
Abstract
Background
Recently, we showed that the c.40_42delAGA (p.Arg14del) mutation in the phospholamban (PLN) gene can be identified in 10–15 % of Dutch patients with dilated cardiomyopathy or arrhythmogenic cardiomyopathy. The arrhythmogenic burden of the p.Arg14del mutation was illustrated by the high rate of appropriate ICD discharges and a positive family history for sudden cardiac death.
Methods
Our goal was to evaluate the geographical distribution and the origin of this specific mutation in the Netherlands and to get an estimation of the prevalence in a Dutch population cohort. Therefore, we investigated the postal codes of the places of residence of PLN p.Arg14del mutation carriers and places of birth of their ancestors. In addition, a large population-based cohort (PREVEND) was screened for the presence of this mutation.
Results
By April 2012, we had identified 101 probands carrying the PLN p.Arg14del mutation. A total of 358 family members were also found to carry this mutation, resulting in a total of 459 mutation carriers. The majority of mutation carriers live in the northern part of the Netherlands and analysing their grandparents’ places of birth indicated that the mutation likely originated in the eastern part of the province of Friesland. In the PREVEND cohort we identified six heterozygous PLN p.Arg14del mutation carriers out of 8,267 subjects (0.07 %).
Conclusion
The p.Arg14del mutation in the PLN gene is the most frequently identified mutation in Dutch cardiomyopathy patients. The mutation that arose 575–825 years ago is likely to have originated from the eastern part of the province of Friesland and is highly prevalent in the general population in the northern part of the Netherlands.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inherited cardiomyopathies are genetically heterogeneous disorders of the heart which often develop during adolescence or early adult life [1]. Cardiomyopathies are grouped into different categories, based on their functional and morphological properties and subdivisions can be made according to the genetic basis [2, 3]. Genetic overlap between cardiomyopathies is well-recognised. Hypertrophic cardiomyopathy (HCM) is characterised by a thickened wall of the left ventricle [4], and mutations in any of nine genes encoding sarcomeric proteins can be found in 30–65 % of HCM patients worldwide [5–7]. However, mutations in these genes can also be identified in 18 % of patients with dilated cardiomyopathy (DCM) [8], which is characterised by left ventricular dilatation and contractile dysfunction. Arrhythmogenic right ventricular cardiomyopathy (ARVC) is another cardiomyopathy subtype, characterised by fibrofatty replacement of cardiomyocytes, primarily in the right ventricle [9]. In addition to this classic right ventricular subtype, biventricular involvement and left ventricular predominance have been described [10]. ARVC is considered to be mainly a ‘disease of the desmosome’, a cell-cell adhesion complex [11]. Screening of desmosomal genes has identified mutations in 40-58 % of patients diagnosed with ARVC, but also in 5 % of patients in a DCM cohort from the United Kingdom [12–15]. The observed clinical and genetic overlap between ARVC and DCM has led to the postulation of arrhythmogenic cardiomyopathy as the encompassing entity [16].
Of the cardiomyopathies, DCM is genetically the most heterogeneous; in addition to the sarcomere genes, more than 30 genes have been identified in DCM families [17]. One of the genes implicated in DCM encodes phospholamban (PLN), a calcium handling protein in the sarcoplasmic reticulum of cardiac muscle [18]. A number of mutations have been identified, leading to a highly variable phenotype, ranging from cardiac death in early adulthood to middle-aged asymptomatic mutation carriers [19, 20].
The yield from screening cardiomyopathy populations for PLN mutations is generally very low, ranging from 0.08 % to 0.38 % in selected cohorts [21–25]. Surprisingly, we identified the PLN p.Arg14del mutation not only in 13 % (31/240) of Dutch patients diagnosed with DCM, but also in 12 % (12/97) of Dutch patients diagnosed with ARVC [26]. The arrhythmogenic burden of the p.Arg14del mutation was illustrated by the high rate of appropriate ICD discharges and a positive family history for sudden cardiac death. Furthermore, p.Arg14del mutation carriers more frequently underwent cardiac transplantation, compared with patients with familial DCM [26]. Cascade screening has identified dozens of family members carrying the same mutation. Both variable expression and age-dependent penetrance, which are hallmarks of all inherited cardiomyopathies, are characteristic of the p.Arg14del mutation.
Recurrent and founder mutations in the Netherlands causing cardiac disease have been described in this series of the Netherlands Heart Journal [7, 27–30]. Here, we report our analysis of the origin of the p.Arg14del mutation, both by geographic region using postal code maps, and by age using haplotype analysis. Furthermore, we screened a large population-based cohort for the presence of the p.Arg14del mutation to get an impression of the potential number of mutation carriers in the Netherlands.
Materials & methods
Genetic evaluation
Sequencing analysis and haplotype analysis for the PLN p.Arg14del mutation has been described elsewhere [26]. To estimate the age of the haplotype, the linkage disequilibrium between the mutation and recombinant microsatellite markers was calculated and the recombination fraction from the distances between the mutation and microsatellite markers was determined, enabling estimation of the number of generations since a mutation had occurred [31].
Postal code analysis
The postal codes of the places of residence of all PLN p.Arg14del mutation carriers were plotted to study their geographical distribution. To study the region where the mutation likely originated, we used a scoring system based on the birthplaces of the grandparents of the proband carrying the PLN p.Arg14del mutation. Without any additional genetic results, each grandparent had a chance of 1 in 4 of being a carrier of the mutation and therefore a score of ¼ was applied to the postal code of each grandparent’s place of birth. If it was known whether the mutation was inherited either paternally or maternally, that pair of grandparents had a chance of 1 in 2 of being a carrier of the mutation and a score of ½ was applied to the postal codes of their places of birth. If it was proven which grandparent was the carrier of the mutation, a score of 1 was applied to the postal code for their place of birth. The sum of all the scores per postal code area was plotted. These analyses were based on the PC2 code (the first two numbers of the postal code) of the mutation carriers and their grandparents. Data were visualised with MapInfo Professional (MapInfo, Toronto, Canada).
Cohort study
A large population-based cohort from the city of Groningen, in the north of the Netherlands, the Prevention of Renal and Vascular ENd-stage Disease (PREVEND) cohort (N = 8,267), was screened for the presence of the PLN p.Arg14del mutation. The PREVEND study was designed to prospectively investigate the association between urinary albumin excretion and renal and cardiovascular outcome in the general population. The study protocol is described elsewhere [32, 33]. In the period 1997–1998, 8592 participants were enrolled in the study and DNA samples were available for 8267 participants. The PREVEND study was approved by the local medical ethics committee and conducted in accordance with the Declaration of Helsinki.
Mutation detection
The prevalence of the PLN p.Arg14del mutation in the PREVEND cohort was evaluated using KASPar® PCR SNP genotyping system (KBiosciences, Herts, UK). All the 8267 available DNA samples were screened for the mutation.
Results
Genetic evaluation
By April 2012, we had identified 101 unique probands carrying the PLN p.Arg14del mutation. A total of 358 family members were found to carry the same mutation, resulting in a total of 459 mutation carriers and an average of 4.5 (range 1–17) mutation carriers per family.
Haplotype analysis was performed in 49 of the 101 Dutch PLN p.Arg14del families and in three families from the United States, Germany and Greece [26]. A shared haplotype for five markers in a 1.2 Mb region surrounding PLN was found, although patients from the Greek family with the p.Arg14del mutation carried another haplotype (Table 1). Allowing 25 years per generation, the age of the ‘Dutch’ haplotype containing the mutation is estimated to be between 575 and 825 years old [26].
Postal code analysis
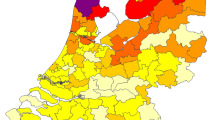
The geographic distribution of the place of residence of the mutation carriers is plotted in Fig. 1, which shows that the majority of the identified carriers live in the northern part of the Netherlands and in the province of Noord-Holland, and that the numbers decrease gradually towards the south.

Postal code map showing the distribution of PLN p.Arg14del mutation carriers in the Netherlands. The number of PLN p.Arg14del mutation carriers per region is shown (in parenthesis: the number of postal code regions, 90 in total). On average, each region contains 180,000 inhabitants
To determine the origin of the Dutch founder haplotype containing the PLN p.Arg14del mutation, we analysed the place of birth of the proband’s grandparents and scored the corresponding postal codes according to their chance of being the grandparent carrying the mutation. Data on these places of birth were available for 60 of the 101 probands and the results are shown in Fig. 2. We found that the majority of ancestors came from the eastern part of the province Friesland.

Postal code map illustrating the likely origin of the founder haplotype containing the PLN p.Arg14del mutation. The number of points based on the grandparents’ birthplaces is shown (in parenthesis: the number of postal code regions, 90 in total). On average, each region contains 180,000 inhabitants. The province of Friesland is enclosed by the bold border
The phenotypes of PLN p.Arg14del mutation carriers vary greatly, not only between families, but also within the same family, as illustrated by the family shown in Fig. 3a and Table 2. The proband III:1 was diagnosed with ARVC at age 21. She sought medical attention after experiencing palpitations. She presented with a low-voltage ECG with ventricular bigeminy and monomorphic ventricular extrasystoles from the right ventricular outflow tract (Fig. 3b). During the course of 9 years follow-up, she developed progressive heart failure and experienced several appropriate ICD discharges because of ventricular tachyarrhythmias. She was placed on the waiting list for a cardiac transplantation, but died at age 30. Her mother, who also carried the PLN p.Arg14del mutation, had recordings of non-sustained VTs on 24 h Holter ECG monitoring, but her evaluation was otherwise unremarkable. The grandfather, also a mutation carrier, died at age 75 without ever seeking medical attention because of possible cardiac problems. The proband’s two sisters (age 30 and 27 when last evaluated) were found to be carriers of the PLN p.Arg14del mutation and are being monitored frequently, but have not experienced any symptoms, although the oldest sister demonstrated 1100 premature ventricular complexes on 24 h Holter monitoring (Table 2).

(a) Pedigree of a family carrying the PLN p.Arg14del mutation, illustrating the variability of the phenotype. Squares indicate male family members, circles indicate female family members, slashes indicate deceased, and the arrow points to the index patient. The solid black symbol indicates proven ARVC, while open symbols indicate clinically unaffected family members. Genotype results are shown by the p.Arg14del mutation present (+) or absent (−). (b) Baseline ECG of patient III:1, diagnosed with ARVC. The ECG shows signs of both DCM and ARVC: low voltage, ventricular bigeminy with left branch, left axis morphology (suggesting right ventricular outflow tract origin) and negative T-waves in the precordial leads
Cohort study
In the PREVEND cohort we identified six heterozygous PLN p.Arg14del mutation carriers out of 8267 subjects (0.07 %). These PLN p.Arg14del mutation carriers (age at baseline 48 ± 16 years; 4 males) did not have a clinically manifested cardiomyopathy at baseline. Three mutation carriers were diagnosed with hypertension at baseline, and three were diagnosed with diabetes mellitus. One subject (male, age at baseline 69 years) developed heart failure during the 10-year follow-up period; the others had not experienced heart failure at a mean age of 53.4 years. None of the six mutation carriers were diagnosed with atrial fibrillation or left ventricular hypertrophy on the ECG. The set-up of the database provided no additional cardiological data.
Discussion
The p.Arg14del mutation in the PLN gene is the most frequently identified mutation in cardiomyopathy patients in the Netherlands and the single most identified mutation in cardiomyopathy patients worldwide. Dutch mutation carriers showed high rates of appropriate ICD discharges, cardiac transplantation and a positive family history for sudden cardiac death [26]. This mutation has also been identified in cohorts in other countries, such as Germany, Spain, Greece, Canada, and the United States [21, 22, 34, 35 and personal communications M. Gollob & R. Hamilton]. The German family and an ARVC patient from the United States carried the same haplotype surrounding the PLN gene as the patients from the Netherlands. The Greek patients carried another haplotype, illustrating that the PLN p.Arg14del mutation is recurrent, with at least two different haplotypes identified. All the Dutch patients carried the same haplotype and we estimated this haplotype to be between 575 and 825 years old. Although we have identified the founder effect of the PLN p.Arg14del mutation in the Netherlands, we cannot exclude that its origin is foreign, due to immigration from Germany, Belgium, France or Spain, for example, especially considering the age of the mutation [36]. The emigration in the 19th and early 20th century of PLN p.Arg14del mutation carriers from the Netherlands or from other Northern European countries to the United States and Canada has likely resulted in the presence of this mutation in North America. Dutch mutation carriers could also have emigrated to South Africa, Australia and New Zealand. The presence of the p.Arg14del mutation needs to be confirmed in genetic studies in these countries.
To estimate the geographical origin of the p.Arg14del mutation, we used a scoring method based on the places of birth of the probands’ grandparents. This scoring system revealed the eastern part of the province Friesland as the most likely area of origin of the p.Arg14del mutation in the Netherlands (Fig. 2). Most current mutation carriers live in the northern part of the Netherlands including the province of Noord-Holland, i.e. close to the origin of the mutation, and a gradual decline towards the southern part of the Netherlands can be seen in Fig. 1. As has been shown for other founder mutations in the Netherlands, including some described in this NHJ series [7, 27–30], the migration of much of the Dutch population, and therefore mutation carriers, has been fairly limited and the distribution of current mutation carriers already gives a clear indication of its origin.
Following the identification of the p.Arg14del mutation in cardiomyopathy patients, we sought to identify whether the mutation was also present in a large population-based cohort from the northern Netherlands that had not been selected for the presence of cardiomyopathy or other cardiac diseases. Six of 8267 subjects (0.07 %) were identified as mutation carriers, suggesting that the mutation could be present in approximately 1:1400 individuals in the northern part of the Netherlands. Postal code analysis suggested a lower frequency towards the south. By April 2012, the population of the three northern provinces of Groningen, Friesland and Drenthe was 1.7 million [37]. Given these numbers, approximately 1250 PLN p.Arg14del mutation carriers are suspected to live in these provinces. The prevalence of the mutation in other provinces is expected to be lower, but since they have larger populations (e.g. 2.7 million in the province of North Holland), the total number of Dutch PLN p.Arg14del carriers is likely to be more than 2000. Cascade family screening is ongoing and will probably identify several hundred more carriers.
The age at inclusion (48 ± 16 years) of these carriers illustrates the age-dependent penetrance and variable expression of the p.Arg14del mutation. Since the onset of mutation-related symptoms (mean age 44.4 years) is well after the start of their reproductive age [26], many mutation carriers had already had children, explaining the continued high prevalence of the mutation. This mean age is likely to be biased, since the index patients usually mark the severe end of the disease spectrum. Some index patients had had a child who had died of sudden cardiac death at a much younger age than their age of onset.
Following the arrhythmogenic profile of the PLN p.Arg14del mutation, primary prevention by implanting an ICD could be beneficial for mutation carriers. We aim to determine the risk factors which will identify individuals at high risk for life-threatening ventricular arrhythmias. Identification of independent risk factors would enable the selection of PLN p.Arg14del mutation carriers who are most likely to benefit from an ICD, while the absence of such risk factors in other carriers would justify alternative treatment options. We have set up the PHOspholamban RElated CArdiomyopathy STudy (PHORECAST, www.phorecast.nl) to evaluate the possible risk factors for life-threatening ventricular arrhythmias. This study will be of great importance for all p.Arg14del mutation carriers as well as their cardiologists, who are faced with a great variability of symptoms and large clinical differences, both between and within affected families.
References
Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011;364:1643–56.
Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–16.
Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–6.
Thierfelder L, Watkins H, MacRae C, et al. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77:701–12.
Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–32.
Van Driest SL, Ommen SR, Tajik AJ, et al. Sarcomeric genotyping in hypertrophic cardiomyopathy. Mayo Clin Proc. 2005;80:463–9.
Christiaans I, Nannenberg EA, Dooijes D, et al. Founder mutations in hypertrophic cardiomyopathy patients in the Netherlands. Neth Heart J. 2010;18:248–54.
Hershberger RE, Norton N, Morales A, et al. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3:155–61.
Basso C, Corrado D, Marcus FI, et al. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–300.
Sen-Chowdhry S, Syrris P, Ward D, et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115:1710–20.
Van Tintelen JP, Hofstra RM, Wiesfeld AC, et al. Molecular genetics of arrhythmogenic right ventricular cardiomyopathy: emerging horizon? Curr Opin Cardiol. 2007;22:185–92.
Sen-Chowdhry S, Syrris P, McKenna WJ. Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50:1813–21.
Den Haan AD, Tan B, Zikusoka M, et al. Comprehensive desmosome mutation analysis in North Americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Cardiovasc Genet. 2009;2:428–35.
Cox MG, van der Zwaag PA, van der Werf C, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation. 2011;123:2690–700.
Elliott P, O’Mahony C, Syrris P, et al. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3:314–22.
Sen-Chowdhry S, Morgan RD, Chambers JC, et al. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med. 2010;61:233–53.
Dellefave L, McNally EM. The genetics of dilated cardiomyopathy. Curr Opin Cardiol. 2010;25:198–204.
MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–77.
Schmitt JP, Kamisago M, Asahi M, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299:1410–3.
Haghighi K, Kolokathis F, Pater L, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–76.
Haghighi K, Kolokathis F, Gramolini AO, et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci USA. 2006;103:1388–93.
DeWitt MM, MacLeod HM, Soliven B, et al. Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy. J Am Coll Cardiol. 2006;48:1396–8.
Chiu C, Tebo M, Ingles J, et al. Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2007;43:337–43.
Landstrom AP, Adekola BA, Bos JM, et al. PLN-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: summary of the literature and implications for genetic testing. Am Heart J. 2011;161:165–71.
Medeiros A, Biagi DG, Sobreira TJ, et al. Mutations in the human phospholamban gene in patients with heart failure. Am Heart J. 2011;162:1088–1095.e1.
Van der Zwaag PA, van Rijsingen IA, Asimaki A, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14:1199–207.
Postema PG, Van den Berg M, Van Tintelen JP, et al. Founder mutations in the Netherlands: SCN5a 1795insD, the first described arrhythmia overlap syndrome and one of the largest and best characterised families worldwide. Neth Heart J. 2009;17:422–8.
Van der Zwaag PA, Cox MG, van der Werf C, et al. Recurrent and founder mutations in the Netherlands: plakophilin-2 pArg79X mutation causing arrhythmogenic right ventricular cardiomyopathy/dysplasia. Neth Heart J. 2010;18:583–91.
Van den Wijngaard A, Volders P, Van Tintelen JP, et al. Recurrent and founder mutations in the Netherlands: cardiac Troponin I (TNNI3) gene mutations as a cause of severe forms of hypertrophic and restrictive cardiomyopathy. Neth Heart J. 2011;19:344–51.
Van Spaendonck-Zwarts KY, van der Kooi AJ, et al. Recurrent and founder mutations in the Netherlands: the cardiac phenotype of DES founder mutations p.S13F and p.N342D. Neth Heart J. 2012;20:219–28.
Machado PM, Brandao RD, Cavaco BM, et al. Screening for a BRCA2 rearrangement in high-risk breast/ovarian cancer families: evidence for a founder effect and analysis of the associated phenotypes. J Clin Oncol. 2007;25:2027–34.
Hillege HL, Janssen WM, Bak AA, et al. Microalbuminuria is common, also in a nondiabetic, nonhypertensive population, and an independent indicator of cardiovascular risk factors and cardiovascular morbidity. J Intern Med. 2001;249:519–26.
Linssen GC, Bakker SJ, Voors AA, et al. N-terminal pro-B-type natriuretic peptide is an independent predictor of cardiovascular morbidity and mortality in the general population. Eur Heart J. 2010;31:120–7.
Posch MG, Perrot A, Geier C, et al. Genetic deletion of arginine 14 in phospholamban causes dilated cardiomyopathy with attenuated electrocardiographic R amplitudes. Hear Rhythm. 2009;6:480–6.
Gomez Milanes I, Garcia-Molina E, Sabater-Molina M, et al. R14Del, a Dutch phospholamban mutation in a Spanish family. Genotype-phenotype aspects. Eur Heart J. 2012;33 Suppl. 1:877. Abstract.
Zeegers MP, van Poppel F, Vlietinck R, et al. Founder mutations among the Dutch. Eur J Hum Genet. 2004;12:591–600.
Statline; Centraal Bureau voor de Statistiek. http://statline.cbs.nl (24/206/2012)
Acknowledgments
We thank all the patients and their relatives who made this work possible, Hennie Bikker, Dennis Dooijes, Marielle E. van Gijn, Arjan Houweling, Jan D.H. Jongbloed, Marjon A. Slegtenhorst, and Arthur van den Wijngaard for providing results of DNA sequence analyses, and Jackie Senior for editing the manuscript.
Funding
This study was supported by a grant from the Netherlands Heart Foundation (2007B132).
Conflict of interests
None declared.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
van der Zwaag, P.A., van Rijsingen, I.A.W., de Ruiter, R. et al. Recurrent and founder mutations in the Netherlands—Phospholamban p.Arg14del mutation causes arrhythmogenic cardiomyopathy. Neth Heart J 21, 286–293 (2013). https://doi.org/10.1007/s12471-013-0401-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12471-013-0401-3