
Abstract
The CCN (cyr61, ctgf, nov) family of modular proteins regulate diverse biological affects including cell adhesion, matrix production, tissue remodelling, proliferation and differentiation. Recent targeted gene disruption studies have demonstrated the CCN family to be developmentally essential for chondrogenesis, osteogenesis and angiogenesis. CCN2 is induced by agents such as angiotensin II, endothelin-1, glucocorticoids, HGF, TGFβ, and VEGF, and by hypoxia and biomechanical and shear stress. Dysregulated expression of CCN2 has also been widely documented in many fibroproliferative diseases. This mini-review will focus on CCN2, and the recent progress in understanding CCN2 gene regulation in health and disease. That CCN2 should be considered a novel and informative surrogate clinical bio-marker for fibroproliferative disease is discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The CCN (cyr61, ctgf, nov) family comprises six members (CCN1–6). These proteins act through integrin- and heparan sulfate proteoglycan-mediated adhesive signaling to directly modulate adhesion and indirectly modulate the functional activities of other extracellular ligands such as cytokines, growth factors, morphogens and matrix components (Leask and Abraham 2006). Of these, connective tissue growth factor (CTGF, CCN2) is perhaps the best studied family member.
CCN2 was initially identified as a protein secreted by cultured human endothelial cells (Bradham et al. 1991). In adult mammals, CCN2 exhibits a restricted expression, produced only by hepatic stellate cells (Paradis et al. 1999; Chen et al. 2001) and kidney mesangial cells (Ito et al. 1998; Chen et al. 2002). Although CCN2 is not normally expressed by other mesenchymal cells, it is rapidly induced during the tissue repair process, for example upon injury (Igarashi et al. 1993; Kapoor et al. 2008). The most potent inducer of CCN2 expression thus far identified is TGFβ. The induction of CCN2 by TGFβ is restricted to the mesenchymal cell lineages (e.g. tissue fibroblasts, myofibroblasts, pericytes) and generally not in epithelial cells (Leask et al. 2001, 2003; Kantarci et al. 2006). However, CCN2 is expressed in response to TGFβ in renal proximal tubule epithelial cells (Phanish et al. 2005). These results indicate that CCN2 gene regulation in response to TGFβ can differ based on the cell type examined.
CCN2 is over-expressed in connective tissue pathologies such as in excessive scarring and fibrosis and in stroma surrounding tumors (Blom et al. 2002). Data examining the location of CCN2 expression and the use of genetically modified cells has led to a hypothesis that CCN2 selectively mediates or sustains the specific actions of TGFβ in mesenchymal cells (Grotendorst 1997; Leask et al. 2004). CCN2 may exert its stimulating effect on TGFβ signaling by decreasing Smad7 availability and increasing Smad2 (Qi et al. 2007). However, CCN2-deficient fibroblasts show only a partial impairment of TGFβ responses and no defect in Smad3-dependent responses (Shi-wen et al. 2006). In this review, we summarize recent key observations concerning the regulation of the expression of the CCN2 gene, insights derived from these observations into the potential contribution that CCN2 may make to cell biology, and evaluate the potential use of CCN2 as a fibrogenic bio-marker.
CCN2 gene expression in health and disease
CCN2 is principally regulated at the level of transcription (Grotendorst et al. 1996). The CCN2 proximal promoter is induced by a number of specific factors such as endothelin-1 (ET-1) and TGFβ in addition to changes such as hypoxia (Holmes et al. 2001; Leask et al. 2001, 2003; Shi-wen et al. 2004; Higgins et al. 2004). TGFβ induction of CCN2 mRNA in fibroblasts is immediate-early, occurring within 30 min of TGFβ treatment, in a fashion that does not involve de novo protein synthesis (Grotendorst et al. 1996). This induction is severely impaired in the fibroblasts deficient in Smad3 (Holmes et al. 2001). Consistent with this observation, a functional Smad element resides within the CCN2 promoter (Holmes et al. 2001; Fig. 1). However, the ability of TGFβ to fully induce the CCN2 promoter and protein also requires protein kinase C and the ras/MEK/ERK MAP kinase cascade (Chen et al. 2002; Leask et al. 2003; Fig. 1).

Elements in the CCN2 promoter involved with its expression in normal mesenchymal cells. HIF-1=hypoxia inducible factor-1; BCE-1=basal control element-1; TEF/ets=transcription enhancer factor; CAESAR=cis-acting element of structure-anchored repression
As for all Smad-responsive promoters, additional basal transcription factors (co-activators/co-repressors) are required for complete coordinated regulation of CCN2 expression by TGFβ. The Smad element of the CCN2 promoter is not sufficient to confer TGFβ-responsiveness to a heterologous promoter (Leask et al. 2001), but rather acts in concert with a tandem repeat of an element similar to an Ets/TEF recognition motif (Leask et al. 2003). The protein(s) which bind to this element are enriched in fibroblasts relative to keratinocytes, suggesting that this protein directly contributes to the specific induction of CCN2 in different cell types (Leask et al. 2003). Recently, we have shown this motif to be a functional ETS binding element; Ets-1 is required for the ability of TGFβ to induce CCN2 expression. siRNA recognizing Ets-1 ablates the ability of TGFβ to induce CCN2 (van Beek et al. 2006; Fig. 1). Moreover, the oncogenic transcriptional co-activator YAP also operates through this site (14). Additional elements, including a BCE-1 (basal control element-1) site and a Sp1 site (Holmes et al. 2001, 2003), have been shown to play a role in basal activity of the CCN2 promoter (Fig. 1) and hence are indirectly required for TGFβ induced CCN2 gene expression. Sp1, similar to factors binding to a TATA box, though required for formation of a functional transcriptional complex is not directly required for TGFβ induced CCN2 expression (Holmes et al. 2003). The BCE-1 site was originally thought to be involved with the TGFβ-induction of CCN2, but has subsequently been shown to be involved with basal promoter activity (Grotendorst et al. 1996; Leask et al. 2001; Holmes et al. 2001) and in response of the CCN2 promoter to ET-1, through ras/MEK/ERK signal transduction cascade (Shi-wen et al. 2004). Since TGFβ induces ET-1 and ET-1 is essential for the ability of TGFβ to induce CCN2 (Shi-wen et al. 2007b), this element appears to be, albeit indirectly, a TGFβ response element (Grotendorst et al. 1996). Finally, Wnt3a induces CCN2 through a β-catenin-dependent mechanism, although the promoter sequences mediating this action are unknown, they reside within in the first 805 base pairs of the CCN2 proximal promoter (Luo et al. 2004; Chen et al. 2007).
It is now appreciated that post-transcriptional controls also contribute to CCN2 gene regulation. The chicken ccn2 gene is regulated not only at the transcriptional level, but also by the interaction between a post-transcriptional element in the 3′-untranslated region (3′-UTR) and a nucleophosmin (NPM/B23) cofactor (Mukudai et al. 2008). The 3′-untranslated region (3′-UTR) has a suppressive effect on CCN2 gene expression via a minimal RNA element in the 3′-UTR, which acts as a cis-acting element of structure-anchored repression (CAESAR) (Kubota et al. 2000). Moreover, hypoxia increases stability of CCN2 mRNA in chondrosarcoma cells (Kondo et al. 2006). Finally, the unusually long 5′UTRs of CCNs 1, 2, and 4 harbor cryptic promoters that show varying degrees of activity in different cell types (Huang et al. 2007).
In fibrotic fibroblasts isolated from the involved skin area of scleroderma patients, CCN2 is over-expressed independent of TGFβ and Smads but is dependent on BCE-1, Sp1 and endothelin-1 (Holmes et al. 2001, 2003; Chen et al. 2006; Shi-wen et al. 2007a). Finally, a functional polymorphism has been found in some scleroderma patients which results in increased Sp1-dependent transcription (Fonseca et al. 2007). However, increased expression of CCN2 in cell-culture activated hepatic stellate cells is dependent on TGFβ (Leask et al. 2008). Intriguingly, elective disruption of TβRII in mouse skin fibroblasts increases constitutive expression CTGF/CCN2 (Denton et al. 2009). It remains to be established whether the rise in CCN2 expression in patients correlates with the increased expression of ET-1 or TGFβ. Overall, these data reveal that the regulation of CCN2 expression in fibrotic conditions is complex and is perhaps disease/organ specific.
Inhibitors of CCN2 expression
It has long been suggested that agents that inhibit CCN2 expression may be used as anti-fibrotic therapies (Blom et al. 2002). As discussed above, inhibitors of TGFβ or endothelin receptors may be used to reduce CCN2 expression in activated hepatic stellate cells or scleroderma fibroblasts, respectively (Leask et al. 2008; Shi-wen et al. 2007a). Moreover, prostaglandins [PGE(2)] and prostacyclins (such as the synthetic prostacyclin Iloprost) can antagonize CCN2 expression, including in scleroderma fibroblasts (Ricupero et al. 1999; Stratton et al. 2001, 2002). It has been shown that 9-cis-retinoic acid reduced CCN2 expression in scleroderma fibroblasts, possibly through its ability to produce PGE(2) production (Xiao et al. 2008). Iloprost works, at least in part, by antagonizing MEK/ERK signaling (Stratton et al. 2002) and, indeed, the MEK inhibitor U0126 reduces CCN2 expression in response to TGFβ and ET-1 in fibroblasts as well as constitutive over-expression of CCN2 in the pancreatic cancer cell line PANC-1 (Pickles and Leask 2007). TNFα suppresses TGFβ-induced gene expression in fibroblasts but has no appreciable effect on the constitutive CCN2 expression in scleroderma fibroblasts (Abraham et al. 2000). Caffeine also reduces TGFβ-induced CCN2 expression in hepatocytes by blocking Smad activation (Gressner et al. 2008; Leask 2008). Finally, it has been shown recently that the addition of the lipid second messenger C2-ceramide can also reduce TGFβ-induced CCN2 expression in human foreskin fibroblasts (Kennedy et al. 2008).
CCN2 as a surrogate marker of fibroproliferative disease
As discussed above, CCN2 is over-expressed in fibrotic disorders. Early studies examining the kinetics of CCN2 induction showed that, in the anti-Thy-1.1 antibody model of rat kidney fibrosis, CCN2 induction paralleled the progression of fibrogenesis and repair (Ito et al. 2001; Table 1). Subsequent studies have established that CCN2 levels in biological fluids correlate with the levels of fibrosis in patient samples (Table 1). For example, urinary CCN2 levels appear at both stage nephropathy and appear to predict those patients who are destined for progressive glomerulosclerosis and end-stage renal disease (Riser et al. 2003). In addition, glomerular basement membrane thickness correlates with tubular and total CCN2 levels (Thomson et al. 2008)
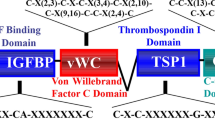
In particular, studies using an enzyme-linked immunosorbent assay (ELISA) to examining the appearance of amino-terminal fragments of CCN2 appear especially promising as potential diagnostic tools to show the severity of fibrosis. An ELISA detecting amino-terminal CCN2 appears to be superior to ELISAs detecting full-length of carboxy-terminal CCN2 as the latter moieties of CCN2 bind the cell surface and are internalized through the heparin- and integrin-binding carboxy-terminal domain of CCN2. The advantage of these ELISAs is that CCN2 can be readily detected in body fluids (e.g., urine, blood, blister fluid). Amino-terminal CCN2 ELISAs may be especially useful in diagnosing fibrosis associated with diabetes or, alternatively, in clinical trials testing the efficacy of anti-fibrotic compounds. For example, in type 1 diabetic patients with incipient and overt diabetic nephropathy, the magnitude of urinary amino-terminal excretion relates to the severity of diabetic nephropathy (Gilbert et al. 2003). Similarly, NH2-terminal CCN2 is also increased in the vitreous of patients with active progressive diabetic proliferative vitreoretinopathy, suggesting that it represents a surrogate marker of fibrosis in the disorder (Hinton et al. 2002). Indeed, plasma CCN2 levels contribute significantly to prediction of end-stage renal disease and mortality in patients with type 1 diabetic nephropathy (Nguyen et al. 2008). Such a diagnostic tool may not merely be limited to diabetes. N-terminal CCN2 levels in plasma and dermal interstitial fluid correlated with severity of skin disease of scleroderma and (negatively) with disease duration (Dziadzio et al. 2005). Moreover in liver fibrosis, the mean concentration of total CCN2 is highest in the fibrosis group (5.2-fold) and in the chronic viral hepatitis group (4.3-fold) but lower in those patients with fully developed cirrhosis (Gressner et al. 2006). Similarly, CCN2 concentration appears to predict myocardial fibrosis in chronic heart failure patients (Koitabashi et al. 2008).
Conclusion
CCN2 was identified over 15 years ago; however, the actual physiological relevance of CCN2 is only just beginning to emerge. However, it remains clear that studies on CCN2 expression have established that CCN2 is an effective marker of fibroproliferative disease. Thus ELISAs examining CCN2 levels in patients are warranted as diagnostic tools for fibrosis as well as in investigating the efficacy of drugs in clinical trials.
References
Abraham DJ, Shiwen X, Black CM, Sa S, Xu Y, Leask A (2000) Tumor necrosis factor α suppresses the induction of connective tissue growth factor by transforming growth factor-β in normal and scleroderma fibroblasts. J Biol Chem 275:15220–15225 doi:10.1074/jbc.275.20.15220
Blom IE, Goldschmeding R, Leask A (2002) Gene regulation of connective tissue growth factor: new targets for antifibrotic therapy? Matrix Biol 21:473–482 doi:10.1016/S0945-053X(02)00055-0
Bradham DM, Igarashi A, Potter RL, Grotendorst GR (1991) Connective tissue growth factor: a cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early geneproduct CEF-10. J Cell Biol 114:1285–1294 doi:10.1083/jcb.114.6.1285
Chen Y, Segarini P, Raoufi F, Bradham D, Leask A (2001) Connective tissue growth factor is secreted through the Golgi and is degraded in the endosome. Exp Cell Res 271:109–117 doi:10.1006/excr.2001.5364
Chen Y, Blom IE, Sa S, Goldschmeding R, Abraham DJ, Leask A (2002) CTGF expression in mesangial cells: involvement of SMADs, MAP kinase, and PKC. Kidney Int 62:1149–1159 doi:10.1111/j.1523-1755.2002.kid567.x
Chen Y, Shi-wen X, Eastwood M, Black CM, Denton CP, Leask A, Abraham DJ (2006) Contribution of activin receptor-like kinase 5 (transforming growth factor beta receptor type I) signaling to the fibrotic phenotype of scleroderma fibroblasts. Arthritis Rheum 54:1309–1316 doi:10.1002/art.21725
Chen S, McLean S, Carter DE, Leask A (2007) The gene expression profile induced by Wnt 3a in NIH 3T3 fibroblasts. J Cell Commun Signal 1:175–183 doi:10.1007/s12079-007-0015-x
Cheng O, Thuillier R, Sampson E, Schultz G, Ruiz P, Zhang X, Yuen PS, Mannon RB (2006) Connective tissue growth factor is a biomarker and mediator of kidney allograft fibrosis. Am J Transplant 6:2292–2306 doi:10.1111/j.1600-6143.2006.01493.x
Denton CP, Khan K, Hoyles RK, Shiwen X, Leoni P, Chen Y, Eastwood M, Abraham DJ (2009) Inducible lineage-specific deletion of TbetaRII in fibroblasts defines a pivotal regulatory role during adult skin wound healing. J Invest Dermatol 129:194–204 doi:10.1038/jid.2008.171
Dziadzio M, Usinger W, Leask A, Abraham D, Black CM, Denton C, Stratton R (2005) N-terminal connective tissue growth factor is a marker of the fibrotic phenotype in scleroderma. QJM 98:485–492 doi:10.1093/qjmed/hci078
Fonseca C, Lindahl GE, Ponticos M, Sestini P, Renzoni EA, Holmes AM, Spagnolo P, Pantelidis P, Leoni P, McHugh N, Stock CJ, Shi-Wen X, Denton CP, Black CM, Welsh KI, du Bois RM, Abraham DJ (2007) A polymorphism in the CTGF promoter region associated with systemic sclerosis. N Engl J Med 357:1210–1220 doi:10.1056/NEJMoa067655
Gilbert RE, Akdeniz A, Weitz S, Usinger WR, Molineaux C, Jones SE, Langham RG, Jerums G (2003) Urinary connective tissue growth factor excretion in patients with type 1 diabetes and nephropathy. Diabetes Care 26:2632–2636 doi:10.2337/diacare.26.9.2632
Gressner AM, Yagmur E, Lahme B, Gressner O, Stanzel S (2006) Connective tissue growth factor in serum as a new candidate test for assessment of hepatic fibrosis. Clin Chem 52:1815–1817 doi:10.1373/clinchem.2006.070466
Gressner OA, Lahme B, Rehbein K, Siluschek M, Weiskirchen R, Gressner AM. (2008) Pharmacological application of caffeine inhibits TGF-beta-stimulated connective tissue growth factor expression in hepatocytes via PPARgamma and SMAD2/3-dependent pathways. J Hepatol. epub
Grotendorst GR (1997) Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev 8:171–179 doi:10.1016/S1359-6101(97)00010-5
Grotendorst GR, Okochi H, Hayashi N (1996) A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ 7:469–480
Higgins DF, Biju MP, Akai Y, Wutz A, Johnson RS, Haase VH (2004) Hypoxic induction of Ctgf is directly mediated by Hif-1. Am J Physiol Renal Physiol 287:F1223–F1232 doi:10.1152/ajprenal.00245.2004
Hinton DR, He S, Jin ML, Barron E, Ryan SJ (2002) Novel growth factors involved in the pathogenesis of proliferative vitreoretinopathy. Eye 16:422–428 doi:10.1038/sj.eye.6700190
Holmes A, Abraham DJ, Sa S, Shiwen X, Black CM, Leask A (2001) CTGF and SMADs, maintenance of scleroderma phenotype is independent of SMAD signaling. J Biol Chem 276:10594–10601 doi:10.1074/jbc.M010149200
Holmes A, Abraham DJ, Chen Y, Denton C, Shi-wen X, Black CM, Leask A (2003) Constitutive connective tissue growth factor expression in scleroderma fibroblasts is dependent on Sp1. J Biol Chem 278:41728–41733 doi:10.1074/jbc.M305019200
Huang BL, Dornbach LM, Lyons KM (2007) The 5′ untranslated regions (UTRs) of CCN1, CCN2, and CCN4 exhibit cryptic promoter activity. J Cell Commun Signal 1:17–32 doi:10.1007/s12079-007-0003-1
Igarashi A, Okochi H, Bradham DM, Grotendorst GR (1993) Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol Biol Cell 4:637–645
Ito Y, Aten J, Bende RJ, Oemar BS, Rabelink TJ, Weening JJ, Goldschmeding R (1998) Expression of connective tissue growth factor in human renal fibrosis. Kidney Int 53:853–861 doi:10.1111/j.1523-1755.1998.00820.x
Ito Y, Goldschmeding R, Bende R, Claessen N, Chand M, Kleij L, Rabelink T, Weening J, Aten J (2001) Kinetics of connective tissue growth factor expression during experimental proliferative glomerulonephritis. J Am Soc Nephrol 12:472–484
Kantarci A, Black SA, Xydas CE, Murawel P, Uchida Y, Yucekal-Tuncer B, Atilla G, Emingil G, Uzel MI, Lee A, Firatli E, Sheff M, Hasturk H, Van Dyke TE, Trackman PC (2006) Epithelial and connective tissue cell CTGF/CCN2 expression in gingival fibrosis. J Pathol 210:59–66 doi:10.1002/path.2000
Kapoor M, Liu S, Huh K, Parapuram S, Kennedy L, Leask A (2008) Connective tissue growth factor promoter activity in normal and wounded skin. Fibrogenesis Tissue Repair 1:1 doi:10.1186/1755-1536-1-3
Kennedy L, Parapuram S, Greenspoon J, Leask A. (2008) Ceramide inhibits CCN2 expression in fibroblasts. J Cell Commun Signal. epub.
Kidd M, Modlin IM, Shapiro MD, Camp RL, Mane SM, Usinger W, Murren JR (2007) CTGF, intestinal stellate cells and carcinoid fibrogenesis. World J Gastroenterol 13:5208–5216
Koitabashi N, Arai M, Niwano K, Watanabe A, Endoh M, Suguta M, Yokoyama T, Tada H, Toyama T, Adachi H, Naito S, Oshima S, Nishida T, Kubota S, Takigawa M, Kurabayashi M (2008) Plasma connective tissue growth factor is a novel potential biomarker of cardiac dysfunction in patients with chronic heart failure. Eur J Heart Fail 10:373–379 doi:10.1016/j.ejheart.2008.02.011
Kondo S, Kubota S, Mukudai Y, Moritani N, Nishida T, Matsushita H, Matsumoto S, Sugahara T, Takigawa M (2006) Hypoxic regulation of stability of connective tissue growth factor/CCN2 mRNA by 3′-untranslated region interacting with a cellular protein in human chondrosarcoma cells. Oncogene 16:1099–1110 doi:10.1038/sj.onc.1209129
Kubota S, Kondo S, Eguchi T, Hattori T, Nakanishi T, Pomerantz RJ, Takigawa M (2000) Identification of an RNA element that confers post-transcriptional repression of connective tissue growth factor/ hypertrophic chondrocyte specific 24 (ctgf/hcs24)gene: similarities to retroviral RNA-protein interactions. Oncogene 19:4773–4786 doi:10.1038/sj.onc.1203835
Kuiper EJ, Van Nieuwenhoven FA, de Smet MD, van Meurs JC, Tanck MW, Oliver N, Klaassen I, Van Noorden CJ, Goldschmeding R, Schlingemann RO (2008) The angio-fibrotic switch of VEGF and CTGF in proliferative diabetic retinopathy. PLoS One 3(7):e2675 doi:10.1371/journal.pone.0002675
Leask A (2008) The Starbuck stops here: it’s a Smad world. J Cell Commun Signal. epub
Leask A, Abraham DJ (2006) All in the CCN family: essential matricellular signaling modulators emerge from the bunker. J Cell Sci 119:4803–4810 doi:10.1242/jcs.03270
Leask A, Sa S, Holmes A, Shiwen X, Black CM, Abraham DJ (2001) The control of ccn2 (ctgf) gene expression in normal and scleroderma fibroblasts. Mol Pathol 54:180–183 doi:10.1136/mp.54.3.180
Leask A, Holmes A, Black CM, Abraham DJ (2003) Connective tissue growth factor gene regulation. Requirements for its induction by transforming growth factor-beta 2 in fibroblasts. J Biol Chem 278:13008–13015 doi:10.1074/jbc.M210366200
Leask A, Denton CP, Abraham DJ (2004) Insights into the molecular mechanism of chronic fibrosis: the role of connective tissue growth factor in scleroderma. J Invest Dermatol 122:1–6 doi:10.1046/j.0022-202X.2003.22133.x
Leask, A., Chen, S., Pala D, Brigstock, DR. (2008) Regulation of CCN2 mRNA expression and promoter activity in activated hepatic stellate cells. J Cell Commun Signal, epub
Luo Q, Kang Q, Si W, Jiang W, Park JK, Peng Y, Li X, Luu HH, Luo J, Montag AG, Haydon RC, He TC (2004) Connective tissue growth factor (CTGF) is regulated by Wnt and bone morphogenetic proteins signaling in osteoblast differentiation of mesenchymal stem cells. J Biol Chem 279:55958–55968 doi:10.1074/jbc.M407810200
Nguyen TQ, Tarnow L, Andersen S, Hovind P, Parving HH, Goldschmeding R, vanNieuwenhoven FA (2006) Urinary connective tissue growth factor excretion correlates with clinical markers of renal disease in a large population of type 1 diabetic patients with diabetic nephropathy. Diabetes Care 29:83–88 doi:10.2337/diacare.29.01.06.dc05-1670
Paradis V, Dargere D, Vidaud M, De Gouville AC, Huet S, Martinez V, Gauthier JM, Ba N, Sobesky R, Ratziu V, Bedossa P (1999) Expression of connective tissue growth factor in experimental rat and human liver fibrosis. Hepatology 30:968–976 doi:10.1002/hep.510300425
Phanish MK, Wahab NA, Hendry BM, Dockrell ME (2005) TGF-beta1-induced connective tissue growth factor (CCN2) expression in human renal proximal tubule epithelial cells requires Ras/MEK/ERK and Smad signalling. Nephron, Exp Nephrol 100:e156–e165 doi:10.1159/000085445
Pickles M, Leask A (2007) Analysis of CCN2 promoter activity in PANC-1 cells: regulation by ras/MEK/ERK. J Cell Commun Signal 1(2):85–90 doi:10.1007/s12079-007-0008-9
Qi W, Chen X, Twigg S, Zhang Y, Gilbert RE, Kelly DJ, Pollock CA (2007) The differential regulation of Smad7 in kidney tubule cells by connective tissue growth factor and transforming growth factor-beta1. Nephrology 12:267–274 doi:10.1111/j.1440-1797.2007.00788.x
Ricupero DA, Rishikof DC, Kuang PP, Poliks CF, Goldstein RH (1999) Regulation of connective tissue growth factor expression by prostaglandin E(2). Am J Physiol 277:L1165–L1171
Riser BL, Cortes P, DeNichilo M, Deshmukh PV, Chahal PS, Mohammed AK, Yee J, Kahkonen D (2003) Urinary CCN2 (CTGF) as a possible predictor of diabetic nephropathy: preliminary report. Kidney Int 264:451–458 doi:10.1046/j.1523-1755.2003.00130.x
Sato S, Nagaoka T, Hasegawa M, Tamatani T, Nakanishi T, Takigawa M, Takehara K (2000) Serum levels of connective tissue growth factor are elevated in patients withsystemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. J Rheumatol 7:149–154
Shi-wen X, Howat SL, Renzoni EA, Holmes A, Pearson JD, Dashwood MR, Bou-Gharios G, Denton CP, du Bois RM, Black CM, Leask A, Abraham DJ (2004) Endothelin-1 induces expression of matrix-associated genes in lung fibroblasts through MEK/ERK. J Biol Chem 279:23098–23103 doi:10.1074/jbc.M311430200
Shi-wen X, Stanton L, Kennedy L, Pala D, Chen Y, Howat SL, Renzoni EA, Carter DE, Bou-Gharios G, Stratton RJ, Pearson JD, Beier F, Lyons KM, Black CM, Abraham DJ, Leask A (2006) CCN2 is necessary for adhesive responses to TGFß1 in embryonic fibroblasts. J Biol Chem 281:10715–10726 doi:10.1074/jbc.M511343200
Shi-Wen X, Renzoni EA, Kennedy L, Howat S, Chen Y, Pearson JD, Bou-Gharios G, Dashwood MR, du Bois RM, Black CM, Denton CP, Abraham DJ, Leask A (2007a) Endogenous endothelin-1 signaling contributes to type I collagen and CCN2 overexpression in fibrotic fibroblasts. Matrix Biol 26:625–632 doi:10.1016/j.matbio.2007.06.003
Shi-wen X, Kennedy L, Renzoni EA, Bou-Gharios G, du Bois RM, Black CM, Denton CP, Abraham DJ, Leask A (2007b) Endothelin is a downstream mediator of profibrotic responses to transforming growth factor beta in human lung fibroblasts. Arthritis Rheum 56:4189–4194 doi:10.1002/art.23134
Stratton R, Rajkumar V, Ponticos M, Nichols B, Shiwen X, Black CM, Abraham DJ, Leask A (2002) Prostacyclin derivatives prevent the fibrotic response to TGF-beta by inhibiting the Ras/MEK/ERK pathway. FASEB J 16:1949–1951
Stratton R, Shiwen X, Martini G, Holmes A, Leask A, Haberberger T, Martin GR, Black CM, Abraham D (2001) Iloprost suppresses connective tissue growth factor production in fibroblasts and in the skin of scleroderma patients. J Clin Invest 108:241–250
Tamatani T, Kobayashi H, Tezuka K, Sakamoto S, Suzuki K, Nakanishi T, Takigawa M, Miyano T (1998) Establishment of the enzyme-linked immunosorbent assay for connective tissue growth factor (CTGF) and its detection in the sera of biliary atresia. Biochem Biophys Res Commun 251:748–752 doi:10.1006/bbrc.1998.9543
Thomson SE, McLennan SV, Kirwan PD, Heffernan SJ, Hennessy A, Yue DK, Twigg SM (2008) Renal connective tissue growth factor correlates with glomerular basement membrane thickness and prospective albuminuria in a non-human primate model of diabetes: possible predictive marker for incipient diabetic nephropathy. J Diabetes Complications 22:284–294 doi:10.1016/j.jdiacomp.2007.07.001
Van Beek JP, Kennedy L, Rockel JS, Bernier SM, Leask A (2006) The induction of CCN2 by TGFbeta1 involves Ets-1. Arthritis Res Ther 8(2):R36 doi:10.1186/ar1890
Xiao R, Kanekura T, Yoshida N, Higashi Y, Yan KL, Fukushige T, Kanzaki T (2008) 9-Cis-retinoic acid exhibits antifibrotic activity via the induction of cyclooxygenase-2 expression and prostaglandin E2 production in scleroderma fibroblasts. Clin Exp Dermatol 33:484–490 doi:10.1111/j.1365-2230.2008.02727.x
Acknowledgements
Our research is supported by the Canadian Institute of Health Research, the Canadian Foundation for Innovation, the Ontario Thoracic Society, the Scleroderma Society, Reynaud’s and Scleroderma Association and the Arthritis Research Campaign. AL is a New Investigator of the Arthritis Society (Scleroderma Society of Canada) and an Early Researcher Award recipient.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article also appears in the newsletter section of the CCN society website: www.ccnsociety.com/.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Leask, A., Parapuram, S.K., Shi-wen, X. et al. Connective tissue growth factor (CTGF, CCN2) gene regulation: a potent clinical bio-marker of fibroproliferative disease?. J. Cell Commun. Signal. 3, 89–94 (2009). https://doi.org/10.1007/s12079-009-0037-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-009-0037-7