
Abstract
The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) is a major glutamate-gated ion channel in the brain and is important for synaptic transmission, synaptic plasticity, and learning. Palmitoylation, a post-translational modification, is a critical process regulating AMPAR trafficking, synaptic function and plasticity, and learning and memory in health and diseases. In this review, we discuss current knowledge on the palmitoylation-dependent regulation of AMPAR trafficking and functions. We focus on the palmitoylation of AMPARs and other synaptic proteins that directly or indirectly interact with AMPARs, including postsynaptic density 95, glutamate receptor-interacting protein/AMPAR-binding protein, A-kinase anchoring protein 79/150, and protein interacting with C kinase 1. Finally, we discuss what future studies should address in the field of palmitoylation-dependent AMPAR trafficking and function with regard to physiology and neurodegenerative diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Synapses are the fundamental bases of brain structure and function and consist of a presynaptic terminal, synaptic cleft, and postsynaptic site. Neurons communicate with each other by releasing neurotransmitters from presynaptic terminals to synaptic clefts, detecting neurotransmitters with postsynaptic neurotransmitter receptors, and translating the detection into electrical and chemical signals in the postsynaptic cells. In addition to synaptic transmission, synapses also possess a remarkable ability to modulate their outputs in response to various synaptic inputs, which is called synaptic plasticity. The most well characterized examples of synaptic plasticity in the brain are long-term potentiation (LTP) and long-term depression (LTD).
The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), a major glutamate-gated ion channel in the mammalian central nervous system, play important roles in synaptic transmission, synaptic plasticity such as LTP and LTD, and ultimately, learning and memory cognitive function in the brain. The synaptic localization and abundance of AMPARs are critical for synaptic function and are regulated by exocytosis, endocytosis, recycling, and lateral mobility on the surface membranes of AMPARs (Lu et al. 2001; Borgdorff and Choquet 2002; Tardin et al. 2003; Park et al. 2004; Heine et al. 2008; Patterson et al. 2010; Wu et al. 2017; Park 2018). Post-translational modifications (PTMs), including phosphorylation (Banke et al. 2000; Lee et al. 2000, 2003, 2010; Lu and Roche 2012; Lussier et al. 2015), glycosylation (Hollmann et al. 1994; Standley and Baudry 2000; Traynelis et al. 2010), ubiquitination (Schwarz et al. 2010; Lussier et al. 2011, 2012; Widagdo et al. 2015; Wei et al. 2016), sumoylation (Craig and Henley 2012; Jaafari et al. 2013; Lee et al. 2013), and palmitoylation (Hayashi et al. 2005; Lin et al. 2009; Thomas et al. 2012, 2013; Diering and Huganir 2018), are also critical regulatory factors for AMPAR function, synaptic plasticity, and learning and memory in health and diseases. Among these PTMs, palmitoylation, which was the most frequently observed in neuronal cells than other lipid modifications and was demonstrated to play crucial roles in synaptic function and neurodegenerative diseases (Fukata and Fukata 2010; Cho and Park 2016), will be discussed in this review.
Palmitoylation is a lipid modification that occurs by a covalent attachment of the 16 carbon-containing saturated fatty acid palmitate to specific cysteine (Cys) residues of target proteins via a thioester bond. This process is catalyzed by conserved Aspartate-Histidine-Histidine-Cys (DHHC) motif-containing palmitoyl acyltransferases (PATs), also known as DHHC enzymes because of their catalytic DHHC motif, and reversed by depalmitoylating enzymes such as acyl protein thioesterases and palmitoyl protein thioesterases. The reversibility of palmitoylation enables precise and dynamic regulation of protein functions in neurons, including synaptic targeting, glutamate receptor trafficking, synaptic transmission, and synaptic plasticity (Kang et al. 2004, 2008; Hayashi et al. 2009; Lin et al. 2009; Noritake et al. 2009; Keith et al. 2012; Thomas et al. 2012). Further, the activities of a variety of PATs and depalmitoylating enzymes have been linked to many neurodegenerative diseases (Han et al. 2015; Cho and Park 2016).
Given the importance of AMPARs and palmitoylation in the physiology of synaptic function, synaptic plasticity and learning, we present the current knowledge on palmitoylation-mediated synaptic regulation of AMPARs and their interacting proteins and discuss possible future studies on palmitoylation-dependent AMPAR trafficking in neurodegenerative diseases in this review. The interacting proteins discussed include postsynaptic density 95 (PSD-95), glutamate receptor-interacting protein (GRIP1)/AMPAR-binding protein (ABP), A-kinase anchoring protein 79/150 (AKAP79/150), and protein interacting with C kinase 1 (PICK1).
AMPAR palmitoylation
AMPARs are heterotetrameric proteins composed of the subunits GluA1-GluA4 (also called GluR1-GluR4 or GluRA-GluRD) (Wisden and Seeburg 1993; Hollmann and Heinemann 1994; Dingledine et al. 1999; Collingridge et al. 2009; Traynelis et al. 2010). In the hippocampus, the composition of AMPARs is developmentally distinct (Wenthold et al. 1996; Zhu et al. 2000), with GluA1/GluA2 heteromeric AMPARs more predominant than GluA2/GluA3 heteromers in mature hippocampal neurons (Wenthold et al. 1996; Lu et al. 2009). Nevertheless, all four AMPAR subunits have similar structures and topologies (Malinow and Malenka 2002; Lu and Roche 2012). Each subunit has a large extracellular N-terminal domain, four hydrophobic transmembrane domains (TMDs), including three that are membrane spanning (TMD1, TMD3, and TMD4) and the membrane-embedded TMD2 that contributes to channel pore formation, and three intracellular domains (loop1, loop2, and a C-terminal tail) (Fig. 1). Although the N-terminal domain and four hydrophobic TMDs are highly homologous among the subunits, their C-terminal intracellular tails are distinct, which confer distinct regulation to the AMPAR subunits through specific interactions with cytoplasmic proteins (Malinow and Malenka 2002; Shepherd and Huganir 2007).

α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) structure and palmitoylation. Schematic diagrams show each of the four AMPAR subunits. In each diagram, the large extracellular N-terminal domain includes S1, which forms the glutamate binding site together with S2 that is located on the extracellular loop linking transmembrane domain 3 (TMD3) and TMD4. Four hydrophobic TMDs including three membrane-spanning TMDs (TMD1, TMD3, and TMD4) and one membrane-embedded TMD (TMD2) and three intracellular domains (intracellular loop1, loop2, and the cytoplasmic tail) are shown. Palmitoylation sites for each subunit are marked in red
All four AMPAR subunits are palmitoylated at two conserved Cys residues: one immediately next to TMD2, and the other next to TMD4 in the C-tail juxtamembrane region (labeled red in Fig. 1) (Hayashi et al. 2005; Diering and Huganir 2018). Palmitoylation at Cys585 in GluA1 and at Cys610 in GluA2 immediately after TMD2, which is mediated by a PAT, DHHC3 (also known as Golgi-specific DHHC zinc finger protein [GODZ]; (Uemura et al. 2002) (Fig. 2), accumulated AMPARs in the Golgi apparatus and reduced AMPAR surface expression (Hayashi et al. 2005). The finding of accumulation of palmitoylated AMPARs at the Golgi apparatus implies that depalmitoylation of AMPARs at the Golgi apparatus would be a releasing signal for AMPARs to traffic forward to the plasma membrane. Palmitoylation at Cys811 in GluA1 and at Cys836 in GluA2 in the C-tail juxtamembrane region did not affect the AMPAR surface expression level at steady state. Rather, it inhibited the interaction of GluA1 with the 4.1N protein and regulated activity-dependent endocytosis of GluA1 (Hayashi et al. 2005), the insertion of GluA1 into the plasma membrane, and LTP (Lin et al. 2009). Interestingly, GluA1 palmitoylation requires anterograde trafficking from the endoplasmic reticulum (ER) to the Golgi apparatus, where DHHC3/GODZ is located (Uemura et al. 2002), whereas GluA2 palmitoylation primarily occurs within the ER (Yang et al. 2009). Because DHHC3/GODZ was reported to be exclusively located on the Golgi apparatus (Uemura et al. 2002), the PAT in the ER that palmitoylates GluA2 remains to be revealed. In addition, because palmitoylation next to TMD2 at Cys585 in GluA1 and at Cys610 in GluA2 occurred only when co-expressed with DHHC3/GODZ (Hayashi et al. 2005), it is very plausible that other PATs palmitoylate AMPAR residues immediately after TMD2 endogenously or that DHHC3/GODZ or other PATs palmitoylate AMPAR residues when some stimuli are applied.

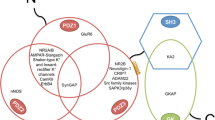
Interaction network between palmitoyl acyltransferases (PATs)/depalmitoylating enzymes and their synaptic substrates. Only the PAT/depalmitoylating enzyme–substrate pairs described in this review are shown. Blue circles, PAT enzymes; red circles, depalmitoylating enzymes; black circles, synaptic substrates. PSD-95, postsynaptic density 95; GRIP1, glutamate receptor-interacting protein; AKAP79/150, A-kinase anchoring protein 79/150; PICK1, protein interacting with C kinase 1; ABHD17, α/β-hydrolase domain-containing protein 17
Neuronal activity also regulates AMPAR palmitoylation dynamically (Hayashi et al. 2005; Yang et al. 2009; Spinelli et al. 2017). Glutamate treatment, which stimulates neurons, induced depalmitoylation of AMPARs (Hayashi et al. 2005; Yang et al. 2009) whereas tetrodotoxin (TTX) treatment, which blocks neuronal activity, enhanced palmitoylation of AMPARs (Yang et al. 2009). Interestingly, a high-fat diet elevated levels of palmitic acid and insulin resistance, which led to increased expression of DHHC3/GODZ in the hippocampus (Spinelli et al. 2017). Elevated levels of palmitic acid and DHHC3/GODZ caused GluA1 palmitoylation, which suppressed its activity-dependent delivery to the plasma membrane and impaired LTP and memory in mice fed a high-fat diet (Spinelli et al. 2017). In addition to the studies on AMPAR palmitoylation in hippocampal and cortical neurons (Hayashi et al. 2005; Lin et al. 2009; Yang et al. 2009; Spinelli et al. 2017), a study performed in the nucleus accumbens demonstrated that intraperitoneal administration of cocaine enhanced palmitoylation of GluA1 and GluA3 and redistributed intracellular GluA1 and GluA3 (Van Dolah et al. 2011). Cocaine-induced palmitoylation and subcellular redistribution of GluA1 and GluA3 were blocked by the application of the palmitoylation inhibitor 2-bromopalmitate (Van Dolah et al. 2011). These findings suggest that the differential roles of AMPAR palmitoylation in various forms of synaptic plasticity should be investigated further in multiple brain regions.
Regulation of AMPARs by palmitoylation of synaptic proteins
In addition to the direct palmitoylation of AMPARs themselves, palmitoylaton of synaptic proteins directly or indirectly interacting with AMPARs (Kim and Sheng 2004) such as PSD-95, GRIP1, GRIP2, PICK1, and AKAP79/150, can also be an important regulatory factor for AMPAR trafficking and function (Fukata and Fukata 2010; Thomas and Huganir 2013; Han et al. 2015).
PSD-95, the major PSD scaffolding protein, regulates synaptic trafficking of AMPARs (El-Husseini et al. 2000; Elias and Nicoll 2007). PSD-95 is palmitoylated at Cys3 and Cys5 by DHHC2, DHHC3/GODZ, DHHC5, DHHC7, DHHC8, and DHHC15 (Fukata et al. 2004) (Figs. 2, 3), which is required for its synaptic targeting (Topinka and Bredt 1998; Craven et al. 1999; El-Husseini et al. 2000; Noritake et al. 2009; Sturgill et al. 2009) and for the synaptic trafficking of AMPARs (El-Husseini et al. 2000; El-Husseini Ael et al. 2002; Schnell et al. 2002; Noritake et al. 2009). Overexpression of PSD-95 increased AMPAR-mediated synaptic transmission (El-Husseini et al. 2000; Schnell et al. 2002) whereas the non-palmitoylatable form of PSD-95 disrupted GluA1 clustering and AMPAR-mediated synaptic transmission (El-Husseini et al. 2000; El-Husseini Ael et al. 2002; Schnell et al. 2002). Glutamate treatment (10 µM) caused PSD-95 depalmitoylation, PSD-95 synaptic cluster dispersal, and AMPAR internalization (El-Husseini Ael et al. 2002). Ca2+/calmodulin (CaM) binding to the N-terminus of PSD-95 was shown to block PSD-95 palmitoylation at Cys3 and Cys5, which triggered PSD-95 dispersal and dissociation from the postsynaptic membrane (Zhang et al. 2014). Conversely, treatment with neuronal activity blockers such as kynurenic acid (an ionotropic glutamate receptor blocker), APV (an N-methyl-d-aspartate receptor [NMDAR] blocker), CNQX (an AMPAR blocker), and TTX (a sodium channel blocker) increased the palmitoylation and synaptic clustering of PSD-95 (Noritake et al. 2009) as TTX increased AMPAR palmitoylation (Yang et al. 2009). This activity-dependent palmitoylation of PSD-95 is mediated by DHHC2, which is translocated from the dendritic shaft to the PSD upon activity blockade (Noritake et al. 2009). The palmitoylation of PSD-95 altered its conformation from a compact to an extended form, which was oriented perpendicular to the PSD membrane (Jeyifous et al. 2016) and associated indirectly with AMPARs via Stargazin, an AMPAR auxiliary protein (Tomita et al. 2005) (Fig. 3), or NMDARs via the GluN2B subunit. Increased PSD-95 palmitoylation also increased the surface AMPAR level with no change in the NMDAR level, indicating the differential regulation of AMPAR and NMDAR trafficking by PSD-95 palmitoylation (Jeyifous et al. 2016).

AMPAR palmitoylation and regulation of AMPARs by palmitoylation of synaptic proteins. Palmitoylation at Cys811 in the GluA1 C-terminus inhibits the interaction of GluA1 with 4.1N and triggers activity-dependent endocytosis of AMPARs. PSD-95 palmitoylations at Cys3 and Cys5 mediated by DHHC2, DHHC3, DHHC5, DHHC7, DHHC8, and DHHC15, stabilize PSD-95-AMPAR interaction via Stargazin. GRIP1b palmitoylation targets GRIP1b to recycling endosomes and enhances activity-dependent recycling of GluA2-containing AMPARs to the plasma membrane. AKAP79 palmitoylations at Cys36 and Cys129 mediated by DHHC2 are necessary for AKAP79 targeting to recycling endosomes and dendritic spines. PSD-95, postsynaptic density 95; GRIP1, glutamate receptor-interacting protein; AKAP79/150, A-kinase anchoring protein 79/150
In addition to controlling the PSD-95 palmitoylation process, direct regulation of the PSD-95 depalmitoylation process is definitely another key regulatory step for synaptic function. Recently, long-awaited PSD-95 depalmitoylating enzymes were identified as α/β-hydrolase domain-containing protein 17 members (ABHD17A, ABHD17B, and ABHD17C) (Yokoi et al. 2016) (Figs. 2, 3). Overexpression of ABHD17B selectively depalmitoylated PSD-95 and decreased the synaptic clustering of PSD-95 and GluA1 (Yokoi et al. 2016). Further investigations on PSD-95 deplamitoylation and palmitoylation mechanisms will help clarify the regulatory mechanisms underlying synaptic function and plasticity.
GRIP1 and GRIP2 (also known as ABP), which have multi-PSD-95/discs large/zona occludens (PDZ) domains, directly interact with the C-termini of the AMPAR GluA2 and GluA3 subunits through its PDZ and stabilize AMPARs (Dong et al. 1997; Srivastava et al. 1998) (Fig. 3). The genes encoding GRIP1 and GRIP2 have multiple splice isoforms (Dong et al. 1997; Wyszynski et al. 1999), and GRIP1b and GRIP2b (also known as pABP-long [pABP-L]) have additional N-terminal sequences, which contain a unique Cys residue that is palmitoylated (Yamazaki et al. 2001; DeSouza et al. 2002; Thomas et al. 2012). Palmitoylatable pABP-L targets the plasma membrane of dendritic spines where it associates with surface GluA2, whereas the non-palmitoylatable ABP-L form (a variant of ABP with seven PDZ domains) localizes with intracellular AMPARs (DeSouza et al. 2002; Misra et al. 2010) (Fig. 3). In addition, palmitoylated pABP-L increased the amplitude and frequency of AMPAR-mediated excitatory postsynaptic currents (Misra et al. 2010). Palmitoylation-deficient pABP-L that is mutated at the 11th amino acid Cys (to alanine) changed its localization to intracellular clusters from the spinal plasma membrane, indicating the requirement of pABP-L palmitoylation in its synaptic localization (DeSouza et al. 2002). Whereas the palmitoylation of GRIP2/ABP induced plasma membrane targeting, the palmitoylation of GRIP1b, which is mediated by DHHC5 and DHHC8 (Thomas et al. 2012) (Figs. 2, 3), targeted intracellular endosomes and enhanced NMDA-induced AMPAR endocytic recycling trafficking. This finding indicates the involvement of GRIP1 palmitoylation in AMPAR endocytic recycling during NMDA-induced LTD (Hanley and Henley 2010; Thomas et al. 2012) (Fig. 3). Although the PATs for GRIP1b, DHHC5 and DHHC8 were identified (Thomas et al. 2012), the PATs for GRIP2b remain to be discovered.
AKAP79/150 (AKAP79 in humans and AKAP150 in rodents), a scaffold protein that is not a direct binding protein with AMPARs, is palmitoylated at Cys36 and Cys129 in AKAP79 by DHHC2 (Keith et al. 2012; Delint-Ramirez et al. 2015; Woolfrey et al. 2015) (Figs. 2, 3). Palmitoylation of AKAP79/150 is required for its targeting to recycling endosomes (Keith et al. 2012; Purkey et al. 2018) and dendritic spines upon LTP-inducing stimulation (Keith et al. 2012; Woolfrey et al. 2015). Conversely, LTD-inducing stimulation, which is accompanied by spine shrinkage, required depalmitoylation and synaptic removal of AKAP79/150 (Keith et al. 2012; Woolfrey et al. 2018). Super-resolution (approximately 40–60 nm) stimulated emission depletion microscopy revealed that palmitoylation-deficient AKAP150 localization to the PSD was significantly reduced in AKAP150 palmitoylation-deficient knockin mice (Purkey et al. 2018). In addition, AKAP150 palmitoylation restricted synaptic localization of Ca2+-permeable AMPARs containing GluA1 but lacking GluA2 in the basal state and was required for Ca2+-permeable AMPAR-dependent LTP (Purkey et al. 2018).
In addition to PSD-95, GRIP/ABP, and AKA79/150, many other synaptic proteins have also been reported to be palmitoylated (Fukata and Fukata 2010; Cho and Park 2016). PICK1 is a PDZ domain-containing protein that directly interacts with GluA2 and GluA3 (Dev et al. 1999; Xia et al. 1999). PICK1 plays critical roles in cerebellar LTD (Steinberg et al. 2006; Anggono et al. 2013; Thomas et al. 2013), hippocampal synaptic plasticity (Terashima et al. 2004, 2008; Volk et al. 2010; Anggono et al. 2011) and cortical synaptic plasticity (Clem et al. 2010) by regulating synaptic abundance, trafficking, and functions of AMPARs. Palmitoylation of PICK1 by DHHC8 was reported to be critical for cerebellar LTD (Thomas et al. 2013). Even though important work has reported the roles of PICK1 in AMPAR trafficking, synaptic plasticity, and learning and memory, very little is known about how PICK1 is palmitoylated and how this contributes to AMPAR trafficking and synaptic function.
Perspectives
Our understanding of the roles of palmitoylation in synaptic targeting, glutamate receptor trafficking, synaptic transmission, synaptic plasticity, and learning and memory has increased with the aid of identifications of pamitoylating enzymes, namely, the DHHC family PATs (Fukata et al. 2004; Fukata and Fukata 2010) and depalmitoylating ABHD enzymes (Yokoi et al. 2016) and development of the non-radioactive palmitoylation assay (Wan et al. 2007) and omics technologies (Kang et al. 2008). Many neuronal substrates for PATs have been discovered with rather broad specificities of the PATs as well as the substrates (Fukata and Fukata 2010; Cho and Park 2016). Future investigations will be required to clarify specific substrate and PAT pairs by dissecting the mechanisms underlying these specificities. In addition, the detailed mechanisms underlying how PATs regulate synaptic function in pathophysiology remain to be revealed. Furthermore, depalmitoylation-mediated regulation of synaptic function remains largely unexplored.
Palmitoylation of AMPARs is affected by another PTM, namely phosphorylation (Lin et al. 2009). Depalmitoylation at Cys811 in the C-tail of GluA1 positively regulates nearby protein kinase C phosphorylation at Ser816 and Ser818, which enhances the interaction of 4.1N with GluA1, and facilitates GluA1 insertion and LTP (Lin et al. 2009). Given that ubiquitination has also been suggested to modulate signaling for fine regulation of AMPAR trafficking via communication with palmitoylation (Yang et al. 2009) or phosphorylation of GluA1 (Kessels et al. 2009), and that synaptic targeting of PSD-95 is also reciprocally controlled by nitrosylation and palmitoylation (Ho et al. 2011), it will be important to further dissect how AMPAR trafficking and synaptic function are regulated through crosstalk among PTMs.
Protein palmitoylation is impaired in various neurodegenerative diseases including Alzheimer’s disease (Mizumaru et al. 2009; Bhattacharyya et al. 2013), Huntington’s disease (Singaraja et al. 2002; Huang et al. 2004; Yanai et al. 2006; Singaraja et al. 2011; Milnerwood et al. 2013), schizophrenia (Liu et al. 2002; Mukai et al. 2004, 2008; Mukai et al. 2015), intellectual disability (Mansouri et al. 2005; Tarpey et al. 2009; Masurel-Paulet et al. 2014; Mitchell et al. 2014), and neuronal ceroid lipofuscinosis (Vesa et al. 1995; Henderson et al. 2016). Altered AMPAR trafficking has also been described in neurological disorders (Ikonomovic et al. 1995, 1997; Muddashetty et al. 2007; Suvrathan et al. 2010; Reinders et al. 2016; Jurado 2017). To our knowledge, however, studies showing direct evidence of palmitoylation-mediated regulation of AMPAR trafficking in neurological diseases are absent. Future studies detailing how the palmitoylation and depalmitoylation of AMPARs and other synaptic proteins affect the pathogeneses of neurodegenerative diseases are needed.
AMPAR trafficking, especially the surface delivery of AMPARs, is crucial for the physiological functioning of synapses and pathogeneses of neurodegenerative diseases. As described in this review, AMPARs themselves and other AMPAR-interacting neuronal proteins are palmitoylated, and palmitoylation plays important roles in the surface delivery of AMPARs, which is necessary for synaptic function. Therefore, future studies in the field of neuronal palmitoylation should focus on the regulation of AMPAR trafficking by specific PATs and depalmitoylating enzymes in pathophysiological conditions. This will provide informative clues for the development of selective pharmacological therapeutics aimed at ameliorating neurodegenerative diseases derived from PATs or depalmitoylating enzymes associated with AMPAR trafficking.
References
Anggono V, Clem RL, Huganir RL (2011) PICK1 loss of function occludes homeostatic synaptic scaling. J Neurosci 31:2188–2196
Anggono V, Koc-Schmitz Y, Widagdo J, Kormann J, Quan A, Chen CM, Robinson PJ, Choi SY, Linden DJ, Plomann M, Huganir RL (2013) PICK1 interacts with PACSIN to regulate AMPA receptor internalization and cerebellar long-term depression. Proc Natl Acad Sci USA 110:13976–13981
Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF (2000) Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci 20:89–102
Bhattacharyya R, Barren C, Kovacs DM (2013) Palmitoylation of amyloid precursor protein regulates amyloidogenic processing in lipid rafts. J Neurosci 33:11169–11183
Borgdorff AJ, Choquet D (2002) Regulation of AMPA receptor lateral movements. Nature 417:649–653
Cho E, Park M (2016) Palmitoylation in Alzheimer’s disease and other neurodegenerative diseases. Pharmacol Res 111:133–151
Clem RL, Anggono V, Huganir RL (2010) PICK1 regulates incorporation of calcium-permeable AMPA receptors during cortical synaptic strengthening. J Neurosci 30:6360–6366
Collingridge GL, Olsen RW, Peters J, Spedding M (2009) A nomenclature for ligand-gated ion channels. Neuropharmacology 56:2–5
Craig TJ, Henley JM (2012) Protein SUMOylation in spine structure and function. Curr Opin Neurobiol 22:480–487
Craven SE, El-Husseini AE, Bredt DS (1999) Synaptic targeting of the postsynaptic density protein PSD-95 mediated by lipid and protein motifs. Neuron 22:497–509
Delint-Ramirez I, Willoughby D, Hammond GR, Ayling LJ, Cooper DM (2015) Palmitoylation targets AKAP79 protein to lipid rafts and promotes its regulation of calcium-sensitive adenylyl cyclase type 8. J Biol Chem 290:13041
Desouza S, Fu J, States BA, Ziff EB (2002) Differential palmitoylation directs the AMPA receptor-binding protein ABP to spines or to intracellular clusters. J Neurosci 22:3493–3503
Dev KK, Nishimune A, Henley JM, Nakanishi S (1999) The protein kinase C alpha binding protein PICK1 interacts with short but not long form alternative splice variants of AMPA receptor subunits. Neuropharmacology 38:635–644
Diering GH, Huganir RL (2018) The AMPA receptor code of synaptic plasticity. Neuron 100:314–329
Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51:7–61
Dong H, O’brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL (1997) GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature 386:279–284
El-Husseini Ael D, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier-Campbell C, Aguilera-Moreno A, Nicoll RA, Bredt DS (2002) Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108:849–863
El-Husseini AE, Craven SE, Chetkovich DM, Firestein BL, Schnell E, Aoki C, Bredt DS (2000) Dual palmitoylation of PSD-95 mediates its vesiculotubular sorting, postsynaptic targeting, and ion channel clustering. J Cell Biol 148:159–172
Elias GM, Nicoll RA (2007) Synaptic trafficking of glutamate receptors by MAGUK scaffolding proteins. Trends Cell Biol 17:343–352
Fukata Y, Fukata M (2010) Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci 11:161–175
Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS (2004) Identification of PSD-95 palmitoylating enzymes. Neuron 44:987–996
Han J, Wu P, Wang F, Chen J (2015) S-palmitoylation regulates AMPA receptors trafficking and function: a novel insight into synaptic regulation and therapeutics. Acta Pharm Sin B 5:1–7
Hanley LJ, Henley JM (2010) Differential roles of GRIP1a and GRIP1b in AMPA receptor trafficking. Neurosci Lett 485:167–172
Hayashi T, Rumbaugh G, Huganir RL (2005) Differential regulation of AMPA receptor subunit trafficking by palmitoylation of two distinct sites. Neuron 47:709–723
Hayashi T, Thomas GM, Huganir RL (2009) Dual palmitoylation of NR2 subunits regulates NMDA receptor trafficking. Neuron 64:213–226
Heine M, Thoumine O, Mondin M, Tessier B, Giannone G, Choquet D (2008) Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc Natl Acad Sci USA 105:20947–20952
Henderson MX, Wirak GS, Zhang YQ, Dai F, Ginsberg SD, Dolzhanskaya N, Staropoli JF, Nijssen PC, Lam TT, Roth AF, Davis NG, Dawson G, Velinov M, Chandra SS (2016) Neuronal ceroid lipofuscinosis with DNAJC5/CSPalpha mutation has PPT1 pathology and exhibit aberrant protein palmitoylation. Acta Neuropathol 131:621–637
Ho GP, Selvakumar B, Mukai J, Hester LD, Wang Y, Gogos JA, Snyder SH (2011) S-nitrosylation and S-palmitoylation reciprocally regulate synaptic targeting of PSD-95. Neuron 71:131–141
Hollmann M, Heinemann S (1994) Cloned glutamate receptors. Annu Rev Neurosci 17:31–108
Hollmann M, Maron C, Heinemann S (1994) N-glycosylation site tagging suggests a three transmembrane domain topology for the glutamate receptor GluR1. Neuron 13:1331–1343
Huang K, Yanai A, Kang R, Arstikaitis P, Singaraja RR, Metzler M, Mullard A, Haigh B, Gauthier-Campbell C, Gutekunst CA, Hayden MR, El-Husseini A (2004) Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmitoylation and trafficking of multiple neuronal proteins. Neuron 44:977–986
Ikonomovic MD, Sheffield R, Armstrong DM (1995) AMPA-selective glutamate receptor subtype immunoreactivity in the hippocampal formation of patients with Alzheimer’s disease. Hippocampus 5:469–486
Ikonomovic MD, Mizukami K, Davies P, Hamilton R, Sheffield R, Armstrong DM (1997) The loss of Glu R2(3) immunoreactivity precedes neurofibrillary tangle formation in the entorhinal cortex and hippocampus of Alzheimer brains. J Neuropathol Exp Neurol 56:1018–1027
Jaafari N, Konopacki FA, Owen TF, Kantamneni S, Rubin P, Craig TJ, Wilkinson KA, Henley JM (2013) SUMOylation is required for glycine-induced increases in AMPA receptor surface expression (ChemLTP) in hippocampal neurons. PLoS ONE 8:e52345
Jeyifous O, Lin EI, Chen X, Antinone SE, Mastro R, Drisdel R, Reese TS, Green WN (2016) Palmitoylation regulates glutamate receptor distributions in postsynaptic densities through control of PSD95 conformation and orientation. Proc Natl Acad Sci USA 113:E8482–E8491
Jurado S (2017) AMPA receptor trafficking in natural and pathological aging. Front Mol Neurosci 10:446
Kang R, Swayze R, Lise MF, Gerrow K, Mullard A, Honer WG, El-Husseini A (2004) Presynaptic trafficking of synaptotagmin I is regulated by protein palmitoylation. J Biol Chem 279:50524–50536
Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey AO, Thompson JX, Roth AF, Drisdel RC, Mastro R, Green WN, Yates JR 3rd, Davis NG, El-Husseini A (2008) Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature 456:904–909
Keith DJ, Sanderson JL, Gibson ES, Woolfrey KM, Robertson HR, Olszewski K, Kang R, El-Husseini A, Dell’acqua ML (2012) Palmitoylation of A-kinase anchoring protein 79/150 regulates dendritic endosomal targeting and synaptic plasticity mechanisms. J Neurosci 32:7119–7136
Kessels HW, Kopec CD, Klein ME, Malinow R (2009) Roles of stargazin and phosphorylation in the control of AMPA receptor subcellular distribution. Nat Neurosci 12:888–896
Kim E, Sheng M (2004) PDZ domain proteins of synapses. Nat Rev Neurosci 5:771–781
Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL (2000) Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405:955–959
Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL (2003) Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112:631–643
Lee HK, Takamiya K, He K, Song L, Huganir RL (2010) Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol 103:479–489
Lee L, Sakurai M, Matsuzaki S, Arancio O, Fraser P (2013) SUMO and Alzheimer’s disease. Neuromol Med 15:720–736
Lin DT, Makino Y, Sharma K, Hayashi T, Neve R, Takamiya K, Huganir RL (2009) Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat Neurosci 12:879–887
Liu H, Abecasis GR, Heath SC, Knowles A, Demars S, Chen YJ, Roos JL, Rapoport JL, Gogos JA, Karayiorgou M (2002) Genetic variation in the 22q11 locus and susceptibility to schizophrenia. Proc Natl Acad Sci USA 99:16859–16864
Lu W, Roche KW (2012) Posttranslational regulation of AMPA receptor trafficking and function. Curr Opin Neurobiol 22:470–479
Lu W, Man H, Ju W, Trimble WS, Macdonald JF, Wang YT (2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29:243–254
Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA (2009) Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62:254–268
Lussier MP, Nasu-Nishimura Y, Roche KW (2011) Activity-dependent ubiquitination of the AMPA receptor subunit GluA2. J Neurosci 31:3077–3081
Lussier MP, Herring BE, Nasu-Nishimura Y, Neutzner A, Karbowski M, Youle RJ, Nicoll RA, Roche KW (2012) Ubiquitin ligase RNF167 regulates AMPA receptor-mediated synaptic transmission. Proc Natl Acad Sci USA 109:19426–19431
Lussier MP, Sanz-Clemente A, Roche KW (2015) Dynamic regulation of N-methyl-d-ASPARTATE (NMDA) and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by posttranslational modifications. J Biol Chem 290:28596–28603
Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25:103–126
Mansouri MR, Marklund L, Gustavsson P, Davey E, Carlsson B, Larsson C, White I, Gustavson KH, Dahl N (2005) Loss of ZDHHC15 expression in a woman with a balanced translocation t(X;15)(q13.3;cen) and severe mental retardation. Eur J Hum Genet 13:970–977
Masurel-Paulet A, Kalscheuer VM, Lebrun N, Hu H, Levy F, Thauvin-Robinet C, Darmency-Stamboul V, El Chehadeh S, Thevenon J, Chancenotte S, Ruffier-Bourdet M, Bonnet M, Pinoit JM, Huet F, Desportes V, Chelly J, Faivre L (2014) Expanding the clinical phenotype of patients with a ZDHHC9 mutation. Am J Med Genet A 164A:789–795
Milnerwood AJ, Parsons MP, Young FB, Singaraja RR, Franciosi S, Volta M, Bergeron S, Hayden MR, Raymond LA (2013) Memory and synaptic deficits in Hip14/DHHC17 knockout mice. Proc Natl Acad Sci USA 110:20296–20301
Misra C, Restituito S, Ferreira J, Rameau GA, Fu J, Ziff EB (2010) Regulation of synaptic structure and function by palmitoylated AMPA receptor binding protein. Mol Cell Neurosci 43:341–352
Mitchell DA, Hamel LD, Reddy KD, Farh L, Rettew LM, Sanchez PR, Deschenes RJ (2014) Mutations in the X-linked intellectual disability gene, zDHHC9, alter autopalmitoylation activity by distinct mechanisms. J Biol Chem 289:18582–18592
Mizumaru C, Saito Y, Ishikawa T, Yoshida T, Yamamoto T, Nakaya T, Suzuki T (2009) Suppression of APP-containing vesicle trafficking and production of beta-amyloid by AID/DHHC-12 protein. J Neurochem 111:1213–1224
Muddashetty RS, Kelic S, Gross C, Xu M, Bassell GJ (2007) Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci 27:5338–5348
Mukai J, Liu H, Burt RA, Swor DE, Lai WS, Karayiorgou M, Gogos JA (2004) Evidence that the gene encoding ZDHHC8 contributes to the risk of schizophrenia. Nat Genet 36:725–731
Mukai J, Dhilla A, Drew LJ, Stark KL, Cao L, Macdermott AB, Karayiorgou M, Gogos JA (2008) Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nat Neurosci 11:1302–1310
Mukai J, Tamura M, Fenelon K, Rosen AM, Spellman TJ, Kang R, Macdermott AB, Karayiorgou M, Gordon JA, Gogos JA (2015) Molecular substrates of altered axonal growth and brain connectivity in a mouse model of schizophrenia. Neuron 86:680–695
Noritake J, Fukata Y, Iwanaga T, Hosomi N, Tsutsumi R, Matsuda N, Tani H, Iwanari H, Mochizuki Y, Kodama T, Matsuura Y, Bredt DS, Hamakubo T, Fukata M (2009) Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J Cell Biol 186:147–160
Park M (2018) AMPA receptor trafficking for postsynaptic potentiation. Front Cell Neurosci 12:361
Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD (2004) Recycling endosomes supply AMPA receptors for LTP. Science 305:1972–1975
Patterson MA, Szatmari EM, Yasuda R (2010) AMPA receptors are exocytosed in stimulated spines and adjacent dendrites in a Ras-ERK-dependent manner during long-term potentiation. Proc Natl Acad Sci USA 107:15951–15956
Purkey AM, Woolfrey KM, Crosby KC, Stich DG, Chick WS, Aoto J, Dell’acqua ML (2018) AKAP150 palmitoylation regulates synaptic incorporation of Ca(2+)-permeable AMPA receptors to control LTP. Cell Rep 25(974–987):e974
Reinders NR, Pao Y, Renner MC, Da Silva-Matos CM, Lodder TR, Malinow R, Kessels HW (2016) Amyloid-beta effects on synapses and memory require AMPA receptor subunit GluA3. Proc Natl Acad Sci USA 113:E6526–E6534
Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA (2002) Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA 99:13902–13907
Schwarz LA, Hall BJ, Patrick GN (2010) Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J Neurosci 30:16718–16729
Shepherd JD, Huganir RL (2007) The cell biology of synaptic plasticity: aMPA receptor trafficking. Annu Rev Cell Dev Biol 23:613–643
Singaraja RR, Hadano S, Metzler M, Givan S, Wellington CL, Warby S, Yanai A, Gutekunst CA, Leavitt BR, Yi H, Fichter K, Gan L, Mccutcheon K, Chopra V, Michel J, Hersch SM, Ikeda JE, Hayden MR (2002) HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum Mol Genet 11:2815–2828
Singaraja RR, Huang K, Sanders SS, Milnerwood AJ, Hines R, Lerch JP, Franciosi S, Drisdel RC, Vaid K, Young FB, Doty C, Wan J, Bissada N, Henkelman RM, Green WN, Davis NG, Raymond LA, Hayden MR (2011) Altered palmitoylation and neuropathological deficits in mice lacking HIP14. Hum Mol Genet 20:3899–3909
Spinelli M, Fusco S, Mainardi M, Scala F, Natale F, Lapenta R, Mattera A, Rinaudo M, Li Puma DD, Ripoli C, Grassi A, D’ascenzo M, Grassi C (2017) Brain insulin resistance impairs hippocampal synaptic plasticity and memory by increasing GluA1 palmitoylation through FoxO3a. Nat Commun 8:2009
Srivastava S, Osten P, Vilim FS, Khatri L, Inman G, States B, Daly C, Desouza S, Abagyan R, Valtschanoff JG, Weinberg RJ, Ziff EB (1998) Novel anchorage of GluR2/3 to the postsynaptic density by the AMPA receptor-binding protein ABP. Neuron 21:581–591
Standley S, Baudry M (2000) The role of glycosylation in ionotropic glutamate receptor ligand binding, function, and trafficking. Cell Mol Life Sci 57:1508–1516
Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, Huganir RL (2006) Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron 49:845–860
Sturgill JF, Steiner P, Czervionke BL, Sabatini BL (2009) Distinct domains within PSD-95 mediate synaptic incorporation, stabilization, and activity-dependent trafficking. J Neurosci 29:12845–12854
Suvrathan A, Hoeffer CA, Wong H, Klann E, Chattarji S (2010) Characterization and reversal of synaptic defects in the amygdala in a mouse model of fragile X syndrome. Proc Natl Acad Sci USA 107:11591–11596
Tardin C, Cognet L, Bats C, Lounis B, Choquet D (2003) Direct imaging of lateral movements of AMPA receptors inside synapses. EMBO J 22:4656–4665
Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, Omeara S, Latimer C, Dicks E, Menzies A, Stephens P, Blow M, Greenman C, Xue Y, Tyler-Smith C, Thompson D, Gray K, Andrews J, Barthorpe S, Buck G, Cole J, Dunmore R, Jones D, Maddison M, Mironenko T, Turner R, Turrell K, Varian J, West S, Widaa S, Wray P, Teague J, Butler A, Jenkinson A, Jia M, Richardson D, Shepherd R, Wooster R, Tejada MI, Martinez F, Carvill G, Goliath R, De Brouwer AP, Van Bokhoven H, Van Esch H, Chelly J, Raynaud M, Ropers HH, Abidi FE, Srivastava AK, Cox J, Luo Y, Mallya U, Moon J, Parnau J, Mohammed S, Tolmie JL, Shoubridge C, Corbett M, Gardner A, Haan E, Rujirabanjerd S, Shaw M, Vandeleur L, Fullston T, Easton DF, Boyle J, Partington M, Hackett A, Field M, Skinner C, Stevenson RE, Bobrow M, Turner G, Schwartz CE, Gecz J, Raymond FL, Futreal PA, Stratton MR (2009) A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet 41:535–543
Terashima A, Cotton L, Dev KK, Meyer G, Zaman S, Duprat F, Henley JM, Collingridge GL, Isaac JT (2004) Regulation of synaptic strength and AMPA receptor subunit composition by PICK1. J Neurosci 24:5381–5390
Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge GL, Mcbain CJ, Isaac JT (2008) An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron 57:872–882
Thomas GM, Huganir RL (2013) Palmitoylation-dependent regulation of glutamate receptors and their PDZ domain-containing partners. Biochem Soc Trans 41:72–78
Thomas GM, Hayashi T, Chiu SL, Chen CM, Huganir RL (2012) Palmitoylation by DHHC5/8 targets GRIP1 to dendritic endosomes to regulate AMPA-R trafficking. Neuron 73:482–496
Thomas GM, Hayashi T, Huganir RL, Linden DJ (2013) DHHC8-dependent PICK1 palmitoylation is required for induction of cerebellar long-term synaptic depression. J Neurosci 33:15401–15407
Tomita S, Adesnik H, Sekiguchi M, Zhang W, Wada K, Howe JR, Nicoll RA, Bredt DS (2005) Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature 435:1052–1058
Topinka JR, Bredt DS (1998) N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel Kv1.4. Neuron 20:125–134
Traynelis SF, Wollmuth LP, Mcbain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496
Uemura T, Mori H, Mishina M (2002) Isolation and characterization of Golgi apparatus-specific GODZ with the DHHC zinc finger domain. Biochem Biophys Res Commun 296:492–496
Van Dolah DK, Mao LM, Shaffer C, Guo ML, Fibuch EE, Chu XP, Buch S, Wang JQ (2011) Reversible palmitoylation regulates surface stability of AMPA receptors in the nucleus accumbens in response to cocaine in vivo. Biol Psychiatry 69:1035–1042
Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P, Hofmann SL, Peltonen L (1995) Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature 376:584–587
Volk L, Kim CH, Takamiya K, Yu Y, Huganir RL (2010) Developmental regulation of protein interacting with C kinase 1 (PICK1) function in hippocampal synaptic plasticity and learning. Proc Natl Acad Sci USA 107:21784–21789
Wan J, Roth AF, Bailey AO, Davis NG (2007) Palmitoylated proteins: purification and identification. Nat Protoc 2:1573–1584
Wei J, Xiong Z, Lee JB, Cheng J, Duffney LJ, Matas E, Yan Z (2016) Histone modification of Nedd4 ubiquitin ligase controls the loss of AMPA receptors and cognitive impairment induced by repeated stress. J Neurosci 36:2119–2130
Wenthold RJ, Petralia RS, Blahos J, Niedzielski AS (1996) Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci 16:1982–1989
Widagdo J, Chai YJ, Ridder MC, Chau YQ, Johnson RC, Sah P, Huganir RL, Anggono V (2015) Activity-dependent ubiquitination of GluA1 and GluA2 regulates AMPA receptor intracellular sorting and degradation. Cell Rep 10:783–795
Wisden W, Seeburg PH (1993) Mammalian ionotropic glutamate receptors. Curr Opin Neurobiol 3:291–298
Woolfrey KM, Sanderson JL, Dell’acqua ML (2015) The palmitoyl acyltransferase DHHC2 regulates recycling endosome exocytosis and synaptic potentiation through palmitoylation of AKAP79/150. J Neurosci 35:442–456
Woolfrey KM, Oleary H, Goodell DJ, Robertson HR, Horne EA, Coultrap SJ, Dellacqua ML, Bayer KU (2018) CaMKII regulates the depalmitoylation and synaptic removal of the scaffold protein AKAP79/150 to mediate structural long-term depression. J Biol Chem 293:1551–1567
Wu D, Bacaj T, Morishita W, Goswami D, Arendt KL, Xu W, Chen L, Malenka RC, Sudhof TC (2017) Postsynaptic synaptotagmins mediate AMPA receptor exocytosis during LTP. Nature 544:316–321
Wyszynski M, Valtschanoff JG, Naisbitt S, Dunah AW, Kim E, Standaert DG, Weinberg R, Sheng M (1999) Association of AMPA receptors with a subset of glutamate receptor-interacting protein in vivo. J Neurosci 19:6528–6537
Xia J, Zhang X, Staudinger J, Huganir RL (1999) Clustering of AMPA receptors by the synaptic PDZ domain-containing protein PICK1. Neuron 22:179–187
Yamazaki M, Fukaya M, Abe M, Ikeno K, Kakizaki T, Watanabe M, Sakimura K (2001) Differential palmitoylation of two mouse glutamate receptor interacting protein 1 forms with different N-terminal sequences. Neurosci Lett 304:81–84
Yanai A, Huang K, Kang R, Singaraja RR, Arstikaitis P, Gan L, Orban PC, Mullard A, Cowan CM, Raymond LA, Drisdel RC, Green WN, Ravikumar B, Rubinsztein DC, El-Husseini A, Hayden MR (2006) Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat Neurosci 9:824–831
Yang G, Xiong W, Kojic L, Cynader MS (2009) Subunit-selective palmitoylation regulates the intracellular trafficking of AMPA receptor. Eur J Neurosci 30:35–46
Yokoi N, Fukata Y, Sekiya A, Murakami T, Kobayashi K, Fukata M (2016) Identification of PSD-95 depalmitoylating enzymes. J Neurosci 36:6431–6444
Zhang Y, Matt L, Patriarchi T, Malik ZA, Chowdhury D, Park DK, Renieri A, Ames JB, Hell JW (2014) Capping of the N-terminus of PSD-95 by calmodulin triggers its postsynaptic release. EMBO J 33:1341–1353
Zhu JJ, Esteban JA, Hayashi Y, Malinow R (2000) Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci 3:1098–1106
Acknowledgements
The work in the Park laboratory was supported by the Original Technology Research Program for Brain Science of the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (No. 2018M3C7A1021848).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sohn, H., Park, M. Palmitoylation-mediated synaptic regulation of AMPA receptor trafficking and function. Arch. Pharm. Res. 42, 426–435 (2019). https://doi.org/10.1007/s12272-019-01134-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-019-01134-z