. Due to the presence of AR splice variants, as well as the possibility of the mutant AR potentially utilizing Dex as a ligand, we began our analysis with the 22Rv1 cells. As anticipated, relative to control IgG ChIP, treatment with R1881 increased AR occupancy at the both PSA and SGK1 promoter regions (Fig. S5A). Despite the presence of ligand-independent splice mutants in the 22Rv1 line, AR promoter occupancy decreased following treatment with the AR antagonist MDV3100 (Fig. S5A). Of note, the addition of Dex to R1881 treatment compared to R1881 treatment alone increased AR chromatin occupancy of the KLK3 promoter, potentially due to mutant AR LBD responding to Dex [3]. However, when AR is antagonized with MDV3100, the addition of Dex to R1881 treatment did not significantly increase the chromatin binding of AR within either promoter region (Fig. S5A). We also tested the chromatin binding to the KLK3 promoter for the 22Rv1 cells when treated with Dex alone. Importantly, there was no recruitment of AR to the KLK3 promoter with Dex alone in comparison to vehicle (Fig. S5B). In the LAPC4 cells, R1881 led to increased AR occupancy of both gene promoter regions, which was reversed with the addition of MDV3100 (Fig. S5C).
Most pertinent to our underlying hypothesis, we next evaluated GR occupancy of known AR/GR binding regions in the PSA and SGK1 gene promoter regions. In the 22Rv1 cells, when AR signaling was antagonized with MDV3100, GR occupancy of target gene promoter regions significantly increased compared to the R1881 + Dex condition (Figs. 5B and 6B). In fact, for SGK1, there was essentially no chromatin enrichment with GR activation when AR signaling was intact, whereas there was a doubling of occupancy when GR was activated following AR antagonism (Fig. 6B). The chromatin enrichment patterns seen in the LAPC4 cells were similar within these regions of DNA. As with the 22Rv1’s, for LAPC4’s, in the setting of glucocorticoid, GR occupancy significantly increased following AR antagonism (Figs. 5B and 6B). The enrichment of chromatin occupancy seen within the LAPC4 cells may be in part due to the increase in GR protein levels following MDV3100 treatment. With respect to GR occupancy in the SGK1 gene promoter following AR antagonism, GR occupancy increased in both cell lines tested. This enrichment was significant in both lines; however, the magnitude of change was not identical with the 22Rv1’s showing a larger fold enrichment. The reason for this difference is not clear; however, in the 22Rv1’s, there was a decrease in AR occupancy with MDV3100, potentially leading to more accessibility within this specific section of the SGK1 promoter for GR binding within this line. Dex alone increased GR occupancy of the KLK3 promoter compared to vehicle alone (Fig. S5B). Notably, as with 3 days of androgen treatment (Fig. 5B), following 1 h of R1881 treatment, GR promoter occupancy after Dex activation was highest with the addition of MDV3100 (Fig. S5B). In sum, these data support a model in which following AR antagonism with MDV3100, chromatin occupancy of activated GR is increased, which leads to maintained expression of AR/GR target genes.
SGK1 Expression Promotes CRPC Progression
We next sought to determine whether SGK1 expression is required for the effects of GR following anti-AR treatment. LAPC4 and 22Rv1 cell lines were grown for 7 days in androgen (R1881), Dex, MDV3100, and/or the SGK1 inhibitor GSK650394 and assayed for total viable cancer cells using trypan blue exclusion assay (Fig. 7A). Of note, GSK650394 is a validated SGK1/2 (SGK1 > SGK2) small molecule inhibitor that has been shown to counteract AR-mediated PC cell survival in vitro with similar effect in comparison to SGK1-targeted siRNA [37]. It has not been tested in PC with respect to mitigating GR activation. For LAPC4 cells, GSK650394 alone decreased tumor cell viability, which is perhaps consistent with the higher baseline AR-regulated SGK1 expression in that cell line (Fig. 4B). In both cell lines, GSK650394 increased MDV3100’s ability to decrease prostate cancer cell survival (Fig. 6A). As GR modulation impacted in vivo castration-resistant tumor progression (Fig. 3C, D), we then tested the hypothesis that the GR-regulated pro-survival gene SGK1 would be sufficient to promote castration resistance. Stable pools of SGK1-Flag overexpressing or empty vector PC cell lines were generated (Fig. 7B, C insets). Of note in the LAPC4 line, the endogenous levels of SGK1 were lower than those of the SGK1-Flag-overexpressing cells. However, the 22Rv1 SGK1-overexpressing cells had levels of protein comparable to parental cells treated with R1881 + Dex + MDV3100 (Fig. S4D). Furthermore, with glucocorticoid treatment in vitro, SGK1 levels persisted despite AR antagonism with MDV3100 (Fig. 4B). Therefore, SGK1-overexpressing cells were appropriate for testing SGK1’s role in castration-resistant progression in vivo. Castrated athymic nude mice were injected bilaterally (one side vector, one site SGK1-Flag-expressing cells) in the subcutaneous tissues and monitored for castration-resistant tumor development with serial tumor measurements. In both cell lines, there was accelerated castration-resistant tumor initiation with SGK1 overexpression (Fig. 7B, C; log-rank LAPC4, p = 0.002; 22Rv1 p = 0.033). Thus, the overexpression of SGK1, a gene that is upregulated by both AR and GR activation, facilitates CRPC establishment in vivo.

The functional role of SGK1 in androgen signaling-deprived PC. A PC cells were treated with SGK1 inhibitor GSK650394 (GSK, 1 μM) for 7 days, and cell viability was assessed with trypan blue exclusion assay. Cell survival results are displayed relative to R1881 plus dexamethasone control (first bar). Error bars represent standard error of the mean, and asterisk denotes p < 0.05 using a paired Student’s t test. B Median time to tumor initiation of LAPC4 cells ectopically expressing SGK1-Flag versus control within a castrate host is 76 days versus not reached (log-rank test p = 0.002). C Time to 22Rv1 xenograft establishment showing median castration-resistant tumor-free survival of mice bearing 22Rv1 SGK1-Flag tumors versus control tumors 22 versus 37 days (p = 0.033)
Discussion
Perhaps the largest unmet need for patients with metastatic PC is the identification and validation of signaling pathways that play a role in enabling resistance to AR-targeted therapy. Understanding the mechanisms responsible for tumor progression despite maximal blockade of AR activity is key to improving therapy of CRPC. Data from several laboratories, using PC model systems and patient samples, have shown that in prostate cancer cells, GR expression can increase following AR blockade. Here, we show that increased GR expression and activity in CRPC maintains prostate cancer cell viability despite AR inhibition and enables CRPC progression in vivo. Although it has been proposed that glucocorticoids may foster PC cell survival through ligand activation of mutant AR [32], our data suggest that glucocorticoids can activate the GR within prostate cancer and increase pro-survival gene expression. Furthermore, the GR antagonists, mifepristone or CORT 122928, were able to reverse the pro-cell survival effects of glucocorticoids, presumably through inhibiting GR signaling. The effects of GR activity in CRPC are complex and may well be dependent on the AR activation state. In the context of functional AR signaling, our data (Figs. 3A and S3A, B), as well as others, suggest that GR activation can slow prostate cancer cell proliferation [13, 50, 51]. Clinically, high dose glucocorticoids can occasionally confer palliative benefits to advanced PC patients. In CRPC, AR signaling is intact, which perhaps explains this observation. In addition to direct antiproliferative effects on prostate cancer cells, glucocorticoids may also have anticancer effects in CRPC through decreasing adrenal steroid production (e.g., androgen production), thereby decreasing AR activity within prostate cancer cells. Although when AR is active glucocorticoids may be antiproliferative in prostate cancer, our data suggest that when AR is inhibited (such as with the second-generation AR antagonist MDV3100) GR signaling may in contrast contribute to CRPC progression. A clinical trial of the GR antagonist mifepristone for the treatment of CRPC has been previously reported [42]. In this trial, when mifepristone was used alone, there were no clinical responses. However, following treatment with the mifepristone, patients experienced a significant increase in adrenal androgen production. The authors postulated that this increase in adrenal androgens promoted prostate cancer cell survival [42]. Based upon our findings, treatment with GR antagonists such as mifepristone [8] or CORT 122928 should be considered in conjunction with an effective AR antagonist such as enzalutamide.
In addition to providing evidence that increased GR activation in prostate cancer can promote failure of AR-targeted therapy, we also found that two GR target genes required for glucocorticoid-mediated cell survival in human breast epithelial cells (SGK1 and MKP1) are upregulated by GR signaling despite AR antagonism. A small molecule SGK1 inhibitor was able to overcome AR and GR activation to inhibit PC growth in vitro, and ectopic SGK1 expression was sufficient to enable castration-resistant progression in vivo. SGK1 is of particular interest as an AR/GR target gene in CRPC as it is activated by phosphorylation downstream of PI3-K/mTOR signaling, which is often aberrantly activated in PC especially in the setting of complete AR blockade [6]. Therefore, SGK1 inhibition may be a viable treatment strategy along with inhibiting AR activity (e.g., abiraterone or enzalutamide) [17].
Although our data demonstrate a potential role for GR signaling in CRPC progression and AR-targeted therapy resistance, it is a beginning step in this line of inquiry, which will open the door to further investigation with respect to GR signaling in prostate cancer. One limitation of this work is that it was focused on established pro-survival AR target genes that could be downstream mediators of GR’s role in CRPC progression. The target genes in prostate cancer for these two receptors are not entirely overlapping [33]. Furthermore, even when the two receptors share the ability to upregulate a single transcriptional target (e.g., SGK1), as we show for the 22Rv1 cells regulation may not be identical in magnitude. AR/GR target gene regulation is undeniably complex; a global understanding of GR’s gene regulation pattern in CRPC, especially in the context of resistance to highly effective second-generation AR antagonists such as MDV3100, will be an essential next step. Further large-scale experiments are underway to characterize more completely how AR and GR DNA occupancy differ depending on the activation of each receptor. These results may identify novel AR gene targets that GR regulates following AR blockade to maintain prostate cancer cell survival.
Our studies focused on the LAPC4 and 22Rv1 cell lines as they express both detectable AR and GR under standard growth conditions yet have distinctly different AR isoforms and AR/GR expression levels as well as different GR changes in response to AR inhibition. With respect to dexamethasone potentially serving as a ligand for the mutated androgen receptor in the 22Rv1 cells, our data show the addition of dexamethasone did not increase AR chromatin occupancy for the genes we studied in the context of MDV3100 (Fig. S5). The 22Rv1s are an established, aggressive, and castration-resistant cell line that potentially utilizes multiple mechanisms in the evasion of AR blockade. Furthermore, 22Rv1s express known ligand-independent AR splice variants that may limit the efficacy of AR antagonism [11, 23, 28]. Interestingly, despite the ligand-independent splice variants in the 22Rv1 cell line, we have shown that MDV3100 robustly decreases AR target gene expression and AR chromatin binding. Critically, we show that regardless of the lower relative potency of MDV3100 in this cell line, GR expression and activation can mitigate the efficacy of AR pathway inhibition. We utilized multiple nuclear hormones and antagonists within this work. It is conceivable that some of the changes observed are due to pleiotropic and nonspecific effects of these agents. With respect to the nuclear receptor antagonists studied, MDV3100 is a specific AR antagonist, and we observed no inhibitory affects on GR signaling or GR chromatin occupancy (in fact, we observed the opposite—an increase in GR activity following treatment with MDV3100). Mifepristone has been described to possess mixed antagonistic and agonistic properties and has established AR antagonist activity [39]. Given that MDV3100 [45] and mifepristone [3] have the same affinity for the AR ligand binding domain, at 10 μM, MDV3100 will likely outcompete 100 nM mifepristone. Furthermore, our data along with the established MDV3100 literature [45] illustrate that 10 μM MDV3100 effectively antagonizes 1 nM R1881, whereas at a 100:1 ratio of R1881 to Mif (e.g., 100 nM Mif and 1 nM R1881), mifepristone had little effect antagonizing the AR agonist [39]. Thus, with concurrent treatment of 10 μM MDV3100, the AR-targeted properties of mifepristone are likely inconsequential. Finally, the novel and highly specific GR antagonist CORT 122928 had effect similar to mifepristone on cell viability by reversing the pro-survival benefits associated with GR activation (Fig. S3C).
Our work suggests an important role for increased GR expression and activity in CRPC progression despite AR antagonism and underscores the need to consider additional nuclear receptors and their role in the pathogenesis of prostate cancer progression. In addition to the work presented herein, the potential importance of GR signaling in accelerating CRPC progression has also been demonstrated by a concurrent publication using alternate PC models [2]. This work demonstrated that GR expression can increase with AR blockade and that decreased GR activity or expression delays CRPC progression in vitro and in vivo, particularly in a PC model with cells engineered to overexpress the AR. Furthermore, as with our work, the work by Arora and colleagues showed that GR can regulate AR target genes following AR antagonism. In contrast, our work utilized several human prostate cancer cell lines, with endogenous AR and GR expression that span the clinical spectrum of prostate cancer with respect to AR expression and AR variants and mutants. Furthermore, we utilized two separate GR antagonists that are FDA approved (mifepristone) or in clinical development (CORT 122928) that mitigated the prostate cancer cell survival advantage conferred by GR activation. Finally, our report uniquely suggests that the GR-regulated pro-survival gene SGK1 may be sufficient to promote CRPC progression.
Further work is necessary to dissect the mechanisms responsible for increased GR expression in prostate cancer. It is not known whether the increased GR expression observed is present globally in a tumor following AR inhibition; our xenograft would suggest that there may be a selection for a subpopulation of PC cells expressing high GR. Work with the estrogen receptor (ER) in breast cancer has suggested that the ER can contribute to proteasome-mediated degradation of GR [21], and this may be one plausible mediator of the GR expression changes seen in PC. A recent report suggests that in certain prostate cancer cell lines, GR alpha and beta isoforms may have different roles: GR-beta stimulates proliferation while GR-alpha inhibits cell growth [24]. Characterizing the gene regulation differences between the numerous GR alpha isoforms in prostate cancer, specifically with respect to the development of AR-targeted therapy resistance, is warranted.
In summary, identifying pro-cell survival signaling networks mediating tumor growth and resistance to AR antagonism is critical for improving CRPC treatment. Several different mechanisms of resistance contributing to progression of CRPC are undoubtedly important. Recently, selection of enzalutamide-resistant clones in the setting of chronic enzalutamide treatment was found to be associated with acquired AR mutations in CRPC [4]. Our data suggest that increased GR signaling may be another endocrine-mediated mechanism mediating tumor cell viability through upregulation of pro-survival genes that would otherwise be downregulated following AR inhibition.
References
Andreu-Vieyra C, Lai J, Berman BP, Frenkel B, Jia L, Jones PA et al (2011) Dynamic nucleosome-depleted regions at androgen receptor enhancers in the absence of ligand in prostate cancer cells. Mol Cell Biol 31:4648–4662
Arora Vivek K, Schenkein E, Murali R, Subudhi Sumit K, Wongvipat J, Balbas Minna D et al (2013) Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 155:1309–1322
Attardi BJ, Burgenson J, Hild SA, Reel JR (2004) Steroid hormonal regulation of growth, prostate specific antigen secretion, and transcription mediated by the mutated androgen receptor in CWR22Rv1 human prostate carcinoma cells. Mol Cell Endocrinol 222:121–132
Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA et al (2013) Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife 2:e00499
Bolton EC, So AY, Chaivorapol C, Haqq CM, Li H, Yamamoto KR (2007) Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev 21:2005–2017
Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S et al (2011) Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19:575–586
Chen Y, Sawyers CL, Scher HI (2008) Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol 8:440–448
Clark RD (2008) Glucocorticoid receptor antagonists. Curr Top Med Chem 8:813–838
Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A et al (2012) ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res 72:1494–1503
Cleutjens CB, Steketee K, van Eekelen CC, van der Korput JA, Brinkmann AO, Trapman J (1997) Both androgen receptor and glucocorticoid receptor are able to induce prostate-specific antigen expression, but differ in their growth-stimulating properties of LNCaP cells. Endocrinology 138:5293–5300
Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ (2008) Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res 68:5469–5477
Duma D, Jewell CM, Cidlowski JA (2006) Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J Steroid Biochem Mol Biol 102:11–21
Fakih M, Johnson CS, Trump DL (2002) Glucocorticoids and treatment of prostate cancer: a preclinical and clinical review. Urology 60:553–561
Gabaglia CR, DeLaney A, Gee J, Halder R, Graham FL, Gauldie J et al (2010) Treatment combining RU486 and Ad5IL-12 vector attenuates the growth of experimentally formed prostate tumors and induces changes in the sentinel lymph nodes of mice. J Transl Med 8:98
Geley S, Fiegl M, Hartmann B, Kofler R (1996) Genes mediating glucocorticoid effects and mechanisms of their regulation. Reviews of physiology biochemistry and pharmacology, vol 128. Springer, Berlin, pp 1–97
Hall BA, Kim TY, Skor MN, Conzen SD (2012) Serum and glucocorticoid-regulated kinase 1 (SGK1) activation in breast cancer: requirement for mTORC1 activity associates with ER-alpha expression. Breast Cancer Res Treat 135:469–479
Hammond M, Washburn DG, Hoang HT, Manns S, Frazee JS, Nakamura H et al (2009) Design and synthesis of orally bioavailable serum and glucocorticoid-regulated kinase 1 (SGK1) inhibitors. Bioorg Med Chem Lett 19:4441–4445
Huggins C, Stevens RJ, Hodges C (1941) Studies on prostatic cancer. II. The effects of castration on advanced carcinoma of the prostate gland. Arch Surg 43:209–223
Jeanneteau F, Garabedian MJ, Chao MV (2008) Activation of Trk neurotrophin receptors by glucocorticoids provides a neuroprotective effect. Proc Natl Acad Sci U S A 105:4862–4867
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61:69–90
Kinyamu HK, Archer TK (2003) Estrogen receptor-dependent proteasomal degradation of the glucocorticoid receptor is coupled to an increase in mdm2 protein expression. Mol Cell Biol 23:5867–5881
Kregel S, Kiriluk KJ, Rosen AM, Cai Y, Reyes EE, Otto KB et al (2013) Sox2 is an androgen receptor-repressed gene that promotes castration-resistant prostate cancer. PLoS One 8:e53701
Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM (2013) Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res 73:483–489
Ligr M, Li Y, Logan SK, Taneja S, Melamed J, Lepor H et al (2012) Mifepristone inhibits GRbeta coupled prostate cancer cell proliferation. J Urol 188:981–988
Lin MF, Kawachi MH, Stallcup MR, Grunberg SM, Lin FF (1995) Growth inhibition of androgen-insensitive human prostate carcinoma cells by a 19-norsteroid derivative agent, mifepristone. Prostate 26:194–204
Litvinov IV, Vander Griend DJ, Xu Y, Antony L, Dalrymple SL, Isaacs JT (2006) Low-calcium serum-free defined medium selects for growth of normal prostatic epithelial stem cells. Cancer Res 66:8598–8607
Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD (2001) Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem 276:16649–16654
Nadiminty N, Tummala R, Liu C, Yang J, Lou W, Evans CP et al (2013) NF-kappaB2/p52 induces resistance to enzalutamide in prostate cancer: role of androgen receptor and its variants. Mol Cancer Ther 12:1629–1637
Nishimura K, Nonomura N, Satoh E, Harada Y, Nakayama M, Tokizane T et al (2001) Potential mechanism for the effects of dexamethasone on growth of androgen-independent prostate cancer. J Natl Cancer Inst 93:1739–1746
Pan D, Kocherginsky M, Conzen SD (2011) Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res 71:6360–6370
Pienta KJ, Abate-Shen C, Agus DB, Attar RM, Chung LW, Greenberg NM et al (2008) The current state of preclinical prostate cancer animal models. Prostate 68:629–639
Richards J, Lim AC, Hay CW, Taylor AE, Wingate A, Nowakowska K et al (2012) Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: a rationale for increasing abiraterone exposure or combining with MDV3100. Cancer Res 72:2176–2182
Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, Hautaniemi S et al (2013) FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res 73:1570–1580
Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller MD et al (2012) Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 367:1187–1197
Scher HI, Sawyers CL (2005) Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol 23:8253–8261
Shanmugam I, Cheng G, Terranova PF, Thrasher JB, Thomas CP, Li B (2007) Serum/glucocorticoid-induced protein kinase-1 facilitates androgen receptor-dependent cell survival. Cell Death Differ 14:2085–2094
Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W et al (2008) Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res 68:7475–7483
Smith RG, Syms AJ, Nag A, Lerner S, Norris JS (1985) Mechanism of the glucocorticoid regulation of growth of the androgen-sensitive prostate-derived R3327H-G8-A1 tumor cell line. J Biol Chem 260:12454–12463
Song LN, Coghlan M, Gelmann EP (2004) Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol Endocrinol 18:70–85
Szmulewitz RZ, Chung E, Al-Ahmadie H, Daniel S, Kocherginsky M, Razmaria A et al (2012) Serum/glucocorticoid-regulated kinase 1 expression in primary human prostate cancers. Prostate 72:157–164
Szmulewitz RZ, Clark R, Lotan T, Otto K, Taylor Veneris J, Macleod K et al (2012) MKK4 suppresses metastatic colonization by multiple highly metastatic prostate cancer cell lines through a transient impairment in cell cycle progression. Int J Cancer 130:509–520
Taplin ME, Manola J, Oh WK, Kantoff PW, Bubley GJ, Smith M et al (2008) A phase II study of mifepristone (RU-486) in castration-resistant prostate cancer, with a correlative assessment of androgen-related hormones. BJU Int 101:1084–1089
Tenbaum S, Baniahmad A (1997) Nuclear receptors: structure, function and involvement in disease. Int J Biochem Cell Biol 29:1325–1341
Tieszen CR, Goyeneche AA, Brandhagen BN, Ortbahn CT, Telleria CM (2011) Antiprogestin mifepristone inhibits the growth of cancer cells of reproductive and non-reproductive origin regardless of progesterone receptor expression. BMC Cancer 11:207
Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V et al (2009) Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324:787–790
Vogelzang N (2006) Comprehensive textbook of genitourinary oncology, 3rd edn. Lippincott Williams & Wilkins, Philadelphia
Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD (2004) Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res 64:1757–1764
Wu W, Pew T, Zou M, Pang D, Conzen SD (2005) Glucocorticoid receptor-induced MAPK phosphatase-1 (MPK-1) expression inhibits paclitaxel-associated MAPK activation and contributes to breast cancer cell survival. J Biol Chem 280:4117–4124
Yan TZ, Jin FS, Xie LP, Li LC (2008) Relationship between glucocorticoid receptor signal pathway and androgen-independent prostate cancer. Urol Int 81:228–233
Yemelyanov A, Bhalla P, Yang X, Ugolkov A, Iwadate K, Karseladze A et al (2012) Differential targeting of androgen and glucocorticoid receptors induces ER stress and apoptosis in prostate cancer cells: a novel therapeutic modality. Cell Cycle 11:395–406
Zoubeidi A, Zardan A, Beraldi E, Fazli L, Sowery R, Rennie P et al (2007) Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Res 67:10455–10465
Acknowledgments
We thank Dr. Walter Stadler, Director of the Genitourinary Oncology Program, and Dr. Carrie Rinker-Schaeffer, Director of Urologic Research at The University of Chicago, for their guidance and support of this work. We thank the expert technical assistance of the Human Tissue Resource Center core facility especially Leslie Martin, Mary Jo Fekete and Dr. Mark Lingen. We would like to thank Dr. Robert Roe of Corcept Therapeutics for providing pharmacologic grade mifepristone for in vivo studies as well CORT 122928, and for expertise shared with these compounds. We also thank the Immunohistochemistry Core Facility run by Terri Li, the Functional Genomics Core facility run by Dr. Pieter W. Faber, and the Clinical Chemistry Lab run by Jerry Yeo and managed by Christine Lee. Finally, we would like to thank Sydeaka Watson and the Biostatistics Core Facility at The University of Chicago.
This study was funded by the American Cancer Society Institutional Research Grant (ACS-IRG no. 58-004-53-IRG), Cancer Research Foundation (CRF-YIA 2011), Partnership for the Cures (Szmulewitz1109UofC), and Department of Defense Physician Research Training Award (W81XW-09-1-0415). S. Kregel is supported by an HHMI: Med-into-Grad Fellowship (56006772) and an NIH Cancer Biology Training Grant (T32-CA09594)
Conflict of Interest
R.Z.S and S.D.C and The University of Chicago have a pending patent application with claims related to the use of concomitant glucocorticoid and androgen receptor antagonism in prostate cancer treatment (US patent application no. PCT/US2013/027150).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplemental Table S1
Primers for q-PCR (DOCX 56 kb)
Fig. S1
Verification of immunoprecipitation. Following immunoprecipitation of chromatin, validation of specific IP was conducted. Immunoblots (IB) were performed with antibodies to different epitopes than those used for ChIP. In addition to the IP conditions, cleared lysates were assayed for depletion of target proteins. Input lysate and whole cell 22Rv1 protein lysate were used as controls. (JPEG 27 kb)
Fig. S2
Ki-67 immunofluorescence within tumor xenografts. A. PC xenograft tumors were stained for GR (as in Fig. 1). In addition, Ki-67 co-staining was conducted on sections from intact and castrate LAPC4 xenografts. B. LAPC4 cells were grown in 10 % CSS along with combinations of R1881 (1 nM), MDV3100 (10 μM) and Casodex (CDX, 10 μM). Westerns were performed for GR and Actin. (JPEG 25 kb)
Fig. S3
In vitro cell viability experimental data. A. VCaP cells grown for seven days of treatment in 5 % CSS along with combinations of R1881 (1 nM), Dex (100 nM), MDV3100 (10 μM) as indicated and assessed for total viable cells using trypan blue exclusion. Error bars represent standard error of the mean (SEM). B. 22Rv1 and LAPC4 cells were grown in charcoal stripped serum (CSS: 10 %-LAPC4, 1 % 22Rv1) for seven days along with either R1881 (1 nM) or Dex (100 nM) and assessed for total viable cells using trypan blue exclusion. C. Prostate cancer cell lines were grown in media supplemented with charcoal stripped serum (as described in B) along with combinations of R1881 (1 nM), Dex (1 μM), MDV3100 (10 μM) and assessed for viable cells using trypan blue exclusion after 7 days of treatment. Error bars represent standard error of the mean (SEM). D. Similar viability study performed with the addition of GR-specific antagonist CORT 122928 after seven days of treatment. Relative number of viable cells plotted with control denominator noted in figure. Names and concentrations of treatments used: R1881 (R, 1 nM), Dex (D, 100 nM), MDV3100 (MDV, 10 μM) and CORT 122928 (CORT, 1 μM). Error bars represent standard error of the mean (SEM). (JPEG 68 kb)
Fig. S4
GR knockdown in vivo. A. As in Fig. 3D, after 22Rv1 xenograft establishment, animals were castrated and treated with and without oral doxycycline to induce GR knockdown. Tumor sizes shown over time. GR knockdown xenografts were smaller overall (2-way ANOVA, p < 0.01) B. Resected tumors showing sustained GR knock-down with GR-targeted sh-RNA within LAPC4 xenografts (left) and within 22Rv1 xenograft (right) from mice on doxycycline/standard. C. As in Fig. 3C, CRPC xenografts were formed, after which, animals were treated with mifepristone (Mif) 12 mg/kg/day or vehicle intraperitoneally. Animals were treated and monitored daily for CRPC progression as defined by doubling of tumor volume. Mif delayed time to progression (Log-Rank p < 0.05, median 12 days to 21 days). B. SGK1 protein immunoblot of SGK1-Flag ectopically expressing cell lines along with parental CWR-22Rv1 and LAPC cell lines treated with R1881 (1 nM), Dex (100 nM), MDV3100 (MDV, 10 μM) as noted in the figure. (JPEG 64 kb)
Fig. S5
Promoter occupancy. A. 22Rv1 cells were treated for 3 days in either vehicle, R1881 (1 nM), or R1881 + MDV3100 (10 μM) containing media. Conditions specifying dexamethasone treatment were stimulated with Dex (100 nM) for 1 h prior to chromatin harvest. Targeted qPCR was performed for the SGK1 and PSA promoter after ChIP, and y-values represent fold chromatin enrichment (relative to IgG controls for each condition). The addition of R1881 compared to vehicle alone resulted in a statistically significant (p < 0.05) increase in AR chromatin occupancy, and a significant (p < 0.05) decrease with the addition of MDV3100 was observed. B. 22Rv1 cells were cultured in 1 % CSS serum for 72 h and then treated with combinations of R1881 (1 nM, 1 h), Dex (100 nM, 1 h), and MDV3100 (10 μM, 1.5 h). Targeted qPCR was performed for the PSA promoter as described above for A. C. LAPC4 cells were treated for 3 days and targeted qPCR was performed as described above in A. The addition of R1881 compared to vehicle alone resulted in a statistically significant (p < 0.05) increase in AR chromatin occupancy, and a significant (p < 0.05) decrease with the addition of MDV3100 was observed. Error bars represent standard deviation of mean cycle threshold values, and * indicates p < 0.05 of performed paired t-tests. (JPEG 67 kb)
Rights and permissions
About this article
Cite this article
Isikbay, M., Otto, K., Kregel, S. et al. Glucocorticoid Receptor Activity Contributes to Resistance to Androgen-Targeted Therapy in Prostate Cancer. HORM CANC 5, 72–89 (2014). https://doi.org/10.1007/s12672-014-0173-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-014-0173-2