
Abstract
Arrhythmogenic cardiomyopathy (AC), also known as arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C), is a hereditary disease characterised by ventricular arrhythmias, right ventricular and/or left ventricular dysfunction, and fibrofatty replacement of cardiomyocytes. Patients with AC typically present between the second and the fourth decade of life with ventricular tachycardias. However, sudden cardiac death (SCD) may be the first manifestation, often at young age in the concealed stage of disease. AC is diagnosed by a set of clinically applicable criteria defined by an international Task Force. The current Task Force Criteria are the essential standard for a correct diagnosis in individuals suspected of AC. The genetic substrate for AC is predominantly identified in genes encoding desmosomal proteins. In a minority of patients a non-desmosomal mutation predisposes to the phenotype. Risk stratification in AC is imperfect at present. Genotype-phenotype correlation analysis may provide more insight into risk profiles of index patients and family members. In addition to symptomatic treatment, prevention of SCD is the most important therapeutic goal in AC. Therapeutic options in symptomatic patients include antiarrhythmic drugs, catheter ablation, and ICD implantation. Furthermore, patients with AC and also all pathogenic mutation carriers should be advised against practising competitive and endurance sports.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Arrhythmogenic right ventricular (RV) dysplasia/cardiomyopathy (ARVD/C) is histopathologically characterised by progressive fibrofatty replacement of cardiomyocytes, primarily in the right ventricle [1–3]. However, histopathologically and functionally the left ventricle is affected in many cases and both ventricles are similarly affected by desmosomal and gap junctional protein redistribution [4, 5]. Because of these observations, at present arrhythmogenic cardiomyopathy (AC) is the preferred terminology [6]. AC can be defined as a structural myocardial disease preceded by ventricular arrhythmias. Typical ARVD/C with predominant RV abnormalities can be considered a large and important subcategory of AC. Clinical diagnosis is made according to international consensus-based Task Force Criteria [7, 8].
The first series of ARVD/C patients was published in 1982 [1]. It was described as a developmental disease of the RV musculature, hence the terminology ‘dysplasia’. In the past 25 years, increased insight into the development of the disease as well as the discovery of pathogenic gene mutations involved led to the current understanding that AC is a genetically determined cardiomyopathy. The molecular genetic substrate for the disease is mainly acknowledged in genes encoding desmosomal adhesion proteins in the intercalated disk [9–14].
This review provides an overview of AC, from phenotypic and genetic features of the disease, to diagnosis, risk stratification and treatment options.
Epidemiology
Estimations of the prevalence of AC in the general population vary from 1:1000 to 1:5000 [15, 16]. The real prevalence of AC, however, is unknown and is presumably higher due to many non-diagnosed and misdiagnosed cases. In one study, features of AC were detected at post-mortem evaluation in as many as 20 % of sudden deaths occurring in people under 35 years of age [17]. In nearly half of them, no prior symptoms had been reported. Furthermore, in the study by Tabib et al. [18], 26 % of forensic autopsy cases following exercise-related sudden cardiac death (SCD) under the age of 30 years revealed AC.
From the genetic point of view, both men and women should be equally affected. However, men are more frequently diagnosed with AC than women. In a large multicentre study, 57 % of affected individuals were male. As many women as men show at least some signs of disease, but women less frequently fulfil criteria to meet the diagnosis [19]. It is speculated that (sports) activity or hormonal factors may play a role in this difference in severity of disease expression [20]. Familial disease, with AC diagnosis in at least one other family member besides the index patient, has been demonstrated in more than one-third of AC cases [21, 22].
Presentation
Patients with AC typically present between the second and the fourth decade of life with palpitations, lightheadedness, or syncope due to ventricular ectopy or (monomorphic) ventricular tachycardia (VT) with left bundle branch block (LBBB) morphology, thus originating from the right ventricle (Fig. 1). However, SCD may be the first clinical manifestation, often at young age in the concealed stage of disease. In a study by Quarta et al. [22], SCD was the presenting symptom in 50 % of index patients, with SCD occurring in 31 % at young age, i.e. between 14 and 20 years. A presentation with (aborted) SCD occurred less often in other studies [19, 23–25]. In the Dutch AC cohort, 2/149 (1.3 %) index patients presented with SCD and 8 % (12/149) with aborted SCD [21].

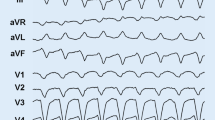
12-lead ECG of a ventricular tachycardia (VT) in an arrhythmogenic cardiomyopathy (AC) index patient with a pathogenic and a most likely pathogenic plakophilin-2 mutation (c.397C>T p.Gln133* and c.2615C>T p.Thr872Ile). The VT has a left bundle branch block morphology, with inferior axis. The QRS complex is predominantly negative in lead aVL and most positive in lead II, suggesting an origin from the right ventricular outflow tract area in the right ventricle
AC can present in four clinical stages which do not necessarily proceed from one into the other: 1) concealed stage without or with minimal structural disease, although SCD may occur, 2) overt stage with structural alterations of primarily the right ventricle, and episodes of monomorphic VT, 3) overt stage with obvious structural biventricular involvement, and 4) the end-stage of the disease with heart failure [1, 26, 27]. Recently, left ventricular (LV) dominant and primarily biventricular variants have also been described, in analogy with the AC concept [4, 28].
Diagnosis
Accurate AC diagnosis is critical due to lifelong implications, not only for the index patient but also for family members. Diagnosis has been facilitated by a set of clinically applicable criteria. These criteria, originally formulated for ARVD/C, were defined by an international Task Force based on consensus in 1994 and were revised in 2010 [7, 8]. In the revised Task Force Criteria (TFC) a minor criterion for (additional) LV involvement, inverted T waves in left precordial leads V4-6, was incorporated. The current TFC are the essential standard for a correct diagnosis in individuals suspected of AC. In addition, its universal acceptance contributes importantly to unambiguous interpretation of clinical studies and facilitates comparison of results. The TFC include six different categories: 1) global and/or regional dysfunction and structural RV alterations, 2) tissue characterisation, 3) depolarisation abnormalities, 4) repolarisation abnormalities, 5) arrhythmias, and 6) family history and genetics. Within these groups, diagnostic criteria are categorised as major or minor according to their disease specificity. A diagnosis of AC is made with the fulfilment of two major, one major and two minor, or with four minor TFC. From each different category, only one criterion can be counted for diagnosis, even when multiple criteria in one group are present. Table 1 provides an overview of the TFC defined in 2010. Figure 2 demonstrates typical repolarisation abnormalities in AC patients. Several studies suggested that this new set of criteria improved the diagnostic yield, with equal specificity [22, 29, 30]. Despite these efforts, AC diagnosis in the concealed stage of disease with subsequent risk for SCD still poses a great challenge for physicians.

12-lead ECG (during atenolol 25 mg once daily) of the same index patient as in Figure 1. The ECG shows sinus rhythm, horizontal axis, and typical negative T waves in the right precordial leads V1-3. The terminal activation duration (from the nadir of the S wave to the end of all depolarisation deflections) in leads V1-3 is normal (≤55 ms)
Specific evaluations are recommended in all patients suspected of AC: detailed history and family history, physical examination, 12-lead ECG (while off medications), signal averaged ECG, 24-hour Holter monitoring, maximal exercise testing, two-dimensional echocardiography with quantitative wall motion analysis, and more detailed imaging by cardiac magnetic resonance imaging (MRI) with delayed enhancement analysis. Invasive tests are also useful for diagnostic purposes: RV and LV cineangiography, electrophysiological testing, and endomyocardial biopsies for histopathological and immunohistochemical analysis [5, 31].
Differential diagnosis
Early and occasionally late stages of AC may show similarities with a few other diseases. In particular, differentiation from idiopathic VT originating from the RV outflow tract (RVOT) can be challenging. However, idiopathic RVOT VT is a benign non-familial condition, in which the ECG shows no depolarisation or repolarisation abnormalities and no RV structural changes can be detected. Furthermore, VT episodes have a single morphology (LBBB morphology with inferior axis) and are based on abnormal automaticity or triggered activity, whereas reentry is the prevalent arrhythmia mechanism in AC [32, 33]. It is important to differentiate idiopathic RVOT VT from AC with regard to screening of family members, prognosis, and outcome of catheter ablation.
Another disease mimicking AC is cardiac sarcoidosis [34]. Clinical symptoms of cardiac involvement are present in about 5 % of all patients with sarcoidosis. In a study by Vasaiwala et al. [35] a remarkably high incidence (15 %) of cardiac sarcoidosis was found in patients with suspected AC. Presence of extracardiac sarcoidosis, mediastinal lymphadenopathy, septal conduction abnormalities, and septal scar on cardiac imaging may be indicative of cardiac sarcoidosis rather than AC as disease aetiology [36]. Histopathological findings from endomyocardial biopsies can differentiate between the two entities. Nonetheless, cardiac sarcoidosis may mimic AC even on the molecular level. In a recent study by Asimaki et al. [37] a markedly reduced immunoreactive signal in the intercalated disk of the desmosomal protein plakoglobin was observed in AC as well as in cardiac sarcoidosis patients.
Myocarditis might also be considered in the differential diagnosis. In general, endomyocardial biopsy is required to distinguish myocarditis from AC. In analogy with cardiac sarcoidosis, giant cell myocarditis may be indistinguishable from AC using immunohistochemical analysis. In contrast, viral myocarditis does not result in plakoglobin redistribution from the intercalated disk. Differential profiles of cytokines are implicated to underlie this difference in disruption of desmosomal proteins [37].
AC may also mimic dilated cardiomyopathy (DCM), especially in the more advanced stages of disease. However, patients with DCM usually present with heart failure rather than arrhythmias. Thus, patients with sustained VT or SCD as the initial symptom of a supposed DCM, should also be screened for AC.
Finally, there are a number of other rare differential diagnoses to consider: Brugada syndrome with similar electrocardiographic or RV arrhythmias, congenital abnormalities such as Uhl’s disease, Ebstein’s anomaly, and atrial-septal defects, RV infarction, and possibly pulmonary hypertension [27].
Molecular genetic background of AC
In 2000, the seminal discovery of mutations in the plakoglobin (JUP) gene as the basis of Naxos disease, an autosomal recessive cardio-cutaneous syndrome with AC, directed the search for the genetic substrate to other genes encoding desmosomal proteins [9]. This candidate gene approach identified mutations first in the desmoplakin (DSP) gene, and thereafter in the plakophilin-2 (PKP2), desmoglein-2 (DSG2), and desmocollin-2 (DSC2) genes [10, 11, 13, 14].
In the Netherlands, as in most European countries, and in North America, mutations are predominantly found in the PKP2 gene [21, 22, 25, 38, 39]. PKP2 mutations are found in 52 % of Dutch AC index patients and even in 90 % of familial cases [21]. This high yield is partly explained by the occurrence of founder mutations in the Netherlands. Haplotype analysis suggested a founder effect of four different PKP2 mutations [38, 40]. On the other hand, there are geographical differences in the prevalence of AC-related gene mutations [13, 41].
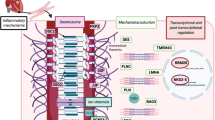
Desmosomes are protein complexes located in the intercalated disk and are amongst others important for mechanical integrity of adjacent cardiomyocytes (Fig. 3) [42]. Desmosomal dysfunction due to a gene mutation may give rise to loss of mechanical cell-cell adhesion, and leads to down-regulation and/or altered distribution of other intercalated disk proteins, i.e. gap junction proteins (Connexin43) and sodium channels (Nav1.5) [43–45]. These alterations give rise to electrical cell-cell uncoupling and slow conduction, respectively, thereby providing a substrate for early activation delay resulting in ventricular tachyarrhythmia, a hallmark of AC [5, 12, 46, 47]. Presumably, at a later stage myocyte loss and fibrofatty replacement will have a major impact on tissue architecture, giving rise to zig-zag conduction pathways and load mismatch, further contributing to enhanced activation delay [32, 48].

Schematic representation of the molecular organisation of cardiac desmosomes. The plasma membrane (PM) spanning proteins desmocollin-2 (DSC2) and desmoglein-2 (DSG2) interact in the extracellular space at the dense midline (DM). At the cytoplasmic side, they interact with plakoglobin (PG) and plakophilin-2 (PKP2) at the outer dense plaque (ODP). The PKP2 and PG also interact with desmoplakin (DSP). At the inner dense plaque (IDP), the C-terminus of DSP anchors the intermediate filament desmin (DES). (Source: Reprint with permission from: Van Tintelen et al. Curr Opin Cardiol. 2007;22:185–92)
Although the function of the desmosome and other components of the intercalated disk becomes increasingly clear, the exact mechanism by which a gene mutation results in the disease remains to be elucidated. Furthermore, not every subject with a mutation and thus a predisposition for AC develops signs and symptoms of the disease. Additional genetic factors, e.g. compound or digenic heterozygosity (carrying more than one mutation in one or more genes), or environmental factors such as exercise, may explain differences in severity of disease evolution within mutation carriers [49, 50].
In a minority of patients a non-desmosomal gene mutation is associated with the AC phenotype. Mutations in the non-desmosomal transforming growth factor β3 (TGFβ3), transmembrane protein 43 (TMEM43), desmin (DES), titin (TTN), lamin A/C (LMNA), αT-catenin (CTNNA3), and phospholamban (PLN) genes have been related to index patients and/or families with AC [51–57]. Of note, the PLN founder mutation c.40_42delAGA has been identified in 13 % of AC index patients in the Netherlands. AC patients with the PLN mutation often have low-voltage electrocardiograms, negative T waves in the left precordial leads V4-6, and additional LV involvement (Fig. 4) [58, 59]. To facilitate interpretation of genetic data, a large web-based database of genes and mutations underlying AC has been created (www.arvcdatabase.info). This database currently contains information on nearly 900 variants [60].

12-lead ECG (while off medications) of a phospholamban founder mutation carrier (c.40_42delAGA, p.Arg14del). The ECG shows sinus rhythm with right axis deviation, low voltages (<0.5 mV in standard leads), and characteristic negative T waves in left precordial leads from V3-6. The terminal activation duration (from the nadir of the S wave to the end of all depolarisation deflections) is 60 ms and therefore prolonged in lead V1 (vertical black lines)
Not all forms of AC are genetically proven. These cases may be explained by mutations in yet unknown genes, by the contribution of genetic variants of unknown significance in the known genes, or epigenetic phenomena. The evaluation of a family history of AC, suggesting a still unknown genetic factor, is crucial in patients without identifiable genetic predisposition. Alternatively, these cases may be due to environmental factors, for example exercise. Exercise was a trigger and accelerator of the AC phenotype in a mouse model with JUP haploinsufficiency [61]. Very recently, the effect of exercise on AC disease expression was corroborated in humans. Of 87 pathogenic AC mutation carriers, 56 were endurance athletes. Endurance athletes were more likely to have symptoms at young age, fulfil the 2010 TFC, and had lower survival free from VT, ventricular fibrillation (VF), and heart failure. A reversible effect of reduction of sports activities, being a significant decrease in risk for VT or VF, was observed in those individuals who exercised the most (top quartile) [20].
Risk stratification
Risk stratification in AC is imperfect at present. The annual mortality rates reported vary from 0.08–3.6 % [23, 24, 62, 63]. Retrospective analysis of clinical and pathological studies identified several risk factors for sudden death or appropriate ICD therapy, such as previously aborted SCD, syncope, young age, severe RV dysfunction, and LV involvement. Patients without VT had the best prognosis [23, 63]. Presence of sustained and non-sustained VT on Holter monitoring or during exercise testing, and sustained VT/VF during electrophysiological study were significant predictors of ICD therapy in a multivariate analysis in 84 patients treated with ICDs for primary prevention (appropriate ICD therapy in 48 %, of which 19 % for VF/ventricular flutter episodes) [64]. More recently, Te Riele et al. [65] showed that sustained arrhythmias in AC patients presenting alive seem to coincide with structural abnormalities and that patients with solely electrical abnormalities have a lower arrhythmic risk, implicating a role for RV structural abnormalities in risk stratification.
Molecular genetic analysis is also of importance in risk stratification in AC since cascade screening allows early detection of presymptomatic disease, identification of individuals at risk, and genetic counselling for this sudden death-predisposing disease. However, incorporating genetic results in AC risk stratification is hampered by incomplete penetrance and an extremely variable clinical expression. Mutation carriers may present with SCD but can also remain without signs and symptoms of the disease into old age. Therefore, genetic screening results should be viewed as probabilistic and as part of the overall clinical assessment.
Genotype-phenotype correlation analysis can provide more insight into risk profiles of index patients and family members. In contrast to index patients, mutation-positive family members have a better prognosis, with signs and symptoms of AC present in 50 % of relatives in their fifth decades of life [22]. Furthermore, family members with more than one genetic variant had a significant fivefold increase in risk of disease expression, suggesting gene-gene interactions and gene-dose effects. A genotype-phenotype correlation study by our group showed that compared with relatives of index patients without mutations, mutation carrying family members had 1) a sixfold higher risk of AC diagnosis, 2) a markedly enhanced risk for ventricular arrhythmias, and 3) earlier onset of AC signs and symptoms [21].
Clinical management
In addition to symptomatic treatment, prevention of SCD is the most important therapeutic goal in AC. As there have been no randomised trials of AC treatment modalities, screening regimens, or medications, most recommendations are based on clinical expertise, results of retrospective registry-based studies, and studies on model systems.
Evidence suggests that, in the absence of sustained and non-sustained VT and/or high number of ventricular premature complexes (>1000 VPC/24 h), asymptomatic patients and healthy mutation carriers do not require treatment with an ICD for primary prevention [64]. These individuals should undergo regular cardiac evaluations (every 1–2 years) including 12-lead ECG, 24-h Holter monitoring, echocardiography, and exercise testing for timely identification of unfavourable signs necessitating ICD implantation.
However, in all patients diagnosed with or having signs or symptoms of AC as well as asymptomatic mutation carriers, specific lifestyle advice is recommended. Sports participation has been shown to increase the risk of SCD fivefold in AC patients [66]. Furthermore, excessive mechanical stress, such as during competitive sports activity and training, may aggravate underlying myocardial abnormalities and accelerate disease progression [20, 61]. Therefore, patients with AC, and in our opinion also all pathogenic mutation carriers, should be advised against practising competitive and endurance sports.
Therapeutic options in symptomatic patients with AC include antiarrhythmic drugs, catheter ablation, and ICD implantation. However, at present ICD implantation is the only proven lifesaving therapeutic modality for fast VT/VF. In patients who have presented with stable sustained VT but also patients and family members with non-sustained VT or >500 VPC on 24-h Holter monitoring, medication may be considered to reduce arrhythmias and symptoms. This pharmacological reduction may be critically important in combination with ICD therapy to reduce shock delivery. Since ventricular arrhythmias and cardiac arrest occur frequently during or after physical exercise or may be triggered by catecholamines, non-class III antiadrenergic beta-blockers are recommended. In the absence of adequate antiarrhythmic response, sotalol in an appropriate dose is the drug of first choice. Alternatively, amiodarone and flecainide have been reported to be useful [67]. Efficacy of drug treatment has to be evaluated by serial Holter monitoring and/or exercise testing.
Catheter ablation is an alternative in patients who are refractory to drug treatment and have frequent VT episodes (with a predominantly single morphology) [68]. Of note, the role of this therapy in AC patients is to improve quality of life by decreasing the frequency of episodes of sustained VT, symptomatic non-sustained VT, and ventricular ectopy. Accordingly, in a recent study by Philips et al. [69] the overall freedom from VT of 175 ablation procedures in 87 AC patients was 47 %, 21 %, and 15 %, at 1, 5, and 10 years, respectively, over a mean follow-up of 88.3 ± 66 months. The outcomes of VT ablation are improved with a combined endocardial and epicardial approach, incorporating a substrate-based strategy [69, 70].
Although antiarrhythmic drugs and catheter ablation may reduce VT burden, there is no proof from prospective trials that these therapies will also prevent SCD. Implantation of an ICD is indicated in AC patients who are intolerant to antiarrhythmic drug therapy and who are at serious risk for SCD, in patients with aborted cardiac arrest, intolerable fast VT and those with risk factors as mentioned above.
AC in the Netherlands
In the Netherlands, a national collaboration of all University Medical Centers participating in ICIN has resulted in a large dataset with genetic and phenotypic characteristics of AC index patients and family members [21]. Since the prevalence of AC is relatively low, collaboration is essential for adequate studies and thereby improvement of insight into AC. This collaboration has expedited studies on AC-related genes, founder mutations, new diagnostic AC parameters, and clinical outcome of index patients and family members. Continuation of inclusion of more individuals, longer follow-up, and implementation of results of next generation DNA sequencing are needed to optimise further risk stratification.
References
Marcus FI, Fontaine GH, Guiraudon G, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65(2):384–98.
Corrado D, Basso C, Thiene G, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 1997;30(6):1512–20.
Basso C, Thiene G, Corrado D, et al. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation. 1996;94(5):983–91.
Sen-Chowdhry S, Syrris P, Prasad SK, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52(25):2175–87.
Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360(11):1075–84.
Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8(8):1308–39.
McKenna WJ, Thiene G, Nava A, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71(3):215–8.
Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121(13):1533–41.
McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 2000;355(9221):2119–24.
Rampazzo A, Nava A, Malacrida S, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71(5):1200–6.
Gerull B, Heuser A, Wichter T, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36(11):1162–4.
Saffitz JE. Dependence of electrical coupling on mechanical coupling in cardiac myocytes: insights gained from cardiomyopathies caused by defects in cell-cell connections. Ann N Y Acad Sci. 2005;1047:336–44.
Pilichou K, Nava A, Basso C, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113(9):1171–9.
Syrris P, Ward D, Evans A, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. 2006;79(5):978–84.
Gemayel C, Pelliccia A, Thompson PD. Arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2001;38(7):1773–81.
Peters S, Trummel M, Meyners W. Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int J Cardiol. 2004;97(3):499–501.
Basso C, Corrado D, Thiene G. Cardiovascular causes of sudden death in young individuals including athletes. Cardiol Rev. 1999;7(3):127–35.
Tabib A, Miras A, Taniere P, et al. Undetected cardiac lesions cause unexpected sudden cardiac death during occasional sport activity. A report of 80 cases. Eur Heart J. 1999;20(12):900–3.
Marcus FI, Zareba W, Calkins H, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia clinical presentation and diagnostic evaluation: results from the North American Multidisciplinary Study. Heart Rhythm. 2009;6(7):984–92.
James CA, Bhonsale A, Tichnell C, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62(14):1290–7.
Cox MG, van der Zwaag PA, van der Werf C, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation. 2011;123(23):2690–700.
Quarta G, Muir A, Pantazis A, et al. Familial evaluation in arrhythmogenic right ventricular cardiomyopathy: impact of genetics and revised task force criteria. Circulation. 2011;123(23):2701–9.
Hulot JS, Jouven X, Empana JP, et al. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2004;110(14):1879–84.
Lemola K, Brunckhorst C, Helfenstein U, et al. Predictors of adverse outcome in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: long term experience of a tertiary care centre. Heart. 2005;91(9):1167–72.
Dalal D, Molin LH, Piccini J, et al. Clinical features of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in plakophilin-2. Circulation. 2006;113(13):1641–9.
Thiene G, Nava A, Corrado D, et al. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318(3):129–33.
Basso C, Corrado D, Marcus FI, et al. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373(9671):1289–300.
Groeneweg JA, van der Zwaag PA, Jongbloed JD, et al. Left-dominant arrhythmogenic cardiomyopathy in a large family: associated desmosomal or nondesmosomal genotype? Heart Rhythm. 2013;10(4):548–59.
Cox MG, van der Smagt JJ, Noorman M, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy diagnostic task force criteria: impact of new task force criteria. Circ Arrhythm Electrophysiol. 2010;3(2):126–33.
Protonotarios N, Anastasakis A, Antoniades L, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia on the basis of the revised diagnostic criteria in affected families with desmosomal mutations. Eur Heart J. 2011;32(9):1097–104.
Basso C, Ronco F, Marcus F, et al. Quantitative assessment of endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy/dysplasia: an in vitro validation of diagnostic criteria. Eur Heart J. 2008;29(22):2760–71.
Ellison KE, Friedman PL, Ganz LI, et al. Entrainment mapping and radiofrequency catheter ablation of ventricular tachycardia in right ventricular dysplasia. J Am Coll Cardiol. 1998;32(3):724–8.
Lerman BB, Stein KM, Markowitz SM. Idiopathic right ventricular outflow tract tachycardia: a clinical approach. Pacing Clin Electrophysiol. 1996;19(12 Pt 1):2120–37.
Ladyjanskaia GA, Basso C, Hobbelink MG, et al. Sarcoid myocarditis with ventricular tachycardia mimicking ARVD/C. J Cardiovasc Electrophysiol. 2010;21(1):94–8.
Vasaiwala SC, Finn C, Delpriore J, et al. Prospective study of cardiac sarcoid mimicking arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol. 2009;20(5):473–6.
Steckman DA, Schneider PM, Schuller JL, et al. Utility of cardiac magnetic resonance imaging to differentiate cardiac sarcoidosis from arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2012;110(4):575–9.
Asimaki A, Tandri H, Duffy ER, et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2011;4(5):743–52.
van Tintelen JP, Entius MM, Bhuiyan ZA, et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113(13):1650–8.
Fressart V, Duthoit G, Donal E, et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace. 2010;12(6):861–8.
van der Zwaag PA, Cox MG, van der Werf C, et al. Recurrent and founder mutations in the Netherlands : Plakophilin-2 p.Arg79X mutation causing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Neth Heart J. 2010;18(12):583–91.
Jacob KA, Noorman M, Cox MG, et al. Geographical distribution of plakophilin-2 mutation prevalence in patients with arrhythmogenic cardiomyopathy. Neth Heart J. 2012;20(5):234–9.
Kaplan SR, Gard JJ, Carvajal-Huerta L, et al. Structural and molecular pathology of the heart in Carvajal syndrome. Cardiovasc Pathol. 2004;13(1):26–32.
Oxford EM, Musa H, Maass K, et al. Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ Res. 2007;101(7):703–11.
Sato PY, Musa H, Coombs W, et al. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009;105(6):523–6.
Noorman M, van der Heyden MA, van Veen TA, et al. Cardiac cell-cell junctions in health and disease: Electrical versus mechanical coupling. J Mol Cell Cardiol. 2009;47(1):23–31.
Basso C, Czarnowska E, Della Barbera M, et al. Ultrastructural evidence of intercalated disc remodelling in arrhythmogenic right ventricular cardiomyopathy: an electron microscopy investigation on endomyocardial biopsies. Eur Heart J. 2006;27(15):1847–54.
Cerrone M, Noorman M, Lin X, et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res. 2012;95(4):460–8.
de Bakker JM, van Capelle FJ, Janse MJ, et al. Slow conduction in the infarcted human heart. ‘Zigzag’ course of activation. Circulation. 1993;88(3):915–26.
Xu T, Yang Z, Vatta M, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55(6):587–97.
Sen-Chowdhry S, Syrris P, Pantazis A, et al. Mutational heterogeneity, modifier genes, and environmental influences contribute to phenotypic diversity of arrhythmogenic cardiomyopathy. Circ Cardiovasc Genet. 2010;3(4):323–30.
Beffagna G, Occhi G, Nava A, et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65(2):366–73.
Merner ND, Hodgkinson KA, Haywood AF, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82(4):809–21.
van Tintelen JP, Van Gelder IC, Asimaki A, et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 2009;6(11):1574–83.
Taylor M, Graw S, Sinagra G, et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation. 2011;124(8):876–85.
Quarta G, Syrris P, Ashworth M, et al. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2012;33(9):1128–36.
van Hengel J, Calore M, Bauce B, et al. Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34(3):201–10.
van der Zwaag PA, van Rijsingen IA, Asimaki A, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14(11):1199–207.
Groeneweg JA, van der Zwaag PA, Olde Nordkamp LR, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy according to revised 2010 task force criteria with inclusion of non-desmosomal phospholamban mutation carriers. Am J Cardiol. 2013;112(8):1197–206.
van der Zwaag PA, van Rijsingen IA, de Ruiter R, et al. Recurrent and founder mutations in the Netherlands-Phospholamban p.Arg14del mutation causes arrhythmogenic cardiomyopathy. Neth Heart J. 2013;21(6):286–93.
van der Zwaag PA, Jongbloed JD, van den Berg MP, et al. A genetic variants database for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Hum Mutat. 2009;30(9):1278–83.
Fabritz L, Hoogendijk MG, Scicluna BP, et al. Load-reducing therapy prevents development of arrhythmogenic right ventricular cardiomyopathy in plakoglobin-deficient mice. J Am Coll Cardiol. 2011;57(6):740–50.
Nava A, Bauce B, Basso C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36(7):2226–33.
Corrado D, Leoni L, Link MS, et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108(25):3084–91.
Bhonsale A, James CA, Tichnell C, et al. Incidence and predictors of implantable cardioverter-defibrillator therapy in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy undergoing implantable cardioverter-defibrillator implantation for primary prevention. J Am Coll Cardiol. 2011;58(14):1485–96.
Te Riele AS, Bhonsale A, James CA, et al. Incremental value of cardiac magnetic resonance imaging in arrhythmic risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62(19):1761–9.
Corrado D, Basso C, Rizzoli G, et al. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol. 2003;42(11):1959–63.
Wichter T, Paul TM, Eckardt L, et al. Arrhythmogenic right ventricular cardiomyopathy. Antiarrhythmic drugs, catheter ablation, or ICD. Herz. 2005;30(2):91–101.
Marchlinski FE, Zado E, Dixit S, et al. Electroanatomic substrate and outcome of catheter ablative therapy for ventricular tachycardia in setting of right ventricular cardiomyopathy. Circulation. 2004;110(16):2293–8.
Philips B, Madhavan S, James C, et al. Outcomes of catheter ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Arrhythm Electrophysiol. 2012;5(3):499–505.
Bai R, Di Biase L, Shivkumar K, et al. Ablation of ventricular arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy: arrhythmia-free survival after endo-epicardial substrate based mapping and ablation. Circ Arrhythm Electrophysiol. 2011;4(4):478–85.
Acknowledgments
We thank the patients and their family members and national research partners for their contribution and collaboration.
Conflict of interest
None declared.
Sources of funding
This work was supported by the Netherlands Heart Foundation (grants 2007B132 and 2007B139); Interuniversity Cardiology Institute of the Netherlands (project 06901); the Heart Lung Foundation Utrecht (J.G., T.vV., R.H.); and the Netherlands Heart Foundation Dr. E. Dekker grant (2013T033, J.G.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Groeneweg, J.A., van der Heijden, J.F., Dooijes, D. et al. Arrhythmogenic cardiomyopathy: diagnosis, genetic background, and risk management. Neth Heart J 22, 316–325 (2014). https://doi.org/10.1007/s12471-014-0563-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12471-014-0563-7