
Abstract
Breast cancer (BC) leads to the most amounts of deaths among women. Chemo-, endocrine-, and targeted therapies are the mainstay drug treatments for BC in the clinic. However, drug resistance is a major obstacle for BC patients, and it leads to poor prognosis. Accumulating evidences suggested that noncoding RNAs (ncRNAs) are intricately linked to a wide range of pathological processes, including drug resistance. Till date, the correlation between drug resistance and ncRNAs is not completely understood in BC. Herein, we comprehensively summarized a dysregulated ncRNAs landscape that promotes or inhibits drug resistance in chemo-, endocrine-, and targeted BC therapies. Our review will pave way for the effective management of drug resistance by targeting oncogenic ncRNAs, which, in turn will promote drug sensitivity of BC in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer (BC) is a significant global health challenge [1]. It is a heterogeneous disease, involving numerous categories. There are five main categories of BC, stratified by the expressions of estrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor receptor 2 (HER2), and Ki-67. The corresponding categories are Luminal A (LA), Luminal B (LB), Human epidermal growth factor receptor 2 (HER2) + , Normal breast-like (NBl) and Basal subtype [Triple negative breast cancer (TNBC)] [2, 3]. LA tumors typically show strong ER and PR levels and scarce HER2 and Ki-67 levels. LB cancers display strong ER and PR levels, strong or weak HER2 levels, and elevated Ki-67 levels. Given their distinct gene expressions, LA and LB tumors are generally more responsive to endocrine therapy, compared to chemotherapy [4]. In contrast, HER2 tumors have no ER and PR expressions, instead, they express HER2 and Ki-67. HER2 tumors are, therefore, better managed with targeted therapies, and adequately respond to neoadjuvant chemotherapy [4, 5]. The NBl form expresses ER and PR, and does not express HER2 and Ki-67. Therefore, these also respond well to chemotherapy. Lastly, TNBC responds well to neoadjuvant chemotherapy, however, the distant recurrence rates are markedly higher than other cancer forms [4]. Despite massive developments in various treatment regimen, a large quantity of patients still experienced disease recurrence and reduced survival due to new or acquired resistance to treatments, which, in turn, enhances metastatic risk [6]. Unfortunately, once metastasis occurs, the five-year overall survival (OS) rate becomes less than 25% [7]. Numerous cancer drug resistance pathways involve modifications in drug efflux, DNA repair, escape from apoptosis, immune system evasion, improvised and differential metabolisms, drug target mutations, and epigenetic alterations [8].
Noncoding RNAs (NcRNAs) are known to regulate drug resistance in BC patients. Hence, it is critical to elucidate the correlation and underlying mechanism of the relationship governing ncRNAs and drug resistance in BC. Scientists reported that > 80% of the entire human genome undergoes transcription [9, 10]. Interestingly, only < 2% of the transcription produces functional proteins, and the rest generates ncRNAs. NcRNAs are largely separated into two categories, depending on their size and function: (1) short ncRNAs: < 200-nucleotides long, include microRNAs (miRNAs), small interfering RNAs (siRNAs), small nucleolar RNAs (snoRNAs), and Piwi-interacting RNAs (piRNAs); and (2) long non-coding RNAs (lncRNAs): > 200-nucleotides long, transcribed via RNA polymerase II, and contains a 5’ cap, transcription start site, and polyadenylation [11]. There is a peculiar class of lncRNAs called circular RNAs (circRNAs), and they are ubiquitously found within mammals [12]. LncRNAs serve essential roles in tumor pathogenesis via both transcriptional and post-transcriptional regulation [13, 14]. In general, cytoplasmic lncRNAs modulate cell signaling, as well as transcript stability or protein translation, while nuclear lncRNAs regulate chromatin associations, as well as transcriptional and mRNA stability regulation [15]. MiRNAs belong to a category of small ncRNA that suppress protein-coding gene expression by targeting respective transcripts [16]. Several studies suggested that ncRNAs modulate gene expression at the epigenetic, transcriptional, post-transcriptional, translational and even sub-cellular localization levels [17]. Therefore, ncRNAs are known to regulate multiple facets of BC progression like cell proliferation, angiogenesis, epithelial-mesenchymal transition (EMT), cancer stem cells (CSCs), drug resistance, and metastasis [17].
In this report, we performed a review of the detailed mechanisms behind the ncRNAs-mediated regulation of chemo-, endocrine-, and targeted therapeutic resistance in BC. Moreover, our review identified possible therapeutic targets that may potentially diminish drug resistance or enhance BC treatment efficacy.
NcRNAs regulate chemotherapeutic resistance in BC
Chemotherapy is a well-known and effective BC treatment that improves prognosis and OS of patients [18]. Chemotherapy includes anthracyclines and/or taxane administration, and in select patients, cyclophosphamide, methotrexate, and/or 5-flurouracil (5-FU) are used [19]. The mechanism underlying chemoresistance likely involves both genetic and epigenetic alterations like drug-driven mutations, drug metabolic enzyme abnormalities, cell-cycle- and apoptosis- related genes, DNA methylation, and histone modifications [20]. Moreover, most chemotherapeutic medications destroy DNA, and in response, cells elicit a DNA damage response (DDR), which may inadvertently induce drug resistance [21]. In addition, drug efflux is a commonly examined mechanism of cancer drug resistance, and enhanced drug efflux is commonly present in multidrug resistance (MDR) [22, 23]. Up-regulations in the levels of ATP-binding cassette (ABC) superfamily members like P-gp (ABCB1), multidrug-resistance-associated protein 1 (MRP1/ABCC1), multidrug-resistance-associated protein 7 (MRP7/ABCC10), and BC resistance protein (BCRP/ABCG2) are frequently observed in drug resistance associated with various forms of cancers [24,25,26].
NcRNAs promote chemoresistance
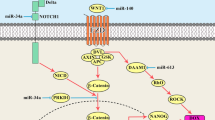
The mechanisms of ncRNAs promoting chemoresistance are summarized based on the following aspects: (i) EMT, (ii) cell cycle, (iii) autophagy, (iv) drug efflux transporters, (v) pro-survival signaling pathways, (vi) apoptosis, and (vii) DNA damage repair (Fig. 1 and Table 1).

NcRNAs promote chemo-resistance
LINK00160
Abnormally expressed trefoil factor 3 (TFF3) enhances oncogenesis of prostate cancer cells [27]. In addition, LINC00160 overexpression was shown to increase TFF3 levels via C/EBPβ regulation. In doing so, MCF-7 cells were made to be resistant to paclitaxel (PTX) and BT474 cells to doxorubicin (DOX) [28].
LncMIAT
Autophagy is a cellular process that is induced by nutrient deprivation, endoplasmic reticulum stress (ERS), and hypoxia [29]. The ERS is intricately linked to drug resistance in BC [30,31,32]. 5-FU induces BC cell resistance via induction of ERS. As a result, the GRP78/OCT4/lncRNA MIAT/AKT pathway is activated [33].
LncNEAT1
HMGA2 is reported to regulate EMT transcription factors (TFs) in BC patients [34]. LncRNA NEAT1 promotes cell proliferation using the miR-211/HMGA2 pathway in BC patients. They also revealed that NEAT1 suppression enhances 5-FU responsiveness to BC [35].
LncRNA BORG
LncRNA BORG levels are very susceptible to cytotoxic medications, and promotes a transcriptional response that mediates survival and chemoresistance of TNBC cells. Mechanically, the chemo-resistant BORG traits depend on the robust activation of the NF-κB axis via a new BORG-based feedback loop, and via its ability to interact with and activate RPA1 [36].
LncRNA NONHSAT101069
Overexpressing lncRNA NONHSAT101069 enhances epirubicin resistance and EMT processing of BC cells. In terms of underlying mechanism, NONHSAT101069 functions as a competing endogenous RNA (ceRNA) and sequesters miR-129-5p, which, in turn, promotes epirubicin resistance, metastasis, and EMT processing of BC cells via the Twist1 axis [37].
LncRNA FTH1P3
FTH1P3 upregulation accelerates cell proliferation, migration, cell cycle and migration via suppression of miR-224-5p in uveal melanoma cell lines [38]. FTH1P3 levels are enriched in PTX-resistant BC tissue specimen and cells. Mechanically, FTH1P3 serves as a ceRNA and sequesters miR-206 to augment ABCB1 protein concentration [39].
Linc00518
MRP1 which originated from the ABCC1 gene, belongs to the ABC transporter superfamily residing on chromosome 16p13.1. Elevated MRP1 levels enhance MDR in BC [40, 41]. Linc00518 induces MDR in BC by modulating the miR-199a/MRP1 network [42].
LncZEB1-AS1
ZEB1-driven BC progression occurs via acceleration of EMT, tumor pathogenesis, and angiogenesis [43, 44]. LncRNA ZEB1-AS1 is ubiquitously expressed in BC. In addition, researchers demonstrated that ZEB1-AS1 deficiency drastically reduces ZEB1 content by up-regulating miR-129-5p, which, ultimately enhances drug sensitivity to cisplatin in BC [45].
LncPTENP1
There is evidence of considerable homology between lncRNA PTENP1 and the upstream section of the 3’untranslated region (UTR) of phosphatase and tensin homologs (PTEN). As such, lncPTENP1 readily modulates PTEN levels, which, in turn, affects cancer pathogenesis [46]. PTENP1 modulates Adriamycin (ADR) chemoresistance by interacting with miR-20a via the PTEN/PI3K/Akt network in BC [47].
LncMat2B
LncMat2B is ubiquitously expressed in the cisplatin-resistant MCF-7 cell line. Moreover, its incorporation into wild type MCF-7 cells reduces sensitivity to cisplatin exposure by diminishing DNA damage and reactive oxygen species (ROS) formation [48].
Linc00668
SND1 is crucial for tumor progression in BC [49,50,51,52]. Linc00668 promotes BC cell resistance to DOX via interaction with SND1. This enables the expression of downstream SND1 targets [53].
LncRNA MAPT-AS1
MAPT is strongly correlated with PTX resistance in BC [54, 55]. MAPT-AS1 is an antisense MAPT transcript, and it is co-expressed with MAPT. Mechanically, MAPT-AS1 overexpression partially protects MAPT transcripts from degradation, and vice versa. Conversely, MAPT-AS1 knock-down makes cancer cells more susceptible to PTX by modulating MAPT levels in ER-negative BC [56].
LncRNA DCST1-AS1
Annexin A1 (ANXA1) modulates cancer cell proliferation, apoptosis, invasion, and metastasis [57]. DCST1-AS1 induces transforming growth factor β (TGF β)-triggered EMT, and augments DOX and PTX resistance in TNBC cells using ANXA1 [58].
LncRNA CBR3-AS1
LncRNA CBR3 antisense RNA 1 (CBR3-AS1) induces chemotherapeutic (ADR) resistance of BC by serving as a ceRNA via the JNK1/MEK4-based mitogen-activated protein kinase (MAPK) network [59].
MIR200CHG
Cellular and animal models revealed that MIR200CHG induces BC cisplatin resistance. Mechanically, MIR200CHG physically interacts with the TF Y-box binding protein-1 (YB-1), and prevents its ubiquitination-mediated destruction. MIR200CHG modulates YB-1-mediated phosphorylation at serine 102, which, in turn, influences expression of tumor cell cisplatin resistance-related genes [60].
LncRNA GAS5
P-gp/ABCB1 overexpression increases energy-based cytotoxic drug efflux from cancer cells, thereby enhancing drug resistance [61, 62]. A recent study revealed that GAS5 restores the ABCB1-induced ADR resistance using the miR-221-3p/DKK2 pathway, and by suppressing the Wnt/β-catenin network [63].
CircCDR1as
CDR1as serves as a miR-7 suppressor in the developing midbrain of zebrafish [64]. Mechanically, circRNACDR1as decreases 5-FU chemo-responsiveness in BC by sequestering miR-7 to modulate CCNE1 [65].
MiR-191-5p
Researchers revealed that miR-191-5p is a negative apoptosis modulator in BC. In addition, SOX4 was shown to influence apoptosis in BC [66]. MiR-191-5p directly targets SOX4. Mechanically, the P53-miR-191-SOX4 axis modulates drug resistance in BC. In contrast, anti-miR-191 treatment makes BC cells more susceptible to the DOX-mediated apoptotic death [67].
MiR-105 and MiR-93-3p
MiR-105 and miR-93-3p are generally elevated and associated with worse outcome in TNBC. Mechanically, miR-105/93-3p promotes cisplatin resistance in BC by activating the Wnt/β-catenin network while down-regulating SFPR1 [68].
MiR-132 and MiR-212
MiR-132/-212 are ubiquitously expressed in DOX-resistant BC. MiR-132/-212 overexpression induces BCRP-induced DOX efflux in MCF-7 cells. Moreover, miR-132/-212 overexpression in MCF-7/ADR cells suppresses PTEN levels, while activating AKT phosphorylation and the NF-κB axis, which, in turn, augments BCRP content [69].
MiR-424 (322)/503
The miR-424 (322)/503 cluster contains both miR-424 (322) and miR-503. MiR-424 (322)/503 is often absent in a subcategory of aggressive BCs. MiR-424(322)/503 deficiency enhances PTX chemoresistance owing to the elevation of pro-apoptotic BCL-2 and insulin-like growth factor-1 receptor (IGF1R) [70].
MiR-125b
MiR-125b is commonly elevated in PTX-resistant cells. Mechanically, miR-125b promotes resistance of BC cells to PTX via inhibition of the pro-apoptotic BCL-2 antagonist killer 1 (Bak1) expression [71].
MiR-671-5p
Forkhead box protein M1 (FOXM1) is a TF that regulates drug resistance in BC cells by activating DNA damage repair networks [72,73,74,75]. MiR-671-5p deficiency, in contrast, activates the FOXM1-triggered EMT progression while enhancing DNA repair, and increasing chemoresistance (cisplatin and PTX) [76].
MiR-1246
MiR-1246 functions as an oncogene in cancer [77, 78]. Cyclin G2 (CCNG2) is modulated via the cell cycle and serves as a tumor-suppressor gene [79] and its expression is drastically diminished in BC [80]. Exosomal miR-1246 incorporation induces drug resistance by regulating CCNG2 expression in BC [81].
NcRNAs promotes chemotherapeutic sensitivity
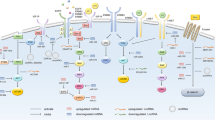
The mechanisms of ncRNAs promoting chemotherapeutic sensitivity using the following factors: (i) EMT, (ii) cell cycle (arrest), (iii) autophagy, (iv) drug efflux transporters, (v) pro-survival signaling pathways, (vi) drug metabolic enzymes, (vii) apoptosis, and (viii) DNA damage repair (Fig. 2 and Table 2).

NcRNAs promote chemo-sensitivity
LncTUG1
NLK is a negative modulator of the WNT network [82]. LncRNA TUG1 mediates its action through the regulation of the miR-197/NLK axis to enhance cisplatin sensitivity in TNBC patients [83].
LncRNA EGOT
Eosinophil granule ontogeny transcript (EGOT) is generated/released by ITPR1, a ligand-gated ion channel involved in the calcium secretion from the intracellular storage [84, 85]. LncRNA EGOT augments autophagy, which, in turn, makes BC more susceptible to PTX cytotoxicity, owing to an elevation in ITPR1 levels [86].
LncRNA-ARA
LncRNA-ARA regulates cell adhesion- and cell cycle progression-linked axes. Jiang et al. reported that ARA deficiency reverses drug resistance, and suppresses cell proliferation, migration, while promoting apoptosis and G2/M arrest in ADR-resistant cells [87].
LINC00968
WNT2 is a major Wnt ligand that regulates placental development [88]. LINC00968 reduces drug resistance (ADR, PTX and Vincristine) in BC by sequestering WNT2 by recruiting HEY1, thereby, suppressing the Wnt/β-catenin axis [89].
LncRNA HCP5
HCP5 was drastically reduced in MDA-MB-231/cisplatin cells, relative to the MDA-MB-231 cells. HCP5 deficiency induces cisplatin resistance in MDA-MB-231 cells by suppressing PTEN levels. Conversely, HCP5 overexpression reverses cisplatin resistance in MDA-MB-231/DDP cells by increasing PTEN levels [90].
LncRNA SNORD3A
Uridine monophosphate synthetase (UMPS) is a 5-FU metabolism-related gene. Mechanically, lncRNA SNORD3A sensitizes BC cells to 5-FU by sequestering miR-185-5p to augment UMPS levels [91].
CircKDM4C
PBLD overexpression is correlated with the suppression of multiple signal networks (Vascular endothelial growth factor A [VEGFA], MAPK, NF-κB, EMT, and angiogenesis) [92]. CircKDM4C abrogates doxorubicin resistance by modulating the miR-548p/PBLD network in BC [93].
MiR-17 and MiR-20b
Nuclear receptor coactivator 3 (NCO3) is a nuclear receptor coactivator which accelerates BC tumor pathogenesis by increasing the ER and PR transcriptional activities [94]. Moreover, miR-17 and miR-20b deficiencies induce PTX resistance in BC by up-regulating NCOA3 levels [95]. In addition, JAB1 is ubiquitously found in BC, and it activates pro-survival cellular networks to confer tamoxifen resistance in ERα-positive BC [96]. MiR-17 also suppresses JAB1’s oncogenic activity, which results in the suppression of tumor development while sensitizing TNBC cells to chemotherapeutic treatments [97].
MiR-708-3p
MiR-708-3p is an anti-cancer miRNA that is inversely associated with BC chemoresistance. MiR-708-3p restoration improves BC chemosensitivity (DOX) by inhibiting EMT via regulating CDH2, ZEB1, and vimentin (EMT stimulators) levels [98].
MiR-20a
MiR-20a overexpression sensitizes BC cells to chemotherapeutic medications (PTX). Mechanically, miR-20a physically interacts with the 3’ UTR of MAPK1, thereby down-regulating levels of P-gp and c-Myc by suppressing the MAPK/ERK network. In the meantime, c-Myc binds to the promoter of the miR-20a gene to induce transcription of the miR-20a gene [99].
MiR-205
VEGFA and fibroblast grow factor-2 (FGF2) are the strongest modulators of angiogenesis [100]. MiR-205 greatly improves chemosensitivity of BC cells to neoadjuvant chemotherapy (docetaxol, DOX, and cyclophosphamide) by diminishing both VEGFA and FGF2 levels, thereby increasing cellular apoptosis evasion [101].
MiR-489
SPIN1 was identified to involve in tumorigenesis [102]. MiR-489 is scarcely expressed in drug resistant BC. Mechanically, miR-489 enhances chemosensitivity (ADR) via the SPIN1/PI3K/Akt network [103].
MiR-199a-3p
MDA-MB-231/cisplatin exhibited a significantly lower expression level of miR-199a-3p compared with its parental cell line MDA-MB-231. MiR-199a-3p regulates mitochondrial transcription factor A (TFAM) levels. TFAM strongly regulates drug resistance (cisplatin) and tumor progression, by suppressing TFAM 3’UTR activity [104].
MiR-181c
Osteopontin (OPN) is excessively expressed in cancer cell lines that are prone to metastasis [105]. MiR-181c increases chemosensitivity (ADR) via diminishing OPN levels, which, in turn, enhances p53-based transactivation and apoptosis in resistant BC cells [106].
MiR-135b-5p
Anterior gradient 2 (AGR2) regulates BC pathogenesis, particularly, growth, drug resistance, and metastasis [107]. Mechanically, miR-135b-5p sequesters AGR2 to augment DOX-responsiveness of BC cells [108].
MiR-302S
BCRP eliminates its substrate anti-cancer drugs to induce MDR in cancer cells [109]. MiR-302a-d is also termed “miR-302S”, owing to the same seed sequence (5’-aagugcu-3’) [110, 111]. MiR-302S down-regulates BCRP expression to enhance chemosensitivity (mitoxantrone) of BC [112].
MiR-140
FEN1 regulates genomic stability and integrity via participation in multiple DNA repair pathways (BER, NHEJ, HRR and NER) [113,114,115,116]. MiR-140 suppresses FEN1 levels via direct interaction with its 3’ UTR, which results in dysfunctional DNA repair and impaired BC progression. MiR-140 overexpression makes BC cells more susceptible to chemotherapeutic drugs (DOX and ADR) targeting BC [117].
MiR-140-5p
Wnt1 belongs to the Wnt family, and accelerates cell cycle, migration, and survival. MiR-140-5p induces chemosensitivity to DOX in BC stem cells (BCSCs) via suppression of ABCB1 levels [118].
MiR-200c-141
The miR-200 family is a critical modulator of EMT [119]. In a study, miR-200c-141 cluster overexpression in an in vivo CSC-enriched claudin-low tumor model, reduced tumor development and stem cell functionality, thus resulting in the absence of EMT characteristics, along with an enhancement of chemotherapeutic (DOX and carboplatin) sensitivity [120].
MiR-770
Stathmin1 (STMN1) induces microtubule depolymerization by sponging tubulin and activating catastrophes [121, 122]. MiR-770 directly targets and diminishes STMN1 levels to suppress chemoresistance (DOX) in TNBC cells [123].
MiR-27b-3p
CBLB is an upstream factor of the PI3K/Akt network. It regulates sensitivity of cetuximab in gastric cancer [124]. GRB2, another essential upstream factor in the MAPK/Erk network is known to resist ovarian cancer therapy by cisplatin. This occurs through the activation of the MAPK/Erk network [125]. Mechanically, miR-27b-3p reverses the PTX-mediated resistance by specifically reducing its target genes (CBLB and GRB2), and thus down-regulating the MAPK/Erk and PI3K/Akt networks [126].
MiR-200b
Arrestin domain containing 3 (ARRDC3) is scarcely expressed in metastatic TNBC cells owing to epigenetic silencing [127]. ARRDC3 inverses EMT characteristics and chemo-resistance (5-FU) of TNBC cells by increasing miR-200b levels [128].
MiR-100
MiR-100 promotes cancer apoptosis [129, 130]. HAX1 (an anti-apoptotic protein) overexpression induces chemoresistance in BC [131,132,133], whereas, miR-100 overexpression enhances responsiveness of MDA-MB-231/R and MCF-7/R cells to cisplatin treatment, while promoting cisplatin-driven mitochondrial apoptosis by regulating HAX1 [134].
MiR-148/152 family
Spindlin (SPIN) is up-regulated in chemo-resistant BC tissues, and participates in the PI3K/Akt-based chemoresistance [103]. The miR-148/152 family targets SPIN1 in BC. As a result, miR-148a-3p, miR-148b-3p, and miR-152-3p enhance ADR responsiveness by modulating SPIN1 in BC [135].
MiR-451
β-catenin is central to the Wnt/β-catenin network. Upon activation of Wnt signaling, β-catenin is rescued from degradation, resulting in its accumulation in the cytoplasm, followed by its translocation to the nucleus, activation of target genes (c-Myc and cyclin D1), which ultimately enhances tumor pathogenesis [136,137,138]. MiR-451 accelerates apoptosis and cell-cycle arrest of PTX-resistant cells via direct binding of the YWHAZ/β-catenin network [139].
PiRNA-36,712
PiRNA-36,712 restrains BC chemoresistance. Mechanically, piRNA-36,712 binds to SEPW1P transcript, thereby decreasing SEPW1 expression via sponging by miR-7 and miR-324. In addition, piRNA-36,712 elicits a combined anticancer effect with PTX and DOX [140].
NcRNAs with endocrine therapy resistance in BC
Approximately 70% of all BC patients exhibit ubiquitous ER expression [141, 142]. As such, it is a promising target for endocrine therapy. Two major ER isoforms (ERα and ERβ), encoded by 2 distinct genes (ESR1 and ESR2), regulate the nuclear and extranuclear ER axes [143, 144]. At present, three forms of endocrine therapies are used in clinics: (a) aromatase inhibitors (AI), (b) selective ER modulators (SERMs) and (c) selective ER degraders (SERDs) that antagonize ER [145]. The first of these SERMs is tamoxifen, a drug used frequently till this day to treat ER-positive patients. However, patients soon become resistant to this drug, which limits its use [146, 147]. AI blocks the enzyme aromatase, which regulates estrogen production. This prevents the development of hormone-receptor-positive BC cells. AI is primarily employed in postmenopausal women, and it performs better than tamoxifen in this demographic [148]. Fulvestrant is the preferred SERD for treating cancer patients. Both preclinical and clinical trials revealed that this is effective even in the tamoxifen-resistant (TR) models, and do not elicit agonistic activity in oestrogen-sensitive tissues like the endometrium [149, 150]. Scientists uncovered several underlying mechanisms that produce endocrine resistance, namely, deregulation of the classical estrogen signaling, activation of growth factor receptor networks, changes in the cell cycle and apoptotic process, and epigenetic modification [151].
Herein, we detailed the ncRNAs-related pathways involved in endocrine therapy resistance and sensitivity, particularly, in terms of the dysregulated signaling pathways: (i) ER signaling pathway, (ii) autophagy signaling pathway, (iii) PI3K/Akt/mTOR signaling pathway, (iv) and other pro-survival signaling pathways (Fig. 3 and Table 3).

NcRNAs regulate response to endocrine therapy
NcRNAs promotes endocrine therapy resistance
LncMIR2052HG
MIR2052HG directly interacts with the early growth response protein 1 (ERG1) protein to increase LMTK3 expression, thereby sustaining ESR1 levels and stabilized ERα protein, thus leading to AI resistance. Mechanistically, LMTK3 regulates ERα stability via the PKC/MEK/ERK/RSK1 pathway and ERα expression via the PKC/AKT/FOXO3 network [152].
LncRNA HOTAIR
HOTAIR is markedly elevated in tumors of TR BC patients, relative to their primary tumors prior to treatment. Direct association between HOTAIR and ER results in high levels of nuclear ER, even under estrogen-depleted conditions. This enables ER genomic targeting and induces transcription of the ER-target genes. Hence, HOTAIR augments the ER axis, and elicits tamoxifen resistance in BC [153].
BCAR4
BCAR4 accelerates BC progression. Godinho et al. reported that BCAR4 levels in BC are strongly correlated with aggressiveness and tamoxifen resistance via regulation of the HER2 axis [154].
LncRNA H19
Autophagy is a potential mechanism for tamoxifen resistance. Beclin1 (a key mediator of autophagy) overexpression makes cells unresponsive to estrogen-based signaling, which leads to tamoxifen resistance in BCs [155]. H19 overexpression augments autophagy and induces tamoxifen resistance in ER-positive BC cells by diminishing methylation in the Beclin 1 promotor region using the H19/SAHH/DNMT3B network [156]. In addition, H19 deficiency makes endocrine therapy resistant (ETR) cells susceptible to tamoxifen and fulvestrant, in an H19-dependent manner. H19 also modulates ERα levels in ETR cells, and protects against fulvestrant-based apoptosis [157].
LincRNA-ROR
LincRNA-ROR regulates BC metastasis [158]. LincRNA-ROR deficiency enhances MDA-MB-231 cell sensitivity to tamoxifen by inhibiting PI3K/Akt/mTOR activity [159].
LncRNA DSCAM‐AS1
Epidermal growth factor receptor pathway substrate 8 (EPS8) modulates cancer cell proliferation and apoptosis [160]. DSCAM‐AS1 induces tamoxifen resistance in BC, and is inversely proportional to miR-137 levels, and directly proportional to EPS8 levels in tamoxifen-resistant BC [161].
LncRNA TMPO-AS1
TMPO-AS1 is ubiquitously expressed in ER-positive BCs from tamoxifen-treated patients. Mechanically, TMPO-AS1 augments the estrogen axis by stabilizing the ESR1 transcript, encoding ERα, and via direct RNA: RNA association with the 3’UTR of ESR1 [162].
DILA1
DILA1 binds to Cyclin D1, and is ubiquitously expressed in tamoxifen-resistant BC. Mechanistically, DILA1 prevents Cyclin D1 phosphorylation at Thr286 via direct association with Thr286, which blocks its degradation, thus enhancing Cyclin D1 levels in BC [163].
LINC ERINA
High lincRNA ERINA levels are strongly associated with worse ER-positive BC patient outcome and responsiveness to CDK inhibitors in BC cell lines. Mechanistically, ERINA is induced by estrogen, and promotes cell cycle progression by regulating the TF E2F1 [164].
MiR-125b
The AKT/mTOR axis regulates AI resistance [165,166,167]. Silencing miR-125b in letrozole-resistant cells prevents the constitutive activation of the AKT/mTOR axis, and overcomes letrozole resistance, by sensitizing cells to the AI treatment [168].
MiR-519a
PTEN, CDKN1/p21, and retinoblastoma protein (RB1) are directly targeted by miR-519a. Mechanically, tamoxifen-resistant cells express high levels of miR-519a, which blocks the expressions of PTEN, RB1, and CDKN1A/p21, thus enabling cells to proliferate, even after tamoxifen exposure [169].
MiR-186-3p
EGFR signaling is also crucial for developing tamoxifen resistance in BC cells [170, 171]. Epiregulin (EREG) induces EGFR homodimerization, which initiates downstream signaling to promote cell proliferation [172]. MiR-186-3p targets EREG in BC. Moreover, the miR-186-3p/EREG network produces tamoxifen resistance and aerobic glycolysis in ER-positive BC [173].
MiR-21
Aberrant expression of miR-21 involved in chemoresistance of tumor [174]. Silencing of miR-21 confers the sensitivity to tamoxifen and fulvestrant by enhancing autophagic cell death through inhibition of the PI3K/AKT/mTOR by targeting PTEN [175].
NcRNAs promotes endocrine therapy sensitivity
LncRNA GAS5
PTEN regulates tamoxifen responsiveness in BC [176]. MiR-222 sequesters GAS5, suppresses PTEN, and enhances BC sensitivity to tamoxifen [177].
CircRNA_0025202
FOXO3a was downregulated in BC [178]. CircRNA_0025202 was significantly downregulated in MCF-7/TR cells. In terms of mechanism, circRNA_0025202 promotes tamoxifen sensitization via miR-182-5p/FOXO3a axis [179].
MiR-449a
A disintegrin and metalloproteinase (ADAM22) promotes ER-positive BC progression [180]. Downregulation of miR-449a promotes ADAM22 expression, which induces tamoxifen resistance in BC cells [181].
MiR-27b-3p
Nuclear receptor subfamily 5 group A member 2 (NR5A2) enhances BC cell proliferation by interacting with the ERα promoter to initiate its expression [182]. cAMP-response element binding protein 1 (CREB1) activates essential factors related to the anti-apoptosis pathway [183, 184]. MiR-27b-3p inhibits NR5A2 and CREB1 expressions. As a result, tamoxifen-induced cytotoxicity is enhanced in BC [185].
MiR-873
Cyclin-dependent kinase 3 (CDK3) phosphorylated ER and enhances ER activity. MiR-873 inhibits ERα transcriptional activity and tamoxifen resistance via targeting CDK3 in BC [186].
MiR-125a-3p
CDK3 is a potential target of miR-125a-3p in ER-positive BC [187]. MiR-125a-3p can function as a novel tumor suppressor in ER-positive BC by targeting CDK3, which may be a potential therapeutic approach for tamoxifen resistant BC therapy [188].
MiR-26a/b
Hu-antigen R (HuR) is an RNA-interacting protein (RBP) which binds to the AU-rich regions in the 3’UTR of transcripts to enhance their stability [189]. Reduced miR-26a/b and enhanced HuR levels post-transcriptionally augments ERBB2 expression, which, in turn, mediates the acquired tamoxifen resistance in ER-positive BC cells [190].
MiR-190
MiR-190 suppresses the Wnt/β-catenin axis to enhance anti-estrogen responsiveness by regulating SRY-related high mobility group box 9 (SOX9). In addition, recent evidences suggest a mechanism involving ZEB1-miR-190-SOX9 that mediates resistance to endocrine therapy in BC. ZEB1 interacts with the miR-190 promoter region to competitively inhibit ERα interaction, which enhances resistance to endocrine therapy [191].
MiR-214
Overexpression of UCP2 conferred drug resistance to chemotherapy and a higher survival through downregulation of ROS [192, 193]. MiR-214 increases the sensitivity of BC cells to tamoxifen and fulvestrant through inhibition of autophagy by targeting UCP2 [194].
MiR-1254
Cell cycle and apoptosis regulator 1 (CCAR1) is an apoptosis mediator or transcriptional coactivator for nuclear receptors or P53. As such, it has multiple roles in regulating cancer cell progression [195, 196]. CCAR1 5’ UTR is a natural miRancer of the endogenous miR-1254, and it makes TR BC cells susceptible to tamoxifen [197].
MiR-135a
MiR-135a was downregulated in BC/TR [198, 199]. The decreased expression of miR-135a resulted in an increased level of the miR-135a target genes (ESR1, ESRRA, NCOA1, PIM2, MRAS, and LCP1), which we have demonstrated to be key mediators of ERK1/2 and AKT1 activation, and subsequent increased ERα transcriptional activity to promote tamoxifen resistance [200].
MiR-375
Metadherin (MTDH) has been involved in BC metastasis. MTDH overexpression could induce EMT and modulate invasion as well as metastasis in BC [201]. Re-expression of miRNA-375 reverses both tamoxifen resistance and accompanying EMT-like properties by targeting MTDH in BC [199].
NcRNAs with targeted therapy resistance in BC
Erb-2/Her-2 is up-regulated in 20–30% of human invasive BCs, and is correlated with a worse patient outcome [202, 203]. In terms of monoclonal antibodies, small molecular inhibitors are used to specifically bind a target molecule. At the present time, trastuzumab, lapatinib, and pertuzumab are commonly employed for HER-2-positive BCs treatment [204]. Trastuzumab is a humanized monoclonal antibody that interacts with the HER2 receptor to suppress HER2 dimer formation, thus interrupting downstream networks, which, in turn, inhibits cell proliferation and apoptosis [148]. Lapatinib is a HER2 kinase inhibitor, which improves prognosis of HER2-amplified BC [205]. Multiple mechanisms produce resistance to targeted therapies. These include, ErbB2 levels, enhanced pro-survival signaling via alternation in tyrosine kinases receptors or intracellular signaling, which markedly enhances cell proliferation [206, 207]. Herein, we detailed the ncRNAs-mediated mechanism governing targeted therapy resistance and BC sensitivity (Fig. 4 and Table 4).

NcRNAs regulate response to targeted therapy
NcRNAs promotes targeted therapeutic resistance
LncSNHG14
Polyadenylate‐binding proteins (PABPs) are special proteins that associate in a sequence-specific fashion with single-stranded poly (A) by RNA recognition motif (RPM). PABPC1 regulates mRNA translation and degradation [208, 209], and facilitates the stability of the 5’ cap of transcripts. Mechanically, SNHG14 induces BC trastuzumab resistance by modulating PABPC1 levels via H3K27 acetylation [210].
LncAGAP2-AS1
AGAP2-AS1 induces trastuzumab resistance of BC via epigenetic modulation of MyD88. Mechanically, AGAP2-AS1 interacts with the CREB-interacting protein to increase H3K27ac levels at the MyD88 promoter region, thereby up-regulating MyD88. Hence, the NF-κB axis is activated by MyD88 and AGAP2-AS1 [211].
Lnc TINCR
TINCR deficiency reverses trastuzumab resistance, and acquired EMT in BC. Mechanically, TINCR remains in the cytoplasm of BC cells and is sequestered by miR-125b. This, in turn, releases HER-2 and induces trastuzumab resistance [212].
LncRNA ZNF649-AS1
Trastuzumab treatment enhances H3K27ac levels at the ZNF649-AS1 promoter region, which elevates ZNF649-AS1, which, in turn, enhances ATG5 levels by associating with polypyrimidine tract binding protein 1 (PTBP1) to initiate its transcription. Subsequently, enhanced autophagy related 5 (ATG5) expression induces autophagy and trastuzumab resistance [213].
LncRNA AFAP1-AS1
AFAP1-AS1 is ubiquitously expressed in trastuzumab-resistant cells, relative to sensitive cells. Enhanced AFAP1-AS1 expression is associated with worse response and reduced survival of BC patients. Exosome-mediated AFAP1-AS1 induces trastuzumab resistance via interaction with AUF1 and activation of ERBB2 translation [214].
MiR-205-5p
MiR-205-5p is up-regulated in BCSCs, and directly diminishes ERBB2 expression, while indirectly reducing EGFR expression to induce to resistance to lapatinib. In addition, miR-205-5p also modulates p63 expression, which, in turn, modulates the miR-205/p63/EGFR axis [215].
TRF-30-JZOYJE22RR33/tRF-27-ZDXPHO53KSN
tRNA derived small RNA fragments (TRFs) regulate human cancers [216, 217]. TRF-30-JZOYJE22RR33 and TRF-27-ZDXPHO53KSN are strongly expressed in trastuzumab-resistant versus -sensitive patients, and ROC analysis revealed a strong correlation with trastuzumab resistance [218].
NcRNAs promotes targeted therapy sensitivity
MiR-129-5p
Dysregulated PI3K/Akt/mTOR/rpS6 axis and PTEN deficiency contributes to trastuzumab resistance in BC [219, 220]. MiR-129-5p makes Her-2-positive BC more susceptible to trastuzumab by reducing rpS6 activity [221].
MiR-182
The PI3K/AKT/mTOR axis is an signaling target of MET, and it modulates multiple physiological processes [222]. MiR-182 overexpression reduces trastuzumab resistance in trastuzumab-resistant cells in part by suppressing the MET/PI3K/AKT/mTOR axis [223].
MiR-16
FUBP1 is a TF and RBP that modulates both transcription and translation of multiple genes [224]. CCNJ is not well characterized in mammals, and it may modulate BC [225]. MiR-16 serves as a tumor suppressor to mediate trastuzumab and lapatinib anti-proliferative effects, and CCNJ and FUBP1 are newly confirmed targets of miR-16 [226].
Targeting oncogenic-NcRNAs to conquer drug resistance
In terms of the aforementioned ncRNAs-mediated drug resistance, multiple ncRNAs also possess great therapeutic target potential in future drug developments. Therefore, several researchers targeted oncogenic ncRNAs to address cancer drug resistance. Herein, we detailed the ncRNAs that are highly expressed in cancer cells, where they serve an oncogenic function to induce BC resistance to anti-cancer therapies (Fig. 5). With advancements in nanotechnology, multiple clinical trials either examined or are examining RNA-guided precision machines [227,228,229]. Among the annotated ncRNAs, miRNAs are most commonly examined. Additionally, lncRNAs and circRNAs were also identified as novel targets [230,231,232]. Double-stranded RNA-mediated interference (RNAi) and single-stranded antisense oligonucleotides (ASOs) are two main strategies that target lncRNAs. Till now, three approaches were proposed for targeting ncRNAs: ASOs, locked nucleic acids (LNAs), and morpholinos [233]. Fortunately, a clinical trial (NCT02950207) was launched to testify whether miR-100 silencing impacts patients’ response rate to hormonal treatment in BC (https://clinicaltrials.gov). Moreover, the researchers also examined miR-10b, and revealed that miR-10b LNAs enhances BC sensitivity to doxorubicin in mouse models, with no further damage to normal tissue. This suggests that reduced toxicity is strongly related to the delivery of this LNA nanoparticle [234].

Oncogenic ncRNAs regulate drug resistance in breast cancer
Conclusions
BC is the most common cancer among women, and the major contributor to cancer-related deaths in women [235]. Technological enhancements in early diagnosis and therapy have markedly reduced BC-related mortality, while improving patient outcome to a certain extent [236]. However, close to 35% of BC patients experience recurrence and metastasis. Moreover, they also experience resistance to chemo-, endocrine-, and radiotherapies [237, 238]. The often encountered drug resistance within BC patients severely restricts therapeutic efficacy, and negatively impacts BC patient prognosis [239]. Emerging evidences revealed that ncRNAs can function as diagnostic indicators for multiple diseases, estimator of drug response, and as targets of new drug development [240].
Herein, we summarized the dysregulated ncRNAs governing drug resistance in BC, thereby providing a comprehensive ncRNAs landscape for drug resistance in BC. Some ncRNAs regulate drug resistance and sensitivity via a complex regulatory network. For instance, lncRNA H19 modulate endocrine resistance by regulating autophagy and ERα in BC. Meanwhile, different ncRNAs also influence drug efficacy by targeting the same target molecule. For instance, PTEN modulates drug resistance by simultaneously regulating lncPTENP1, miR-132, miR-212, lncHCP5, miR-519a, GAS5, and miR-129-5p levels in BC. Several studies demonstrated a concrete mechanism of ncRNAs modulating drug resistance, however, some reports only suggested a role of few ncRNAs in regulating drug resistance. This review highlights the direction of future anti-cancer drug development, particularly, approaches that weaken drug resistance by inhibiting drug resistance-related oncogenic ncRNAs. Other studies demonstrated that ncRNAs possess great potential in treating tumor. For example, small molecules were recently shown to abrogate HOTAIR activity by interrupting the HOTAIR/EZH2 scaffold association. This offers a novel approach of inhibition with enhanced applicability in humans. EZH2 inhibitor compounds like DZNep was previously suggested as potential medications targeting solid tumors in clinics [241]. Dysregulated ncRNAs are widely present in tumor drug resistance. A clinical trial must also be launched to enhance drug sensitivity by targeting ncRNAs, as mentioned above. Hence, given the significance of ncRNAs in drug resistance, additional investigations are warranted to identify potential therapeutic targets and approaches that enhance drug sensitivity in BC.
In conclusion, we recommend an extensive investigation, involving clinical trials, to examine the mechanisms behind drug resistance, and subsequently, develop ncRNAs-based therapies to fight BC. Additionally, miRNA, circRNA and TRFs, and other ncRNAs were not reported to modulate drug resistance. However, additional investigations are needed to confirm their association, if any, with drug resistance.
Data availability
Not applicable.
Abbreviations
- ABC:
-
ATP-binding cassette
- ADAM:
-
A disintegrin and metalloproteinase
- ADR:
-
Adriamycin
- AI:
-
Aromatase inhibitors
- Ai-lncRNA:
-
Antisense intronic long noncoding RNA
- AGR2:
-
Anterior gradient 2
- ANXA1:
-
Annexin A1
- ARRDC3:
-
Arrestin domain containing 3
- ATG5:
-
Autophagy related 5
- Bak1:
-
BCL-2 antagonist killer 1
- BCRP:
-
Breast cancer resistance protein
- BCSCs:
-
Breast cancer stem cells
- CCAR1:
-
Cell cycle and apoptosis regulator 1
- CCNG2:
-
Cyclin G2
- CDK3:
-
Cyclin-dependent kinase 3
- ceRNA:
-
Competing endogenous RNA
- circRNA:
-
Circular RNAs
- CREB1:
-
CAMP-response element binding protein 1
- CSCs:
-
Cancer stem cells
- DDR:
-
DNA damage response
- DOX:
-
Doxorubicin
- EGOT:
-
Eosinophil granule ontogeny transcript
- EMT:
-
Epithelial-mesenchymal transition
- EPS8:
-
Epidermal growth factor receptor pathway substrate 8
- ER:
-
Estrogen receptor
- EREG:
-
Epiregulin
- ERG1:
-
Early growth response protein 1
- ETR:
-
Endocrine therapy resistance
- FGF2:
-
Fibroblast grow factor-2
- FOXM1:
-
Forkhead box protein M1
- HER2:
-
Human epidermal growth factor receptor 2
- H3K27ac:
-
H3K27 acetylation
- HuR:
-
Hu-antigen R
- IGF1R:
-
Insulin-like growth factor-1 receptor
- LA:
-
Luminal A
- LB:
-
Luminal B
- lncRNA:
-
Long non-coding RNA
- MAPK:
-
Mitogen-activated protein kinase
- MDR:
-
Multidrug resistance
- miRNA:
-
MicroRNA
- MRP:
-
Multidrug-resistance-associated protein
- MTDH:
-
Metadherin
- NBL:
-
Normal breast-like
- NCO3:
-
Nuclear receptor coactivator 3
- NR5A2:
-
Nuclear receptor subfamily 5 group A member 2
- OPN:
-
Osteopontin
- OS:
-
Overall survival
- PABPs:
-
Polyadenylate‐binding proteins
- piRNAs:
-
Piwi-interacting RNAs
- PR:
-
Progesterone receptor
- PTBP1:
-
Polypyrimidine tract binding protein 1
- PTEN:
-
Phosphatase and tensin homologs
- RB1:
-
Retinoblastoma protein
- RBP:
-
RNA-binding protein
- ROS:
-
Reactive oxygen species
- RPM:
-
RNA recognition motif
- SERDs:
-
Selective estrogen receptor degraders
- SERMs:
-
Selective estrogen receptor modulators
- siRNAs:
-
Small interfering RNAs
- snoRNA:
-
Small nucleolar RNAs
- SOX9:
-
SRY-related high mobility group box 9
- SPIN:
-
Spindlin
- STMN1:
-
Stathmin1
- TF:
-
Transcription factor
- TFAM:
-
Mitochondrial transcription factor A
- TFF3:
-
Trefoil factor 3
- TGF-β:
-
Transforming growth factor β
- TNBC:
-
Triple negative breast cancer
- TR:
-
Tamoxifen-resistant
- TRFs:
-
TRNA derived small RNA fragments
- UTR:
-
Untranslated region
- UMPS:
-
Uridine monophosphate synthetase
- VEGFA:
-
Vascular endothelial growth factor A
- ZEB1:
-
Zinc-finger E-box binding homeobox 1
References
Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends–an update. Cancer Epidemiol Biomarkers Prev. 2016;25(25):16–27.
Bertos NR, Park M. Breast cancer - one term, many entities? J Clin Invest. 2011;121(121):3789–96.
Sorlie T. Molecular portraits of breast cancer: tumour subtypes as distinct disease entities. Eur J Cancer. 2004;40(40):2667–75.
Amelio I., Bernassola F., Candi E., Emerging roles of long non-coding RNAs in breast cancer biology and management, Semin Cancer Biol. (2020).
Perez EA, Romond EH, Suman VJ, Jeong JH, Sledge G, Geyer CE, Martino S, Rastogi P, Gralow J, Swain SM, Winer EP, Colon-Otero G, Davidson NE, Mamounas E, Zujewski JA, Wolmark N. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol. 2014;32(32):3744–52.
Sledge GW, Mamounas EP, Hortobagyi GN, Burstein HJ, Goodwin PJ, Wolff AC. Past, present, and future challenges in breast cancer treatment. J Clin Oncol. 2014;32(32):1979–86.
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(63):11–30.
Leary M, Heerboth S, Lapinska K, Sarkar S. Sensitization of drug resistant cancer cells: a matter of combination therapy. Cancers (Basel). 2018;10:10.
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Roder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See LH, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigo R, Gingeras TR. Landscape of transcription in human cells. Nature. 2012;489(489):101–8.
Hangauer MJ, Vaughn IW, Mcmanus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013;9(9):e1003569.
Evans JR, Feng FY, Chinnaiyan AM. The bright side of dark matter: lncRNAs in cancer. J Clin Invest. 2016;126(126):2775–82.
Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32(32):453–61.
Chen X, Sun Y, Cai R, Wang G, Shu X, Pang W. Long noncoding RNA: multiple players in gene expression. BMB Rep. 2018;51(51):280–9.
Youness RA, Gad MZ. Long non-coding RNAs: functional regulatory players in breast cancer. Noncoding RNA Res. 2019;4(4):36–44.
Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29(29):452–63.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(136):215–33.
Tomar D, Yadav AS, Kumar D, Bhadauriya G, Kundu GC. Non-coding RNAs as potential therapeutic targets in breast cancer. Biochim Biophys Acta Gene Regul Mech. 2020;1863:194378.
Rampurwala MM, Rocque GB, Burkard ME. Update on adjuvant chemotherapy for early breast cancer. Breast Cancer (Auckl). 2014;8(8):125–33.
Senkus E, Kyriakides S, Ohno S, Penault-Llorca F, Poortmans P, Rutgers E, Zackrisson S, Cardoso F. Committee esmo guidelines, primary breast cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(26):8–30.
Kutanzi KR, Yurchenko OV, Beland FA, Checkhun VF, Pogribny IP. MicroRNA-mediated drug resistance in breast cancer. Clin Epigenetics. 2011;2(2):171–85.
Kansara S, Pandey V, Lobie PE, Sethi G, Garg M, Pandey AK. Mechanistic involvement of long non-coding RNAs in Oncotherapeutics resistance in triple-negative breast cancer. Cells. 2020;9:9.
Perez EA. Impact, mechanisms, and novel chemotherapy strategies for overcoming resistance to anthracyclines and taxanes in metastatic breast cancer. Breast Cancer Res Treat. 2009;114(114):195–201.
Yardley DA. Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int J Breast Cancer. 2013;2013:137414.
Wong ST, Goodin S. Overcoming drug resistance in patients with metastatic breast cancer. Pharmacotherapy. 2009;29(29):954–65.
Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55(55):3–29.
Assaraf YG. Molecular basis of antifolate resistance. Cancer Metastasis Rev. 2007;26(26):153–81.
Perera O, Evans A, Pertziger M, Macdonald C, Chen H, Liu DX, Lobie PE, Perry JK. Trefoil factor 3 (TFF3) enhances the oncogenic characteristics of prostate carcinoma cells and reduces sensitivity to ionising radiation. Cancer Lett. 2015;361(361):104–11.
Wu H..Gu J..Zhou D..Cheng W..Wang Y..Wang Q..Wang X., LINC00160 mediated paclitaxel-And doxorubicin-resistance in breast cancer cells by regulating TFF3 via transcription factor C/EBPbeta. J Cell Mol Med. (2020).
Ueno T, Masuda N, Kamigaki S, Morimoto T, Saji S, Imoto S, Sasano H, Toi M. Differential Involvement of autophagy and apoptosis in response to chemoendocrine and endocrine therapy in breast cancer: JBCRG-07TR. Int J Mol Sci. 2019;20:20.
Zhong JTYuJ, Wang HJ, Shi Y, Zhao TS, He BX, Qiao B, Feng ZW. Effects of endoplasmic reticulum stress on the autophagy, apoptosis, and chemotherapy resistance of human breast cancer cells by regulating the PI3K/AKT/mTOR signaling pathway. Tumour Biol. 2017;39:1010428317697562.
Gopisetty MK, Kovacs D, Igaz N, Ronavari A, Belteky P, Razga Z, Venglovecz V, Csoboz B, Boros IM, Konya Z, Kiricsi M. Endoplasmic reticulum stress: major player in size-dependent inhibition of P-glycoprotein by silver nanoparticles in multidrug-resistant breast cancer cells. J Nanobiotechnology. 2019;17:9.
Nikesitch N, Lee JM, Ling S, Roberts TL. Endoplasmic reticulum stress in the development of multiple myeloma and drug resistance. Clin Transl Immunology. 2018;7:e1007.
Yao X, Tu Y, Xu Y, Guo Y, Yao F, Zhang X. Endoplasmic reticulum stress confers 5-fluorouracil resistance in breast cancer cell via the GRP78/OCT4/lncRNA MIAT/AKT pathway. Am J Cancer Res. 2020;10(10):838–55.
Tan EJ, Kahata K, Idas O, Thuault S, Heldin CH, Moustakas A. The high mobility group A2 protein epigenetically silences the Cdh1 gene during epithelial-to-mesenchymal transition. Nucleic Acids Res. 2015;43(43):162–78.
Li X, Wang S, Li Z, Long X, Guo Z, Zhang GZuJ, Chen Y, Wen L. The lncRNA NEAT1 facilitates cell growth and invasion via the miR-211/HMGA2 axis in breast cancer. Int J Biol Macromol. 2017;105(105):346–53.
Gooding AJ, Zhang B, Gunawardane L, Beard A, Valadkhan S, Schiemann WP. The lncRNA BORG facilitates the survival and chemoresistance of triple-negative breast cancers. Oncogene. 2019;38(38):2020–41.
Yao NFuY, Chen L, Liu Z, He J, Zhu Y, Xia T, Wang S. Long non-coding RNA NONHSAT101069 promotes epirubicin resistance, migration, and invasion of breast cancer cells through NONHSAT101069/miR-129-5p/Twist1 axis. Oncogene. 2019;38(38):7216–33.
Zheng X, Tang H, Zhao X, Sun Y, Jiang Y, Liu Y. Long non-coding RNA FTH1P3 facilitates uveal melanoma cell growth and invasion through miR-224–5p. PLoS One. 2017;12:e0184746.
Wang R, Zhang T, Yang Z, Jiang C, Seng J. Long non-coding RNA FTH1P3 activates paclitaxel resistance in breast cancer through miR-206/ABCB1. J Cell Mol Med. 2018;22(22):4068–75.
Munoz M, Henderson M, Haber M, Norris M. Role of the MRP1/ABCC1 multidrug transporter protein in cancer. IUBMB Life. 2007;59(59):752–7.
Lu JF, Pokharel D, Bebawy M. MRP1 and its role in anticancer drug resistance. Drug Metab Rev. 2015;47(47):406–19.
Chang L, Hu Z, Zhou Z, Zhang H. Linc00518 contributes to multidrug resistance through regulating the MiR-199a/MRP1 axis in breast cancer. Cell Physiol Biochem. 2018;48(48):16–28.
Graham TR, Yacoub R, Taliaferro-Smith L, Osunkoya AO, Odero-Marah VA, Liu T, Kimbro KS, Sharma D, O’regan RM. Reciprocal regulation of ZEB1 and AR in triple negative breast cancer cells. Breast Cancer Res Treat. 2010;123(123):139–47.
Liu L, Tong Q, Liu S, Cui J, Zhang Q, Sun W, Yang S. ZEB1 upregulates VEGF expression and stimulates angiogenesis in breast cancer. PLoS One. 2016;11:e0148774.
Gao J, Yuan Y, Zhang L, Yu S, Lu J, Feng J, Hu S. Inhibition of ZEB1-AS1 confers cisplatin sensitivity in breast cancer by promoting microRNA-129–5p-dependent ZEB1 downregulation. Cancer Cell Int. 2020;20:90.
Karreth FA, Tay Y, Perna D, Ala U, Tan SM, Rust AG, Denicola G, Webster KA, Weiss D, Perez-Mancera PA, Krauthammer M, Halaban R, Provero P, Adams DJ, Tuveson DA, Pandolfi PP. In vivo identification of tumor- suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell. 2011;147(147):382–95.
Gao X, Qin T, Mao J, Zhang J, Fan S, Lu Y, Sun Z, Zhang Q, Song B, Li L. PTENP1/miR-20a/PTEN axis contributes to breast cancer progression by regulating PTEN via PI3K/AKT pathway. J Exp Clin Cancer Res. 2019;38:256.
Garcia-Venzor A., Mandujano-Tinoco E. A.,Ruiz-Silvestre A., Sanchez J. M., Lizarraga F., Zampedri C., Melendez-Zajgla J.,Maldonado V., LncMat2B regulated by severe hypoxia induces cisplatin resistance by increasing DNA damage repair and tumor-initiating population in breast cancer cells. Carcinogenesis. (2020).
Wan L, Lu X, Yuan S, Wei Y, Guo F, Shen M, Yuan M, Chakrabarti R, Hua Y, Smith HA, Blanco MA, Chekmareva M, Wu H, Bronson RT, Haffty BG, Xing Y, Kang Y. MTDH-SND1 interaction is crucial for expansion and activity of tumor-initiating cells in diverse oncogene- and carcinogen-induced mammary tumors. Cancer Cell. 2014;26(26):92–105.
Kannan N, Eaves CJ. Tipping the balance: MTDH-SND1 curbs oncogene-induced apoptosis and promotes tumorigenesis. Cell Stem Cell. 2014;15(15):118–20.
Di Yu L, Xin Y, Ren L, Liu Y, Sun X, Zhang X, Yao W. JSND1 acts as a novel gene transcription activator recognizing the conserved Motif domains of Smad promoters, inducing TGFbeta1 response and breast cancer metastasis. Oncogene. 2017;36(36):3903–14.
Yu L, Liu X, Cui K, Di Y, Xin L, Sun X, Zhang W, Yang X, Wei M, Yao Z, Yang J. SND1 acts downstream of TGFbeta1 and upstream of smurf1 to promote breast cancer metastasis. Cancer Res. 2015;75(75):1275–86.
Qian W, Zhu YWuM, Guo QWuZ, Lobie PE, Zhu T. Linc00668 promotes invasion and stem cell-like properties of breast cancer cells by interaction With SND1. Front Oncol. 2020;10:88.
Koo DH, Lee HJ, Ahn JH, Yoon DH, Kim SB, Gong G, Son BH, Ahn SH, Jung KH. Tau and PTEN status as predictive markers for response to trastuzumab and paclitaxel in patients with HER2-positive breast cancer. Tumour Biol. 2015;36(36):5865–71.
Matrone MA, Whipple RA, Thompson K, Cho EH, Vitolo MI, Balzer EM, Yoon JR, Ioffe OB, Tuttle KC, Tan M, Martin SS. Metastatic breast tumors express increased tau, which promotes microtentacle formation and the reattachment of detached breast tumor cells. Oncogene. 2010;29(29):3217–27.
Pan Y, Pan Y, Cheng Y, Yang F, Yao Z, Wang O. Knockdown of LncRNA MAPT-AS1 inhibites proliferation and migration and sensitizes cancer cells to paclitaxel by regulating MAPT expression in ER-negative breast cancers. Cell Biosci. 2018;8(8):7.
Sheikh MH, Solito E. Annexin A1: uncovering the many talents of an old protein. Int J Mol Sci. 2018;19:19.
Tang L, Chen Y, Chen H, Jiang P, Yan L, Mo D, Tang X, Yan F. DCST1-AS1 promotes TGF-beta-induced epithelial-mesenchymal transition and enhances chemoresistance in triple-negative breast cancer cells via ANXA1. Front Oncol. 2020;10:280.
Zhang M, Wang Y, Jiang L, Song X, Zheng A, Gao H, Wei M, Zhao L. LncRNA CBR3-AS1 regulates of breast cancer drug sensitivity as a competing endogenous RNA through the JNK1/MEK4-mediated MAPK signal pathway. J Exp Clin Cancer Res. 2021;40:41.
Tang L, Wei X, Mao X, Mo D, Yan W, Yan F. Long non-coding RNA MIR200CHG promotes breast cancer proliferation, invasion, and drug resistance by interacting with and stabilizing YB-1. NPJ Breast Cancer. 2021;7(7):94.
Chen Z, Ma T, Huang C, Zhang L, Lv XXuTHuT, Li J. MiR-27a modulates the MDR1/P-glycoprotein expression by inhibiting FZD7/beta-catenin pathway in hepatocellular carcinoma cells. Cell Signal. 2013;25(25):2693–701.
Tian F, Dahmani FZ, Qiao J, Ni J, Xiong H, Liu T, Zhou J, Yao J. A targeted nanoplatform co-delivering chemotherapeutic and antiangiogenic drugs as a tool to reverse multidrug resistance in breast cancer. Acta Biomater. 2018;75(75):398–412.
Chen Z, Pan T, Jiang D, Jin L, Geng Y, Feng X, Shen A, Zhang L. The lncRNA-GAS5/miR-221-3p/DKK2 axis modulates ABCB1-mediated adriamycin resistance of breast cancer via the Wnt/beta-catenin signaling pathway. Mol Ther Nucleic Acids. 2020;19(19):1434–48.
Xu H, Guo S, Li W, Yu P. The circular RNA Cdr1as, via miR-7 and its targets, regulates insulin transcription and secretion in islet cells. Sci Rep. 2015;5(5):12453.
Yang WGuJ, Wang X, Wang Y, Feng M, Zhou D, Guo J, Zhou M. Inhibition of circular RNA CDR1as increases chemosensitivity of 5-FU-resistant BC cells through up-regulating miR-7. J Cell Mol Med. 2019;23(23):3166–77.
Song GD, Sun Y, Shen H, Li W. SOX4 overexpression is a novel biomarker of malignant status and poor prognosis in breast cancer patients. Tumour Biol. 2015;36(36):4167–73.
Sharma S, Nagpal N, Ghosh PC, Kulshreshtha R. P53-miR-191-SOX4 regulatory loop affects apoptosis in breast cancer. RNA. 2017;23(23):1237–46.
Li HY, Liang JL, Kuo YL, Lee HH, Calkins MJ, Chang HT, Lin FC, Chen YC, Hsu TI, Hsiao M, Ger LP, Lu PJ. miR-105/93–3p promotes chemoresistance and circulating miR-105/93–3p acts as a diagnostic biomarker for triple negative breast cancer. Breast Cancer Res. 2017;19(19):133.
Xie MFuZ, Cao J, Liu YWuJ, Li Q, Chen Y. MicroRNA-132 and microRNA-212 mediate doxorubicin resistance by down-regulating the PTEN-AKT/NF-kappaB signaling pathway in breast cancer. Biomed Pharmacother. 2018;102(102):286–94.
Rodriguez-Barrueco R, Nekritz EA, Bertucci F, Yu J, Sanchez-Garcia F, Zeleke TZ, Gorbatenko A, Birnbaum D, Ezhkova E, Cordon-Cardo C, Finetti P, Llobet-Navas D, Silva JM. miR-424(322)/503 is a breast cancer tumor suppressor whose loss promotes resistance to chemotherapy. Genes Dev. 2017;31(31):553–66.
Zhou M, Liu Z, Zhao Y, Ding Y, Liu H, Xi Y, Xiong W, Li G, Lu J, Fodstad O, Riker AI, Tan M. MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1 (Bak1) expression. J Biol Chem. 2010;285(285):21496–507.
Bao B, Wang Z, Ali S, Kong D, Banerjee S, Ahmad A, Li Y, Azmi AS, Miele L, Sarkar FH. Over-expression of FoxM1 leads to epithelial-mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J Cell Biochem. 2011;112(112):2296–306.
Saba R, Alsayed A, Zacny JP, Dudek AZ. The role of forkhead box protein M1 in breast cancer progression and resistance to therapy. Int J Breast Cancer. 2016;2016:9768183.
Xue YJ, Xiao RH, Long DZ, Zou XF, Wang XN, Zhang GX, Yuan YH, Wu GQ, Yang J, Wu YT, Xu H, Liu FL, Liu M. Overexpression of FoxM1 is associated with tumor progression in patients with clear cell renal cell carcinoma. J Transl Med. 2012;10:200.
Zona S, Bella L, Burton MJ, Nestal De Moraes G, Lam EW. FOXM1: an emerging master regulator of DNA damage response and genotoxic agent resistance. Biochim Biophys Acta. 2014;1839:1316–22.
Tan X, Li Z, Ren S, Rezaei K, Pan Q, Goldstein AT, Macri CJ, Cao D, Brem RFFSW. Dynamically decreased miR-671–5p expression is associated with oncogenic transformation and radiochemoresistance in breast cancer. Breast Cancer Res. 2019;21(21):89.
Zhang WC, Chin TM, Yang H, Nga ME, Lunny DP, Lim EK, Sun LL, Pang YH, Leow YN, Malusay SR, Lim PX, Lee JZ, Tan BJ, Shyh-Chang N, Lim EH, Lim WT, Tan DS, Tan EH, Tai BC, Soo RA, Tam WL, Lim B. Tumour-initiating cell-specific miR-1246 and miR-1290 expression converge to promote non-small cell lung cancer progression. Nat Commun. 2016;7:11702.
Sakha S, Muramatsu T, Ueda K, Inazawa J. Exosomal microRNA miR-1246 induces cell motility and invasion through the regulation of DENND2D in oral squamous cell carcinoma. Sci Rep. 2016;6:38750.
Bates S, Rowan S, Vousden KH. Characterisation of human cyclin G1 and G2: DNA damage inducible genes. Oncogene. 1996;13(13):1103–9.
Montagner M, Enzo E, Forcato M, Zanconato F, Parenti A, Rampazzo E, Basso G, Leo G, Rosato A, Bicciato S, Cordenonsi M, Piccolo S. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature. 2012;487(487):380–4.
Li XJ, Ren ZJ, Tang JH, Yu Q. Exosomal MicroRNA MiR-1246 promotes cell proliferation, invasion and drug resistance by targeting CCNG2 in breast cancer. Cell Physiol Biochem. 2017;44(44):1741–8.
Masoumi KC, Daams R, Sime W, Siino V, Ke H, Levander F, Massoumi R. NLK-mediated phosphorylation of HDAC1 negatively regulates Wnt signaling. Mol Biol Cell. 2017;28(28):346–55.
Tang T, Cheng Y, She Q, Jiang Y, Chen Y, Yang W, Li Y. Long non-coding RNA TUG1 sponges miR-197 to enhance cisplatin sensitivity in triple negative breast cancer. Biomed Pharmacother. 2018;107(107):338–46.
Echevarria W, Leite MF, Guerra MT, Zipfel WR, Nathanson MH. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat Cell Biol. 2003;5(5):440–6.
Adkins CE, Morris SA, De Smedt H, Sienaert I, Torok K, Taylor CW. Ca2+-calmodulin inhibits Ca2+ release mediated by type-1, -2 and -3 inositol trisphosphate receptors. Biochem. 2000;345(2):357–63.
Xu S, Wang P, Zhang J, Wu H, Sui S, Zhang J, Wang Q, Qiao K, Yang W, Xu H, Pang D. Ai-lncRNA EGOT enhancing autophagy sensitizes paclitaxel cytotoxicity via upregulation of ITPR1 expression by RNA-RNA and RNA-protein interactions in human cancer. Mol Cancer. 2019;18:89.
Jiang M, Huang O, Xie Z, Wu S, Zhang X, Shen A, Liu H, Chen X, Wu J, Lou Y, Mao Y, Sun K, Hu S, Geng M, Shen K. A novel long non-coding RNA-ARA: adriamycin resistance-associated. Biochem Pharmacol. 2014;87(87):254–83.
Li N, Li S, Wang Y, Wang J, Wang K, Liu X, Li Y, Liu J. Decreased expression of WNT2 in villi of unexplained recurrent spontaneous abortion patients may cause trophoblast cell dysfunction via downregulated Wnt/beta-catenin signaling pathway. Cell Biol Int. 2017;41(41):898–907.
Xiu DH, Liu GF, Li LY, Zhao GQ, Liu L, Li XF. Long non-coding RNA LINC00968 attenuates drug resistance of breast cancer cells through inhibiting the Wnt2/beta-catenin signaling pathway by regulating WNT2. J Exp Clin Cancer Res. 2019;38:94.
Wu J..Chen H..Ye M..Wang B..Zhang Y..Sheng J..Meng T..Chen H., Corrigendum to "Downregulation of long noncoding RNA HCP5 contributes to cisplatin resistance in human triple-negative breast cancer via regulation of PTEN expression" [Biomed. Pharmacother. 115 (2019) 108869], Biomed Pharmacother. 122 (2020). 122, 109789.
Luo L, Zhang J, Tang H, Zhai D, Huang D, Ling L, Wang X, Liu T, Zhang Q, Zhang Z, He Z, Zheng G. LncRNA SNORD3A specifically sensitizes breast cancer cells to 5-FU by sponging miR-185–5p to enhance UMPS expression. Cell Death Dis. 2020;11:329.
Li A, Yan Q, Zhao X, Zhong J, Yang H, Feng Z, Du Y, Wang Y, Wang Z, Wang H, Zhou Y, Liu S, Nie Y. Decreased expression of PBLD correlates with poor prognosis and functions as a tumor suppressor in human hepatocellular carcinoma. Oncotarget. 2016;7(7):524–37.
Liang Y, Song X, Li Y, Su P, Han D, Ma T, Guo R, Chen B, Zhao W, Sang Y, Zhang N, Li X, Zhang H, Liu Y, Duan Y, Wang L, Yang Q. circKDM4C suppresses tumor progression and attenuates doxorubicin resistance by regulating miR-548p/PBLD axis in breast cancer. Oncogene. 2019;38(38):6850–66.
Bautista S, Valles H, Walker RL, Anzick S, Zeillinger R, Meltzer P, Theillet C. In breast cancer, amplification of the steroid receptor coactivator gene AIB1 is correlated with estrogen and progesterone receptor positivity. Clin Cancer Res. 1998;4(4):2925–9.
Ao X, Nie P, Zhang T, Wang S, Chang H, Zou Z. Decreased expression of microRNA-17 and microRNA-20b promotes breast cancer resistance to taxol therapy by upregulation of NCOA3. Cell Death Dis. 2016;7:e2463.
Lu R, Hu X, Zhou J, Sun J, Zhu AZ, Xu X, Zheng H, Gao X, Wang X, Jin H, Zhu P, Guo L. COPS5 amplification and overexpression confers tamoxifen-resistance in ERalpha-positive breast cancer by degradation of NCoR. Nat Commun. 2016;7:12044.
Wang S, Leventaki V, Drakos E, Zhang R, Sahin AA, Resetkova E, Edgerton M, Claret FX. MicroRNA-17 acts as a tumor chemosensitizer by targeting JAB1/CSN5 in triple-negative breast cancer. Cancer Lett. 2019;465(465):12–23.
Lee JW, Guan W, Han S, Hong DK, Kim LS, Kim H. MicroRNA-708-3p mediates metastasis and chemoresistance through inhibition of epithelial-to-mesenchymal transition in breast cancer. Cancer Sci. 2018;109(109):1404–13.
Si W, Shen J, Du C, Chen D, Gu X, Li C, Yao M, Pan J, Cheng J, Jiang D, Xu L, Bao C, Fu P, Fan W. A miR-20a/MAPK1/c-Myc regulatory feedback loop regulates breast carcinogenesis and chemoresistance. Cell Death Differ. 2018;25(25):406–20.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(473):298–307.
Hu Y, Qiu Y, Yague E, Ji W, Liu J, Zhang J. miRNA-205 targets VEGFA and FGF2 and regulates resistance to chemotherapeutics in breast cancer. Cell Death Dis. 2016;7:e2291.
Yue W, Sun LY, Li CH, Zhang LX, Pei XT. Screening and identification of ovarian carcinomas related genes. Ai Zheng. 2004;23(23):141–5.
Chen X, Wang YW, Xing AY, Xiang S, Shi DB, Liu L, Li YX, Gao P. Suppression of SPIN1-mediated PI3K-Akt pathway by miR-489 increases chemosensitivity in breast cancer. J Pathol. 2016;239(239):459–72.
Fan X, Zhou S, Zheng M, Deng X, Yi Y, Huang T. MiR-199a-3p enhances breast cancer cell sensitivity to cisplatin by downregulating TFAM (TFAM). Biomed Pharmacother. 2017;88(88):507–14.
Laohaviroj M, Chamgramol Y, Pairojkul C, Mulvenna J, Sripa B. Clinicopathological significance of osteopontin in cholangiocarcinoma cases. Asian Pac J Cancer Prev. 2016;17(17):201–5.
Han B, Huang J, Han Y, Hao J, Wu X, Song H, Chen X, Shen Q, Dong X, Pang H, Cai L. The microRNA miR-181c enhances chemosensitivity and reduces chemoresistance in breast cancer cells via down-regulating osteopontin. Int J Biol Macromol. 2019;125(125):544–56.
Chevet E, Fessart D, Delom F, Mulot A, Vojtesek B, Hrstka R, Murray E, Gray T, Hupp T. Emerging roles for the pro-oncogenic anterior gradient-2 in cancer development. Oncogene. 2013;32(32):2499–509.
Zhang Y, Xia F, Zhang F, Cui Y, Wang Q, Liu H, Wu Y. miR-135b-5p enhances doxorubicin-sensitivity of breast cancer cells through targeting anterior gradient 2. J Exp Clin Cancer Res. 2019;38:26.
Mao Q, Unadkat JD. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport–an update. AAPS J. 2015;17(17):65–82.
Lin SL, Chang DC, Ying SY, Leu D, Wu DT. MicroRNA miR-302 inhibits the tumorigenecity of human pluripotent stem cells by coordinate suppression of the CDK2 and CDK4/6 cell cycle pathways. Cancer Res. 2010;70(70):9473–82.
Dolezalova D, Mraz M, Barta T, Plevova K, Vinarsky V, Holubcova Z, Jaros J, Dvorak P, Pospisilova S, Hampl A. MicroRNAs regulate p21(Waf1/Cip1) protein expression and the DNA damage response in human embryonic stem cells. Stem Cells. 2012;30(30):1362–72.
Wang Y, Zhao L, Xiao Q, Jiang L, He M, Bai X, Ma M, Jiao X, Wei M. miR-302a/b/c/d cooperatively inhibit BCRP expression to increase drug sensitivity in breast cancer cells. Gynecol Oncol. 2016;141(141):592–601.
He L, Luo L, Zhu H, Yang H, Zhang Y, Wu H, Sun H, Jiang F, Kathera CS, Liu L, Zhuang Z, Chen H, Pan F, Hu Z, Zhang J, Guo Z. FEN1 promotes tumor progression and confers cisplatin resistance in non-small-cell lung cancer. Mol Oncol. 2017;11(11):1302–3.
Senkevich TG, Koonin EV, Moss B. Predicted poxvirus FEN1-like nuclease required for homologous recombination, double-strand break repair and full-size genome formation. Proc Natl Acad Sci U S A. 2009;106(106):17921–6.
Wu X, Wilson TE, Lieber MR. A role for FEN-1 in nonhomologous DNA end joining: the order of strand annealing and nucleolytic processing events. Proc Natl Acad Sci U S A. 1999;96(96):1303–8.
Yoon JH, Swiderski PM, Kaplan BE, Takao M, Yasui A, Shen B, Pfeifer GP. Processing of UV damage in vitro by FEN-1 proteins as part of an alternative DNA excision repair pathway. Biochemistry. 1999;38(38):4809–17.
Lu X, Liu R, Wang M, Kumar AK, Pan F, He LHuZ, Guo Z. MicroRNA-140 impedes DNA repair by targeting FEN1 and enhances chemotherapeutic response in breast cancer. Oncogene. 2020;39(39):234–47.
Wu D, Zhang JLuY, Bo S, Li L, Wang L, Zhang Q, Mao J. miR-140-5p inhibits the proliferation and enhances the efficacy of doxorubicin to breast cancer stem cells by targeting Wnt1. Cancer Gene Ther. 2019;26(26):74–82.
Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop–a motor of cellular plasticity in development and cancer? EMBO Rep. 2010;11(11):670–7.
Knezevic J, Pfefferle AD, Petrovic I, Greene SB, Perou CM, Rosen JM. Expression of miR-200c in claudin-low breast cancer alters stem cell functionality, enhances chemosensitivity and reduces metastatic potential. Oncogene. 2015;34(34):5997–6006.
Marklund U, Larsson N, Gradin HM, Brattsand G, Gullberg M. Oncoprotein 18 is a phosphorylation-responsive regulator of microtubule dynamics. EMBO J. 1996;15(15):5290–8.
Rubin CI, Atweh GF. The role of stathmin in the regulation of the cell cycle. J Cell Biochem. 2004;93(93):242–50.
Li Y, Liang Y, Sang Y, Song X, Zhang H, Liu Y, Jiang L, Yang Q. MiR-770 suppresses the chemo-resistance and metastasis of triple negative breast cancer via direct targeting of STMN. Cell Death Dis. 2018;9:14.
Yu P, Fan YQuX, Zhang J, Song N, Liu J, Liu Y. Cbl-b regulates the sensitivity of cetuximab through ubiquitin-proteasome system in human gastric cancer cells. J BUON. 2016;21(21):867–73.
Van Jaarsveld MT, Van Kuijk PF, Boersma AW, Helleman J, Van Ijcken WF, Mathijssen RH, Pothof J, Berns EM, Verweij J, Wiemer EA. miR-634 restores drug sensitivity in resistant ovarian cancer cells by targeting the Ras-MAPK pathway. Mol Cancer. 2015;14:196.
Chen D, Si W, Shen J, Du C, Lou W, Bao C, Zheng H, Pan J, Zhong G, Xu L, Fu P, Fan W. miR-27b-3p inhibits proliferation and potentially reverses multi-chemoresistance by targeting CBLB/GRB2 in breast cancer cell. Cell Death Dis. 2018;9(9):188.
Soung YH, Pruitt K, Chung J. Epigenetic silencing of ARRDC3 expression in basal-like breast cancer cells. Sci Rep. 2014;4:3846.
Soung YH, Chung H, Yan C, Ju J, Chung J. Arrestin domain containing 3 reverses epithelial to mesenchymal transition and chemo-resistance of TNBC cells by Up-regulating expression of miR-200b. Cells. 2019;8:8.
Bi Y, Jing Y, Cao Y. Overexpression of miR-100 inhibits growth of osteosarcoma through FGFR3. Tumour Biol. 2015;36(36):8405–11.
Luan Y, Zhang S, Zuo L, Zhou L. Overexpression of miR-100 inhibits cell proliferation, migration, and chemosensitivity in human glioblastoma through FGFR3. Onco Targets Ther. 2015;8(8):3391–400.
Yang J, Wu Y, Wang X, Xu L, Zhao X, Yang Y. Chemoresistance is associated with overexpression of HAX-1, inhibition of which resensitizes drug-resistant breast cancer cells to chemotherapy. Tumour Biol. 2017;39:1010428317692228.
Sun SJ, Feng L, Zhao GQ, Dong ZM. HAX-1 promotes the chemoresistance, invasion, and tumorigenicity of esophageal squamous carcinoma cells. Dig Dis Sci. 2012;57(57):1838–46.
Suzuki Y, Demoliere C, Kitamura D, Takeshita H, Deuschle U, Watanabe T. HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family tyrosine kinases. J Immunol. 1997;158(158):2736–44.
Wu G, Zhou W, Pan X, Sun YXuH, Shi P, Li J, Gao L, Tian X. miR-100 reverses cisplatin resistance in breast cancer by suppressing HAX-1. Cell Physiol Biochem. 2018;47(47):2077–87.
Chen X, Wang YW, Gao P. SPIN1, negatively regulated by miR-148/152, enhances Adriamycin resistance via upregulating drug metabolizing enzymes and transporter in breast cancer. J Exp Clin Cancer Res. 2018;37:100.
Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014;26(26):570–9.
Zhang J, Gill A, J. Issacs J. D. Atmore B. Johns A. Delbridge L. W. Lai R. Mcmullen T. P.,. The Wnt/beta-catenin pathway drives increased cyclin D1 levels in lymph node metastasis in papillary thyroid cancer. Hum Pathol. 2012;43:1044–50.
Guo Y, Xiao L, Sun L, Liu F. Wnt/beta-catenin signaling: a promising new target for fibrosis diseases. Physiol Res. 2012;61(61):337–46.
Wang W, Zhang L, Wang Y, Ding Y, Chen T, Wang Y, Wang H, Li Y, Duan K, Chen S, Yang Q, Chen C, Involvement of miR-451 in resistance to paclitaxel by regulating YWHAZ in breast cancer, Cell Death Dis. 8 (2017). 8, e3071.
Tan L, Mai D, Zhang B, Jiang X, Zhang J, Bai R, Ye Y, Li M, Pan L, Su J, Zheng Y, Liu Z, Zuo Z, Zhao Q, Li X, Huang X, Yang J, Tan W, Zheng J, Lin D, PIWI-interacting RNA-36712 restrains breast cancer progression and chemoresistance by interaction with SEPW1 pseudogene SEPW1P RNA, Mol Cancer. 18 (2019). 18, 9.
Vargo-Gogola T, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer. 2007;7(7):659–72.
Tamoxifen for early breast cancer. an overview of the randomised trials. Early Breast Cancer Trialists’ Collaborative Group, Lancet. 1998;351(351):1451–67.
Campos-Parra A. D..Lopez-Urrutia E..Orozco Moreno L. T..Lopez-Camarillo C..Meza-Menchaca T..Figueroa Gonzalez G..Bustamante Montes L. P..Perez-Plasencia C., Long Non-Coding RNAs as New Master Regulators of Resistance to Systemic Treatments in Breast Cancer, Int J Mol Sci. 19 (2018). 19,
Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11(11):597–608.
Mcguire WL. Hormone receptors: their role in predicting prognosis and response to endocrine therapy. Semin Oncol. 1978;5(5):428–33.
Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9(9):631–43.
Shen Y, Costantino JP, Qin J. Tamoxifen chemoprevention treatment and time to first diagnosis of estrogen receptor-negative breast cancer. J Natl Cancer Inst. 2008;100(100):1448–53.
Waks AG, Winer EP. Breast Cancer Treatment: A Review. JAMA. 2019;321(321):288–300.
Howell A, Osborne CK, Morris C, Wakeling AE. ICI 182,780 (Faslodex): development of a novel, “pure” antiestrogen. Cancer. 2000;89(89):817–25.
Robertson JF. Faslodex (ICI 182, 780), a novel estrogen receptor downregulator–future possibilities in breast cancer. J Steroid Biochem Mol Biol. 2001;79(79):209–12.
Zhao M, Ramaswamy B. Mechanisms and therapeutic advances in the management of endocrine-resistant breast cancer. World J Clin Oncol. 2014;5(5):248–62.
Cairns J..Ingle J. N..Kalari K. R..Shepherd L. E..Kubo M..Goetz M. P..Weinshilboum R. M..Wang L., The lncRNA MIR2052HG regulates ERalpha levels and aromatase inhibitor resistance through LMTK3 by recruiting EGR1, Breast Cancer Res. 21 (2019). 21, 47.
Xue X, Yang YA, Zhang A, Fong KW, Kim J, Song B, Li S, Zhao JCYuJ. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene. 2016;35(35):2746–55.
Godinho MF, Sieuwerts AM, Look MP, Meijer D, Foekens JA, Van DLC, Agthoven T. Relevance of BCAR4 in tamoxifen resistance and tumour aggressiveness of human breast cancer. Br J Cancer. 2010;103(103):1284–91.
John S, Nayvelt I, Hsu HC, Yang P, Liu W, Das GM, Thomas T, Thomas TJ. Regulation of estrogenic effects by beclin 1 in breast cancer cells. Cancer Res. 2008;68(68):7855–63.
Wang J, Xie S, Yang J, Xiong H, Jia Y, Zhou Y, Chen Y, Ying X, Chen C, Ye C, Wang L, Zhou J. The long noncoding RNA H19 promotes tamoxifen resistance in breast cancer via autophagy. J Hematol Oncol. 2019;12:81.
Basak P, Chatterjee S, Bhat VSuA, Jin H, Lee-Wing V, Liu QHuP, Murphy LC, Raouf A. Long non-coding RNA H19 ACTS as an estrogen receptor modulator that is required for endocrine therapy resistance in ER+ breast cancer cells. Cell Physiol Biochem. 2018;51(51):1518–32.
Li LGuM, You B, Shi S, Shan Y, Bao L, You Y. Long non-coding RNA ROR promotes proliferation, migration and chemoresistance of nasopharyngeal carcinoma. Cancer Sci. 2016;107(107):1215–22.
Lu PW, Li L, Wang F, Gu YT. Inhibitory role of large intergenic noncoding RNA-ROR on tamoxifen resistance in the endocrine therapy of breast cancer by regulating the PI3K/Akt/mTOR signaling pathway. J Cell Physiol. 2019;234(234):1904–12.
Ding X, Zhou F, Wang F, Yang Z, Zhou C, Zhou J, Zhang B, Yang J, Wang G, Wei Z, Hu X, Xiang S, Zhang J. Eps8 promotes cellular growth of human malignant gliomas. Oncol Rep. 2013;29(29):697–703.
Ma Y, Bu D, Long J, Chai W, Dong J. LncRNA DSCAM-AS1 acts as a sponge of miR-137 to enhance tamoxifen resistance in breast cancer. J Cell Physiol. 2019;234(234):2880–94.
Mitobe Y, Ikeda K, Suzuki T, Takagi K, Kawabata H, Horie-Inoue K, Inoue S. ESR1-Stabilizing long noncoding RNA TMPO-AS1 promotes hormone-refractory breast cancer progression. Mol Cell Biol. 2019;39:39.
Shi Q, Li Y, Li S, Jin L, Lai H, Wu Y, Cai Z, Zhu M, Li Q, Li Y, Wang J, Liu Y, Wu Z, Song E, Liu Q. LncRNA DILA1 inhibits Cyclin D1 degradation and contributes to tamoxifen resistance in breast cancer. Nat Commun. 2020;11(1):5513.
Fang Z, Wang Y, Wang ZXuM, Ren S, Yang D, Hong M, Xie W. ERINA Is an estrogen-responsive LncRNA that drives breast cancer through the E2F1/RB1 pathway. Cancer Res. 2020;80(80):4399–413.
Cavazzoni A, Bonelli MA, Fumarola C, La Monica S, Airoud K, Bertoni R, Alfieri RR, Galetti M, Tramonti S, Galvani E, Harris AL, Martin LA, Andreis D, Bottini A, Generali D, Petronini PG. Overcoming acquired resistance to letrozole by targeting the PI3K/AKT/mTOR pathway in breast cancer cell clones. Cancer Lett. 2012;323(323):77–87.
Macedo LF, Sabnis GJ, Goloubeva OG, Brodie A. Combination of anastrozole with fulvestrant in the intratumoral aromatase xenograft model. Cancer Res. 2008;68(68):3516–22.
Vilquin P, Villedieu M, Grisard E, Ben LS, Ghayad SE, Heudel PE, Bachelot T, Corbo L, Treilleux I, Vendrell JA, Cohen PA. Molecular characterization of anastrozole resistance in breast cancer: pivotal role of the Akt/mTOR pathway in the emergence of de novo or acquired resistance and importance of combining the allosteric Akt inhibitor MK-2206 with an aromatase inhibitor. Int J Cancer. 2013;133(133):1589–602.
Vilquin P, Donini CF, Villedieu M, Grisard E, Corbo L, Bachelot T, Vendrell JA, Cohen PA. MicroRNA-125b upregulation confers aromatase inhibitor resistance and is a novel marker of poor prognosis in breast cancer. Breast Cancer Res. 2015;17:13.
Ward A, Shukla K, Balwierz A, Soons Z, Konig R, Sahin O, Wiemann S. MicroRNA-519a is a novel oncomir conferring tamoxifen resistance by targeting a network of tumour-suppressor genes in ER+ breast cancer. J Pathol. 2014;233(233):368–79.
Santen RJ, Fan P, Zhang Z, Bao Y, Song RX, Yue W. Estrogen signals via an extra-nuclear pathway involving IGF-1R and EGFR in tamoxifen-sensitive and -resistant breast cancer cells. Steroids. 2009;74(74):586–94.
Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JM, Harper ME, Barrow D, Wakeling AE, Nicholson RI. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology. 2003;144(144):1032–44.
Shelly M, Pinkas-Kramarski R, Guarino BC, Waterman H, Wang LM, Lyass L, Alimandi M, Kuo A, Bacus SS, Pierce JH, Andrews GC, Yarden Y. Epiregulin is a potent pan-ErbB ligand that preferentially activates heterodimeric receptor complexes. J Biol Chem. 1998;273(273):10496–505.
He M, Jin Q, Chen C, Liu Y, Ye X, Jiang Y, Ji F, Qian H, Gan D, Yue S, Zhu W, Chen T. The miR-186-3p/EREG axis orchestrates tamoxifen resistance and aerobic glycolysis in breast cancer cells. Oncogene. 2019;38(38):5551–65.
Pan X, Wang ZX, Wang R. MicroRNA-21: a novel therapeutic target in human cancer. Cancer Biol Ther. 2010;10(10):1224–32.
Yu X, Li R, Shi W, Jiang T, Wang Y, Li CQuX. Silencing of MicroRNA-21 confers the sensitivity to tamoxifen and fulvestrant by enhancing autophagic cell death through inhibition of the PI3K-AKT-mTOR pathway in breast cancer cells. Biomed Pharmacother. 2016;77(77):37–44.
Tanic N, Milovanovic Z, Tanic N, Dzodic R, Juranic Z, Susnjar S, Plesinac-Karapandzic V, Tatic S, Dramicanin T, Davidovic R, Dimitrijevic B. The impact of PTEN tumor suppressor gene on acquiring resistance to tamoxifen treatment in breast cancer patients. Cancer Biol Ther. 2012;13(13):1165–74.
Gu J, Wang Y, Wang X, Zhou D, Shao C, Zhou M, He Z. Downregulation of lncRNA GAS5 confers tamoxifen resistance by activating miR-222 in breast cancer. Cancer Lett. 2018;434(434):1–10.
Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117(117):421–6.
Sang Y, Chen B, Song X, Li Y, Liang Y, Han D, Zhang N, Zhang H, Liu Y, Chen T, Li C, Wang L, Zhao W, Yang Q. circRNA_0025202 regulates tamoxifen sensitivity and tumor progression via regulating the miR-182-5p/FOXO3a axis in breast cancer. Mol Ther. 2019;27(27):1638–52.
Mccartan D, Bolger JC, Fagan A, Byrne C, Hao Y, Qin L, Mcilroy M, Xu J, Hill AD, Gaora PO, Young LS. Global characterization of the SRC-1 transcriptome identifies ADAM22 as an ER-independent mediator of endocrine-resistant breast cancer. Cancer Res. 2012;72(72):220–9.
Li J, Lu M, Jin J, Lu X, Xu T, Jin S. miR-449a suppresses TAMOXIFEN resistance in human breast cancer cells by targeting ADAM22. Cell Physiol Biochem. 2018;50(50):136–49.
Thiruchelvam PT, Lai CF, Hua H, Thomas RS, Hurtado A, Hudson W, Bayly AR, Kyle FJ, Periyasamy M, Photiou A, Spivey AC, Ortlund EA, Whitby RJ, Carroll JS, Coombes RC, Buluwela L, Ali S. The liver receptor homolog-1 regulates estrogen receptor expression in breast cancer cells. Breast Cancer Res Treat. 2011;127(127):385–96.
Mueller CR, Roskelley CD. Regulation of BRCA1 expression and its relationship to sporadic breast cancer. Breast Cancer Res. 2003;5(5):45–52.
Shankar DB, Cheng JC, Kinjo K, Federman N, Moore TB, Gill A, Rao NP, Landaw EM, Sakamoto KM. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell. 2005;7(7):351–62.
Zhu J, Zou Z, Nie P, Kou X, Wu B, Wang S, Song Z, He J. Downregulation of microRNA-27b-3p enhances tamoxifen resistance in breast cancer by increasing NR5A2 and CREB1 expression. Cell Death Dis. 2016;7:e2454.
Cui J, Yang Y, Li H, Leng Y, Qian K, Huang Q, Zhang C, Lu Z, Chen J, Sun T, Wu R, Sun Y, Song H, Wei X, Jing P, Yang X, Zhang C. MiR-873 regulates ERalpha transcriptional activity and tamoxifen resistance via targeting CDK3 in breast cancer cells. Oncogene. 2015;34:4018.
Zheng L, Li X, Meng X, Chou JHuJ, Zhang F, Zhang Z, Xing Y, Liu Y, Xi T. Competing endogenous RNA networks of CYP4Z1 and pseudogene CYP4Z2P confer tamoxifen resistance in breast cancer. Mol Cell Endocrinol. 2016;427(427):133–42.
Zheng L, Meng X, Li X, Zhang Y, Li C, Xiang C, Xing Y, Xia Y, Xi T. miR-125a-3p inhibits ERalpha transactivation and overrides tamoxifen resistance by targeting CDK3 in estrogen receptor-positive breast cancer. FASEB J. 2018;32(32):588–600.
Brennan CM, Steitz JA. HuR and mRNA stability. Cell Mol Life Sci. 2001;58(58):266–77.
Tan S, Ding K, Chong QY, Zhao J, Liu Y, Shao Y, Zhang Y, Yu Q, Xiong Z, Zhang W, Zhang M, Li G, Li X, Kong X, Ahmad A, Wu Z, Wu Q, Zhao X, Lobie PE, Zhu T. Post-transcriptional regulation of ERBB2 by miR26a/b and HuR confers resistance to tamoxifen in estrogen receptor-positive breast cancer cells. J Biol Chem. 2017;292(292):13551–64.
Yu Y, Yin W, Yu ZH, Zhou Y, Chi JR, Ge J, Cao XC. miR-190 enhances endocrine therapy sensitivity by regulating SOX9 expression in breast cancer. J Exp Clin Cancer Res. 2019;38:22.
Kowaltowski AJ, De Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med. 2009;47(47):333–43.
Derdak Z, Mark NM, Beldi G, Robson SC, Wands JR, Baffy G. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 2008;68(68):2813–9.
Yu X, Luo A, Liu Y, Wang S, Li Y, Shi W, Liu ZQuX. MiR-214 increases the sensitivity of breast cancer cells to tamoxifen and fulvestrant through inhibition of autophagy. Mol Cancer. 2015;14:208.
Kim JH, Yang CK, Heo K, Roeder RG, An W, Stallcup MR. CCAR1, a key regulator of mediator complex recruitment to nuclear receptor transcription complexes. Mol Cell. 2008;31(31):510–9.
Rishi AK, Zhang LYuY, Jiang Y, Nautiyal J, Wali A, Fontana JA, Levi E, Majumdar AP. Cell cycle- and apoptosis-regulatory protein-1 is involved in apoptosis signaling by epidermal growth factor receptor. J Biol Chem. 2006;281(281):13188–98.
Li G, Wu X, Qian W, Cai H, Sun X, Zhang W, Tan S, Wu Z, Qian P, Ding K, Lu X, Zhang X, Yan H, Song H, Guang S, Wu Q, Lobie PE, Shan G, Zhu T. CCAR1 5’ UTR as a natural miRancer of miR-1254 overrides tamoxifen resistance. Cell Res. 2016;26(26):655–73.
Joshi T, Elias D, Stenvang J, Alves CL, Teng F, Lyng MB, Lykkesfeldt AE, Brunner N, Wang J, Gupta R, Workman CT, Ditzel HJ. Integrative analysis of miRNA and gene expression reveals regulatory networks in tamoxifen-resistant breast cancer. Oncotarget. 2016;7(7):57239–53.
Ward A, Balwierz A, Zhang JD, Kublbeck M, Pawitan Y, Hielscher T, Wiemann S, Sahin O. Re-expression of microRNA-375 reverses both tamoxifen resistance and accompanying EMT-like properties in breast cancer. Oncogene. 2013;32(32):1173–82.
Zhang W, Wu M, Chong QY, Zhang M, Zhang X, Hu L, Zhong Y, Qian P, Kong X, Tan S, Li G, Ding K, Lobie PE, Zhu T. Loss of estrogen-regulated MIR135A1 at 3p211 promotes tamoxifen resistance in breast cancer. Cancer Res. 2018;78:4915–28.
Li X, Kong X, Huo Q, Guo H, Yan S, Yuan C, Moran MS, Shao C, Yang Q. Metadherin enhances the invasiveness of breast cancer cells by inducing epithelial to mesenchymal transition. Cancer Sci. 2011;102(102):1151–7.
Krishnamurti U, Silverman JF. HER2 in breast cancer: a review and update. Adv Anat Pathol. 2014;21(21):100–7.
Pan Z, Jing W, He K, Zhang L, Long X. SATB1 is correlated with progression and metastasis of breast cancers: a meta-analysis. Cell Physiol Biochem. 2016;38(38):1975–83.
Pecero ML, Salvador-Bofill J, Molina-Pinelo S. Long non-coding RNAs as monitoring tools and therapeutic targets in breast cancer. Cell Oncol (Dordr). 2019;42(42):1–12.
Viani GA, Afonso SL, Stefano EJ, De Fendi LI, Soares FV. Adjuvant trastuzumab in the treatment of her-2-positive early breast cancer: a meta-analysis of published randomized trials. BMC Cancer. 2007;7:153.
Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2011;9(9):16–32.
Garrett JT, Arteaga CL. Resistance to HER2-directed antibodies and tyrosine kinase inhibitors: mechanisms and clinical implications. Cancer Biol Ther. 2011;11(11):793–800.
Wells SE, Hillner PE, Vale RD, Sachs AB. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell. 1998;2(2):135–40.
Tharun S, Parker R. Targeting an mRNA for decapping: displacement of translation factors and association of the Lsm1p-7p complex on deadenylated yeast mRNAs. Mol Cell. 2001;8(8):1075–83.
Dong H, Wang W, Mo S, Liu Q, Chen X, Chen R, Zhang Y, Zou K, Ye M, He X, Zhang F, Han J, Hu J. Long non-coding RNA SNHG14 induces trastuzumab resistance of breast cancer via regulating PABPC1 expression through H3K27 acetylation. J Cell Mol Med. 2018;22(22):4935–47.
Dong H, Wang W, Mo S, Chen R, Zou K, Han J, Zhang F, Hu J. SP1-induced lncRNA AGAP2-AS1 expression promotes chemoresistance of breast cancer by epigenetic regulation of MyD88. J Exp Clin Cancer Res. 2018;37:202.
Dong H, Hu J, Zou K, Ye M, Chen Y, Wu C, Chen X, Han M. Activation of LncRNA TINCR by H3K27 acetylation promotes Trastuzumab resistance and epithelial-mesenchymal transition by targeting MicroRNA-125b in breast Cancer. Mol Cancer. 2019;18:3.
Han M, Qian X, Cao H, Wang F, Li X, Han N, Yang X, Yang Y, Dou D, Hu J, Wang W, Han J, Zhang F, Dong H. lncRNA ZNF649-AS1 induces trastuzumab resistance by promoting ATG5 expression and autophagy. Mol Ther. 2020;28(28):2488–502.
Han M, Gu Y, Lu P, Li J, Cao H, Li X, Qian X, Yu C, Yang Y, Yang X, Han N, Dou D, Hu J, Dong H. Exosome-mediated lncRNA AFAP1-AS1 promotes trastuzumab resistance through binding with AUF1 and activating ERBB2 translation. Mol Cancer. 2020;19:26.
De Cola A, Volpe S, Budani MC, Ferracin M, Lattanzio R, Turdo A, D’agostino D, Capone E, Stassi G, Todaro M, Di Ilio C, Sala G, Piantelli M, Negrini M, Veronese A, De Laurenzi V. miR-205–5p-mediated downregulation of ErbB/HER receptors in breast cancer stem cells results in targeted therapy resistance. Cell Death Dis. 2015;6(6):e1823.
Green D, Fraser WD, Dalmay T. Transfer RNA-derived small RNAs in the cancer transcriptome. Pflugers Arch. 2016;468(468):1041–7.
Anderson P, Ivanov P. tRNA fragments in human health and disease. FEBS Lett. 2014;588(588):4297–304.
Sun C, Yang F, Zhang Y, Chu J, Wang J, Wang Y, Zhang Y, Li J, Li Y, Fan R, Li W, Huang X, Wu H, Fu Z, Jiang Z, Yin Y. tRNA-derived fragments as novel predictive biomarkers for trastuzumab-resistant breast cancer. Cell Physiol Biochem. 2018;49(49):419–31.
Nahta R., O'regan R. M., Evolving strategies for overcoming resistance to HER2-directed therapy: targeting the PI3K/Akt/mTOR pathway, Clin Breast Cancer. 10 Suppl 3 (2010). 10 Suppl 3, S72–8.
O'brien N. A., Mcdonald K., Tong L., Von Euw E., Kalous O., Conklin D., Hurvitz S. A., Di Tomaso E., Schnell C., Linnartz R., Finn R. S., Hirawat S., Slamon D. J., Targeting PI3K/mTOR overcomes resistance to HER2-targeted therapy independent of feedback activation of AKT, Clin Cancer Res. 20 (2014). 20, 3507-20.
Lu X, Ma J, Chu J, Shao Q, Zhang Y, Lu G, Li J, Huang X, Li W, Li Y, Ling Y, Zhao T. MiR-129-5p sensitizes the response of Her-2 positive breast cancer to Trastuzumab by reducing Rps6. Cell Physiol Biochem. 2017;44(44):2346–56.
Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Ther Adv Med Oncol. 2011;3(3):S7–19.
Yue D, Qin X. miR-182 regulates trastuzumab resistance by targeting MET in breast cancer cells. Cancer Gene Ther. 2019;26(26):1–10.
Zhang J, Chen QM. Far upstream element binding protein 1: a commander of transcription, translation and beyond. Oncogene. 2013;32(32):2907–16.
Feliciano A, Castellvi J, Artero-Castro A, Leal JA, Romagosa C, Hernandez-Losa J, Peg V, Fabra A, Vidal F, Kondoh H, Ramon Y, Cajal S, Lleonart ME. miR-125b acts as a tumor suppressor in breast tumorigenesis via its novel direct targets ENPEP CK2-alpha, CCNJ, and MEGF9. PLoS One. 2013;8:e76247.
Venturutti L, Cordo Russo RI, Rivas MA, Mercogliano MF, Izzo F, Oakley RH, Pereyra MG, De Martino M, Proietti CJ, Yankilevich P, Roa JC, Guzman P, Cortese E, Allemand DH, Huang TH, Charreau EH, Cidlowski JA, Schillaci R, Elizalde PV, MiR-16 mediates trastuzumab and lapatinib response in ErbB-2-positive breast and gastric cancer via its novel targets CCNJ and FUBP1, Oncogene. 35 (2016). 6189-6202
Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20(20):460–9.
Bader AG. miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet. 2012;3:120.
Herrera VL, Colby AH, Ruiz-Opazo N, Coleman DG, Grinstaff MW. Nucleic acid nanomedicines in Phase II/III clinical trials: translation of nucleic acid therapies for reprogramming cells. Nanomedicine (Lond). 2018;13(13):2083–98.
Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. 2017;16(16):167–79.
Dong Y, He D, Peng Z, Peng W, Shi W, Wang J, Li B, Zhang C, Duan C. Circular RNAs in cancer: an emerging key player. J Hematol Oncol. 2017;10:2.
Li X, Yang L, Chen LL. The biogenesis, functions, and challenges of circular RNAs. Mol Cell. 2018;71(71):428–42.
Yu AM, Jian C, Yu AH, Tu MJ. RNA therapy: are we using the right molecules? Pharmacol Ther. 2019;196(196):91–104.
El Fatimy R, Subramanian S, Uhlmann EJ, Krichevsky AM. Genome editing reveals glioblastoma addiction to MicroRNA-10b. Mol Ther. 2017;25(25):368–78.
Carter D. New global survey shows an increasing cancer burden. Am J Nurs. 2014;114:17.
Desantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Goding SA, Jemal A, Siegel RL. Breast cancer statistics 2019. CA Cancer J Clin. 2019;69:438–51.
Sharma R, Sharma R, Khaket TP, Dutta C, Chakraborty B, Mukherjee TK. Breast cancer metastasis: putative therapeutic role of vascular cell adhesion molecule-1. Cell Oncol (Dordr). 2017;40(40):199–208.
Ellis LM, Hicklin DJ. Resistance to Targeted Therapies: Refining Anticancer Therapy in the Era of Molecular Oncology. Clin Cancer Res. 2009;15(15):7471–8.
Du T, Shi Y, Xu S, Wan X, Sun H, Liu B. Long non-coding RNAs in drug resistance of breast cancer. Onco Targets Ther. 2020;13(13):7075–87.
Vicentini C, Galuppini F, Corbo V, Fassan M. Current role of non-coding RNAs in the clinical setting. Noncoding RNA Res. 2019;4(4):82–5.
Sun F, Lee L, Zhang Z, Wang XYuQ, Duan X, Chan E. Preclinical pharmacokinetic studies of 3-deazaneplanocin a, a potent epigenetic anticancer agent, and its human pharmacokinetic prediction using GastroPlus. Eur J Pharm Sci. 2015;77(77):290–302.
Funding
This study was supported by Yantai Yuhuangding Hospital Youth Research Launch Fund (202208).
Author information
Authors and Affiliations
Contributions
Yujuan Kang wrote the manuscript and participated in the conception and revision of the manuscript draft.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, Y. Landscape of NcRNAs involved in drug resistance of breast cancer. Clin Transl Oncol 25, 1869–1892 (2023). https://doi.org/10.1007/s12094-023-03189-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-023-03189-3