
Abstract
Immune and inflammatory pathways have a central role in the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Both the innate and adaptive immune systems contribute to the development of NAFLD. Pathogen-associated molecular patterns and danger-associated molecular patterns are known to activate a variety of pattern-recognition receptors that result in inflammation. The key features of the immune system and inflammatory pathways in the development of NAFLD are discussed in this review.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most common liver disease worldwide, affecting around one third of the Western world with an incidence that continues to grow even in other parts of the world [1, 2]. NAFLD includes a variety of histopathological findings ranging from simple steatosis with no inflammation to steatosis with varying degrees of inflammation (steatohepatitis, NASH) to cirrhosis [1]. It is thought that around 10–20 % of patients with NAFLD have NASH [3]. Patients with NASH have the potential to develop fibrosis and cirrhosis leading to portal hypertension, liver decompensation, and hepatocellular carcinoma [4].
The liver is composed of a diverse array of cell types, most of which have the potential to be involved in inflammation. Hepatocytes make up around 60–80 % of all liver cells, and they are responsible for the metabolic, biosynthetic, biliary secretion, and detoxification roles of the liver. The liver is also enriched in various immune cells. The resident innate immune cells in the liver are comprised of Kupffer cells (KCs), the resident macrophages, and natural killer (NK) cells. Upon liver injury, other innate immune cells, including neutrophils, leukocytes, monocytes, and inflammatory macrophages, are rapidly recruited to the liver. The adaptive immune cells consist primarily of natural killer T (NKT) cells, B cells, and T cells. Other cell types that may contribute to inflammation and fibrosis are the sinusoidal endothelial cells (SECs) and hepatic stellate cells (HSCs).
The cells in the liver contribute to the inflammation that results in the development and progression of steatohepatitis, which is secondary to the immune response of the liver to danger signals, likely from a combination of exogenous and endogenous danger signals, which include injured self-molecules, i.e., DNA and RNA. There are multiple “hits” needed for NASH to develop and progress. In addition to metabolic factors, innate immune alterations, including inflammation caused by free fatty acids (FFA), bacterial lipopolysaccharide (LPS), chemokines, cytokines, and adipokines, all contribute [3].
Contribution of immune cells
Innate immune cells, consisting of KCs, neutrophils, dendritic cells (DCs), and NK cells, all play a role in the pathogenesis of NASH. In the setting of acute or chronic liver disease, KCs are activated by both pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). This activation results in the release of proinflammatory cytokines, such as tumor-necrosis factor α (TNFα), interleukin-6 (IL-6), and interleukin-1β (IL-1β), which lead to T cell activation and induction of apoptosis and HSCs [5]. There are two types of macrophages, M1 or “classically activated” macrophages, which play an important role in humoral immunity and response to pathogens, and M2 or “alternatively activated” macrophages, which have anti-inflammatory properties. There is an increase in M1 relative to M2 macrophages in adipose tissue and in the liver in response to high-fat diet (HFD) feeding and obesity [6–8]. The imbalance between M1 and M2 macrophages may have an important role in the pathogenesis of NASH. Depletion of KCs attenuates methionine-choline-deficient (MCD) and HFD-induced steatohepatitis [9, 10].
Activated neutrophils release proinflammatory cytokines and myeloperoxidase, resulting in oxidative damage to hepatocytes [11, 12]. Furthermore, an increase in the ratio of neutrophils to lymphocytes can increase the likelihood of NASH progression [13]. DCs also appear to play an important role in NASH, as recent reports have shown that DC depletion results in worsening NASH severity, suggesting a regulator role of DCs in NASH [14].
NK cells are an important component of the immune system and play a role in linking the innate and adaptive immune responses [15]. NK cells are abundant in the liver, and in disease they have a role in the development of liver injury and fibrosis. It is thought that NK cells could play a role in the development of NAFLD/NASH. NK cell-activating ligands are increased in human NASH patients and in mice following an MCD diet [16]. Obese patients have both a decrease in circulating levels and impaired cytotoxic function of NK cells as compared to lean controls [17]. Similar impairment in cytotoxic activity was determined to be present in rats with HFD-induced obesity [18]. NK cells may have an anti-fibrotic effect in the liver [19], and the reduction in their levels and activity may contribute to the increased susceptibility of obese patients to develop cirrhosis.
Natural killer T (NKT) cells are a subset of lymphocytes that express NK cell markers, such as CD161 and CD94, as well as a T cell receptor. NKT cells exhibit features of both innate and adaptive immune cells and act as a bridging system between innate and adaptive immunity [20]. NKT cells are enriched within the liver and regulate the immune response by secreting both TH1 and TH2 cytokines after stimulation [21]. Biopsies in patients with NAFLD showed that NKT cell population was decreased when there was moderate to severe steatosis [22]. Both obese leptin-deficient ob/ob mice and HFD-induced obesity showed reduced liver NKT cells [23, 24]. Upregulation of hepatic NKT cells results in improvement of HFD-induced hepatic steatosis [25]. Depletion of NKT cells leads to chronic inflammatory conditions that contribute to hepatic steatosis [23]. On the other hand, there is an increase of NKT cells in MCD diet-induced steatohepatitis and in cirrhotic livers [26]. NKT cells appear to drive production of osteopontin and of hedgehog ligands leading to fibrogenesis, which may explain the increase in NKT cells in the MCD diet model and in cirrhotic livers [27].
Regulatory T cells (Tregs) are a subset of T cells that are either naturally occurring or inducible. An important role of the CD25+ subset of Tregs is their suppression of CD4 and CD8 T cells and thus in a decrease in inflammation. Mouse models have displayed that depleting CD25+ Tregs results in precipitation of autoimmune disease. In the liver, it has been reported that there are increased Tregs in tumor tissue in HCC patients and in patients with either chronic HBV or HCV infection [28–30], suggesting that Tregs have a role in either suppressing or maintaining tumor cells or viral infection. In NASH livers, there are increased numbers of Tregs [31, 32]. Mice fed a HFD showed a gradual decrease of Tregs over time, which may be a key event in the progression from steatosis to steatohepatitis, as Tregs may help control hepatic inflammation [33].
T helper type 17 cells (TH17 cells) are another subset of T cells that produce interleukin-17 (IL-17) and mediate various immune responses. TH17 cells play a role in mediating pathogen clearance and in inflammatory responses in tissues. Studies have shown the importance of TH17 cells in autoimmune liver diseases, chronic viral hepatitis, alcoholic liver disease, and hepatocellular carcinoma [34–36]. Tang et al. [37] show that there are increased numbers of TH17 cells in the liver after an 8-week HFD feeding and that these cells are associated with the progression of hepatic steatosis and inflammation through the production of IL-17. The same group suggests that the balance between TH17 cells and Tregs could play a role in the progression of NASH; however, further studies need to be done to answer this question.
Pathogen-associated molecular patterns (PAMPs)
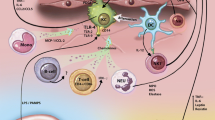
PAMPs are exogenous danger signals derived from microbes that result in inflammation. PAMPs are known to contribute to inflammation in NASH by signaling through PRRs, leading to the activation of both the innate and adaptive arms of the immune system (Fig. 1). PAMPs are recognized by a diverse array of pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), RIG-I like receptors (RLRs), and other non-specific sensors, e.g., C-type lectin receptors (CLRs) [38, 39] (Fig. 2). PAMPs are expressed by bacteria, viruses, parasites, and fungi, and they include lipids, lipoproteins, nucleic acids, and proteins [40].

Immune response in NASH. The immune response in NASH is initiated by both pathogen-associated molecular danger signals and danger-associated molecular signals. This response involves both the innate and adaptive branches of the immune system and results in inflammation

Pattern recognition receptors in NASH. A variety of pattern-associated molecular patterns and danger-associated molecular patterns are involved in the pathogenesis of NASH. PAMP and DAMP signaling can occur through various PRRs, including Toll-like receptors, nucleotide-binding and oligomerization domain (NOD)-like receptors, and RIG-I like receptors. PRR signaling can result in steatosis, insulin resistance, and inflammatory cell activation and recruitment
Toll-like receptors (TLRs)
There are 10 and 13 known TLRs in humans and mice, respectively. In the liver, TLRs are expressed on KCs, hepatocytes, SECs, and HSCs. TLRs recognize a wide variety of PAMPs and are found in different cellular locations: TLRs 2, 4, 5, 6, and 11 are found on extracellular surfaces, whereas TLRs 3, 7, 8, and 9 are expressed exclusively in intracellular vesicles, such as endosomes, lysosomes, and the endoplasmic reticulum [39]. Once a molecular pattern has been recognized by the TLR, downstream signaling is initiated, resulting in inflammatory and antiviral responses. Each TLR is associated with a different set of adaptor proteins. For example, the adaptor myeloid differentiation factor 88 (MyD88) is shared by all TLRs, except TLR3, and leads to recruitment of the IL-1 receptor (IL-1R)-associated kinase family. TLR signaling results in the activation of transcription factors nuclear factor kappa B (NF-κB), activator protein-1 (AP-1), or interferon regulatory factors, leading to the transcription of inflammatory cytokines, chemokines, or type I interferons, respectively.
TLR2/TLR1/TLR6
TLR2 recognizes a variety of PAMPs from bacteria, fungi, parasites, and viruses. These include bacterial lipoprotein, peptidoglycan, and lipoteichoic acid [41]. Dimerization of TLR2 with TLR1 or TLR6 provides additional specificity in ligand recognition. TLR2 deficiency has been shown to accelerate MCD diet-induced steatohepatitis and fibrosis [42, 43]. However, another group found that TLR2-deficient mice fed a choline-deficient amino acid (CDAA) diet had similar steatosis to wild-type mice, but decreased inflammatory cell infiltration and hepatocyte ballooning, suggesting that while steatosis is independent of TLR2 signaling, TLR2 appears to play a role in inflammation [44]. The same group showed that a TLR2 ligand activates both KCs and HSCs in vitro, but in vivo it was determined that KCs were responsible for TLR2-mediated liver inflammation and fibrosis. TLR2 knockout mice fed a HFD showed improvement in glucose tolerance and a decrease in steatosis [45]. The differences seen between the various studies could be secondary to different diets that induce steatohepatitis, as the CDAA and HFD diet result in weight gain and insulin resistance, which is not seen in the MCD diet. Another possibility is differences in intestinal microflora between the mice that could be a source of TLR2 ligands.
TLR3
TLR3 is present exclusively in intracellular vesicles, such as endosomes and lysosomes, and it recognizes double-stranded RNA (dsRNA) and poly(I:C), a synthetic analog of dsRNA, along with genomic RNA of a variety of other viruses [40]. Activation of TLR3 results in type I interferon and inflammatory cytokine production and thus produces an antiviral immune response. TLR3 activation has been implicated in a number of autoimmune diseases, including type I diabetes mellitus and Hashimoto’s thyroiditis [46, 47]. In NAFLD, Wu et al. [48] showed that TLR3 knockout mice fed a HFD displayed an improvement in glucose tolerance and an improvement in lipid and cholesterol metabolism, with no protection from HFD-induced obesity. It is yet to be established what the ligand leading to TLR3 activation is in NAFLD.
TLR4
TLR4 is the most widely studied TLR, and it recognizes a diverse array of PAMPs that include LPS, Pseudomonas exoenzyme, taxol, and other components of microbes [41]. TLR4 signaling occurs in a slightly different manner than in the other TLRs, as it requires an accessory molecule, glycoprotein MD2, to effectively bind LPS. Signaling is initiated through two different pathways, the MyD88-dependent pathway and the MyD88-independent pathway, which involves TIR-domain-containing adaptor-inducing interferon-β (TRIF) and the TRIF-related adaptor protein [39].
There have been numerous studies elucidating the role of TLR4 in NASH. Increased serum LPS levels are detected in patients with NASH and in animal models of NASH [49, 50], suggesting increased TLR4 activation. Furthermore, fructose, which is abundant in the Western diet, results in increased hepatic steatosis and inflammation potentially by causing increased intestinal bacterial overgrowth and intestinal permeability resulting in increased TLR4 signaling [51]. In MCD and HFD-induced steatohepatitis and fibrosis, TLR4 deficiency attenuates steatohepatitis [9, 52, 53]. In the MCD model, there is increased susceptibility to LPS stimulation, further implicating the TLR4 pathway in being involved in the pathogenesis of NASH [42]. MD2 knockouts also display a partial protection from MCD diet-induced steatohepatitis [52]. Both TLR4 and MD2 knockout mice display reduced NADPH oxidase activation and lipid peroxidation compared to WT mice, suggesting reduced free radical levels and improved lipid content. Furthermore, it has been suggested that TLR4 activation and sensitization of HSCs may be the link between inflammation and fibrosis in NAFLD and other forms of chronic liver disease, as HSCs are involved in the regulation of the extracellular matrix and tissue remodeling [54].
TLR9
TLR9 is expressed in intracellular vesicles and recognizes bacterial/viral DNA and CpG DNA. TLR9 ligands directly activate DCs, macrophages, and B cells, and they result in a strong T helper 1 (TH1) response. In murine NASH models, there are elevations in bacterial DNA in the plasma, which is a ligand for TLR9 [44]. The same group showed that TLR9 deficiency attenuates CDAA diet-induced steatohepatitis and fibrosis, suggesting a critical role of TLR9 in NASH [44].
NOD-like receptors
The NLRs are a family of intracellular pattern-recognition receptors that are involved in the innate immune response to microbes by recognizing PAMPs (Fig. 2). NLRs also play an important role in recognizing host-derived signals, known as DAMPs. The NLR family is characterized by the presence of a central nucleotide-binding and oligomerization domain, flanked by C-terminal leucine-rich repeats and N-terminal caspase recruitment or pyrin domains. Further details of NLR complexes and their molecular structure are described elsewhere [55]. To date, four NLR prototypes have been identified, NLRP1 (NALP1), NLRP3 (NALP3, cryoporin), NLRC4 (IPAF), and AIM2 [56].
NLRs are part of intracellular multi-protein complexes known as inflammasomes that are assembled and activated following cellular infection or stress and result in inflammation. Inflammasome activation is thought to involve two steps, in which the first signal upregulates inflammasome expression and is generally from TLR activation, and the second step involves a ligand triggering inflammasome activation [56]. Inflammasome activation occurs in innate immune cells, including macrophages, neutrophils, and DCs, as well as non-immune cells, including hepatocytes, HSCs, endothelial cells, and myofibroblasts [57]. Furthermore, inflammasome activation results in the production of proinflammatory cytokines, including IL-1β and interleukin-18 (IL-18) [55]. The importance of the contribution of each cell type has not been entirely elucidated; however, one study showed that IL-1β or IL-1α depletion in hepatocytes, but not bone marrow cells, resulted in protection from diet-induced steatohepatitis [58].
IL-1β signaling plays an important role in NASH. IL-1R knockout mice showed attenuated liver steatosis, injury, and fibrosis following either a CDAA or HFD feeding [59]. In contrast, a different group found that both IL-1R knockout mice and WT mice treated with IL-1R antagonist (IL-1Ra) that received an MCD diet showed decreased liver steatosis but no improvement in inflammation or fibrosis (Csak et al., unpublished data). IL-1Ra-deficient mice showed severe hepatic steatosis and fibrosis on an atherogenic diet compared to WT mice fed the same diet [60]. Similarly to the IL-1R knockout mice, IL-1β knockout mice displayed attenuated hepatocellular damage, steatosis, and fibrosis following an atherogenic diet feeding [58]. The same group found that IL-1α knockout mice also displayed attenuated liver damage and fibrosis after an HFD feeding.
Thus far, mainly NALP3 has been found to have a potential role in the development of NASH. Further studies need to be completed with the other NLRs to determine whether they have a role in NASH.
NALP3
The NALP3 inflammasome is the most widely studied inflammasome and consists of the NLRP3 scaffold, the apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) adaptor, and the effector molecule, a proinflammatory protease, pro-caspase-1.
NALP3 is involved in antibacterial, viral, fungal, and parasitic immune responses [61]. Components of the NALP3 inflammasome and resultant caspase-1 activation are seen in the livers of mice fed both a HFD and a MCD diet [52, 62]. Increased expression of NALP3 and caspase-1 is also seen in the liver of humans with NASH [52], and the levels of expression in adipose tissue are directly correlated with the extent of type 2 diabetes mellitus in obese individuals [62]. Inflammasome upregulation has been found to occur in innate immune cells [56] as well as in primary hepatocytes of MCD diet-fed mice [52], suggesting that there are multiple cell types in the liver that undergo inflammasome activation and contribute to inflammation.
Mice lacking NALP3, ASC, or caspase-1 have been shown to display a decrease in weight gain and fat mass, and improved insulin resistance following a HFD feeding. However, there were differences among the three knockouts, with ASC knockout mice displaying decreased hepatic steatosis and liver triglyceride levels, whereas NALP3 and caspase-1 knockout mice did not show this change [63]. In contrast, a different group showed that ASC knockout mice are prone to steatosis, obesity, and increased glucose resistance [64]. This group suggested that the difference they find is due to alterations in the intestinal microbiome.
NLRP6 and NLRP12
NLRP6 and NLRP12 have mainly been studied in the context of colonic microbiota and inflammation [61]. NLRP6 knockout mice co-housed with WT mice display increased hepatic inflammation. On the other hand, NLRP12 mice co-housed with WT mice did not display any change in hepatic pathology. The group concludes that a transmissible colonic microbiota present in inflammasome-deficient mice results in NASH in the co-housed WT mice [64].
RLRs and viral infections
Patients with NASH have been shown to have accelerated progression of liver disease and fibrosis and decreased efficacy of treatment with antiviral therapy in the setting of co-infection with hepatitis C virus (HCV) and human immunodeficiency virus [65, 66].
The mechanism behind the increased susceptibility to HCV infection in the setting of NASH has been attributed to a defect in the TLR3 signaling pathway. HCV is recognized in its dsRNA form by TLR3 and the RLRs, which include helicase receptors retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5. RLRs are molecules that sense viral RNA molecules, resulting in the triggering of a danger signal. The adaptor mitochondrial antiviral signaling protein (MAVS) associates with the outer mitochondrial membrane and is crucial for inflammatory cytokine and interferon production after dsRNA binding to the helicases [67]. Whether endogenous RNA triggers RLR activation in NASH has yet to be studied.
Our group has shown that in the setting of MCD diet-induced steatohepatitis, mice challenged with the synthetic dsRNA, poly(I:C) show an inability to mount an efficient anti-viral response as demonstrated by impaired type I IFN production [16]. This deficiency was found to be secondary to dissociation of MAVS from the mitochondria to the cytosol and necrosis. Despite a decrease in inflammatory cytokine production, there was still an increase in liver damage in mice with MCD-diet-induced steatohepatitis.
Microbiome
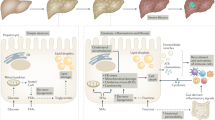
Recently, the role of the intestinal microbiota and probiotics in liver diseases has been a popular area [64, 68]. It is well known that patients with NAFLD have increased intestinal permeability and a higher incidence of bacterial overgrowth in the small intestine compared to patients with normal livers [49]. The translocation of bacterial components promotes hepatic inflammation through TLR signaling and KC activation [9] (Fig. 3). An increase in TLR agonists in the intestine has been shown to drive the progression of NASH in WT mice [64].

Cross-talk in NASH. The immune and inflammatory response in NASH involves an interaction among the liver, gut, and adipose tissue. Various adipokines, including adiponectin, interleukin-6 (IL-6), leptin, tumor necrosis factor α (TNFα), and monocyte chemoattractant protein-1 (MCP-1), are released by adipocytes and macrophages in adipose tissue and contribute to inflammation in the liver. Increased gut permeability, bacterial overgrowth, and changes in microbiota can result in the production of LPS, fatty acids, and other factors that contribute to liver inflammation and steatosis
In mice lacking various inflammasome components, a significant expansion of the intestinal bacteria Porphyromonadacae was detected following a HFD and MCD diet feeding. These bacteria have been implicated in the development and progression of metabolic syndrome in both mice and humans. Treatment with antibiotics abolished the bacteria [64, 69]. Rats fed a HFD and treated with probiotics displayed a change in intestinal microflora and a reduced level of hepatic cholesterol and triglycerides [70]. Treatment with the probiotic VSL no. 3 in mice fed an MCD diet resulted in attenuation of fibrosis without protecting from inflammation or steatosis [71].
Danger-associated molecular patterns
DAMPs are endogenous molecules released from damaged cells that trigger sterile inflammation [72]. Sterile inflammation is a response that occurs in all tissues in response to injury and cellular damage. In NASH there is a chronic state of sterile inflammation resulting from the presence of DAMPs. Similarly to PAMPS, DAMPs activate both the innate and adaptive branches of the immune response (Fig. 1). A wide variety of DAMPs have thus far been identified in many different diseases and are reviewed elsewhere [73]. DAMPs activate PRRs, of which the best characterized are TLRs (Fig. 2). In NASH, it has been a challenge to show an increase in the levels of serum DAMPs, and further research is needed to elucidate whether DAMPs that play a role in other diseases also contribute to the pathogenesis of NASH [73]. Some of the DAMPs that have been identified, such as high-mobility group-1 (HMGB1) and FFAs, signal through several TLRs and, in the case of FFAs, a NLR.
Toll-like receptors (TLRs)
TLR2
It has not yet been determined whether there are DAMPs that directly activate the TLR2 ligand in NASH. Stimulation of TLR2 in KCs with a TLR2 ligand in the presence of the FFA palmitic acid (PA) results in inflammasome activation and increased IL-1β production, suggesting a cross-talk between PAMPs and DAMPs [44]. It has been hypothesized that HMGB1 may also activate TLR2 in addition to TLR4. As discussed earlier, there are opposing phenotypes published by different groups in the setting of TLR2 knockout mice in a diet-induced steatohepatitis model, suggesting that differences in intestinal microflora could be responsible for TLR2 ligand production [44].
TLR4
A variety of DAMPs have been described that activate TLR4, including HMGB1, hyaluronic acid, heat shock proteins, defensins, and fibrinogen [41]. The role, if any, of most of these DAMPs has not yet been conclusively determined in NASH.
HMGB1 is a highly conserved nuclear protein that facilitates regulatory protein binding to DNA [74]. It is constitutively expressed by most cells and is released upon cell injury or death. Yanai et al. [75] describe HMGB proteins as acting as universal intracellular sensors of cytosolic nucleic acids. For example, HMGB1 is released from necrotic cells leading to KC and monocyte stimulation by acting as an endogenous TLR4 ligand either alone or with other molecules, such as endotoxin or ssDNA [76]. Furthermore, increased hepatocyte expression and release of HMGB1 triggers TLR4 and MyD88 signaling and inflammation in response to FFA infusion or HFD [53]. The interaction between HMGB1 and FFAs in TLR4 signaling in NASH is yet to be studied in depth. It has not yet been determined whether HMGB1 directly binds to TLR4 or forms complexes with other molecules.
FFAs have also been described to contribute to the pathogenesis of NASH [1, 77]. FFA levels have been reported to be elevated in mice with HFD-induced, MCD diet-induced, or leptin-deficiency diet-induced steatohepatitis [78–80]. Saturated FFAs result in the activation of the TLR4 pathway in adipocytes and macrophages resulting in inflammation through an unclear mechanism [73]. However, it does not appear that FFAs are ligands for TLR4 in either KCs or HSCs [44, 81].
TLR5
Although not expressed in mammalian livers, TLR5 knockout mice have altered gut flora. These mice develop obesity, steatosis, and insulin resistance secondary to hyperphagia. Transmission of the gut flora from these mice to WT mice resulted in transmission of the disease phenotype [82].
TLR9
Bacterial DNA is often detected in the blood of both rodents and humans with chronic liver diseases [83, 84]. DNA from dying hepatocytes in NASH can act as an endogenous ligand for TLR9 [85]. The DNA from apoptotic cells is modified by caspase-activated DNAse-mediated cleavage and methylation and oxidative damage, resulting in the ability to activate TLR9 [85]. The combination of both exogenous and endogenous bacterial DNA could potentially lead to an exponential increase in TLR9 activation in NASH. Mitochondrial dysfunction has also been implicated in the pathogenesis of NASH, resulting in increased oxidative stress, cytokine production, and cell death [86]. Mitochondrial DNA, which resembles bacterial DNA in having non-methylated CpG motifs, released by injured cells results in the activation of neutrophils through TLR9 signaling. Furthermore, HMGB1 has also been described to interact with TLR9, likely involved in the signal response to CpG DNA [75], but its role in NASH is not yet clear.
NOD-like receptors (NLRs)
NALP3
The NALP3 inflammasome can be activated by a variety of host-derived molecules. These activators include extracellular ATP, hyaluronic acid, amyloid-β, and uric acid crystals [55].
Both experimental and clinical studies have shown a link between IL-1β and IL-18, which are produced by inflammasome activation, to the development of metabolic syndrome [87, 88]. The underlying mechanism has not yet been elucidated.
Obesity has been shown to result in adipocyte hypertrophy and preferential M1 macrophage infiltration [89]. The NALP3 inflammasome is activated in these macrophages by the saturated fatty acids PA and lipotoxic ceramics [62, 90]. Hepatocytes treated with PA displayed upregulation of the NALP3 inflammasome and caspase-1 activation. Hepatocytes stimulated with PA have been shown to activate liver mononuclear cells, suggesting that hepatocytes can transfer inflammasome activation to surrounding immune cells [52]. Furthermore, pre-treatment of hepatocytes with PA resulted in sensitization to stimulation with LPS and a significantly higher level of caspase-1 than treatment with either PA or LPS alone [52]. In addition to leading to inflammation, fatty acids also induce apoptosis and the subsequent release of DAMPs. The release of mitochondrial DNA into the cytosol in response to LPS and ATP stimulation is dependent on NALP3 and is necessary to achieve robust caspase-1 activation and IL-1β production [91]. Mitochondrial DNA appears to also act by directly binding to and activating NALP3 [92] and likely contributes to the pathogenesis of NASH.
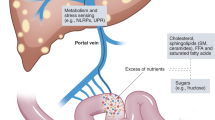
Role of adipose tissue
Adipose tissue is a highly active tissue that has both endocrine and immune functions, and has been found to have a role in mediating disease. In obesity, there is dysfunctional adipose tissue resulting from hypertrophic adipocytes and macrophage infiltration into adipose tissue [93]. The liver is an important target for adipose tissue, and its dysregulation in obesity can contribute to steatohepatitis (Fig. 3). The increase in FFAs seen in obesity results in TLR4 activation in adipocytes and macrophages, resulting in inflammation. Furthermore, there is an increase in circulating FFAs secondary to an increase in adipose FFAs, resulting in an increase in portal FFAs, thus contributing to NAFLD and eventually NASH [94]. Activation of adipose tissue macrophages also results in ectopic fat deposition in the liver.
Hypertrophic adipocytes behave similarly to macrophages and produce adipokines similarly to foam cell fat-loaded macrophages in arterial plaques [6]. The production of insulin-sensitizing adipokines is reduced, such as adiponectin, resulting in decreased insulin sensitivity. Overexpression of adiponectin in genetically obese leptin-deficient ob/ob mice results in a reversal of the diabetic phenotype and reduced liver fat content, despite retaining the obese phenotype, suggesting that adiponectin plays a critical role in preventing both local and systemic inflammation [95].
Other adipokines that have a proinflammatory role are the cytokines interleukin-6 (IL-6) and TNFα as well as the chemokine monocyte chemoattractant protein-1 (MCP-1). IL-6 levels are elevated in the serum and adipocytes of obese patients, and the level of expression is significantly higher in adipose tissue than in liver, suggesting that adipose tissue is the main source of IL-6 in NASH [96, 97]. Furthermore, TNFα expression is profoundly increased in adipose tissue of obese patients [96]. Adipose tissue-derived IL-6 and TNFα likely target the liver, as exposure to both of these cytokines has an effect on hepatic insulin resistance [98]. IL-6-deficient mice fed a MCD diet or HFD develop worsening inflammation and liver damage [99, 100]. However, blockade of IL-6 signaling by a neutralizing antibody against the IL-6 receptor resulted in enhanced liver steatosis and improved liver injury in MCD-fed mice [101]. The balance in the levels of IL-6 appears to play an important role in the development of liver injury.
Conclusions
NAFLD is a disease that encompasses a wide range of pathology, from steatosis to steatosis with inflammation to fibrosis; it is commonly seen in the presence of the metabolic syndrome. It is clear that there are complex immune and inflammatory pathways that result in the progression of NASH, involving signaling in various cell types that are stimulated by PAMPs and DAMPs as well as interaction between different cell types and tissues. While there has been a significant amount of research examining the immune and inflammatory pathways in NAFLD, many questions remain. The reason why certain people develop steatohepatitis and progress to cirrhosis while others only develop steatosis has not yet been determined. There are likely undiscovered PAMPs and DAMPs that contribute to NAFLD, as well as unknown interactions between various PAMPs and DAMPs. Furthermore, understanding cell specificity in both NLR and TLR activation in NASH is an area that deserves further investigation and could be important as a target for future therapeutics. In addition, more studies looking at the interactions of the components of the adaptive immune system are warranted.
Despite the significant advancement in knowledge about the development of NAFLD, a dearth of effective treatments remains, aside from weight loss and treating associated conditions, such as hyperlipidemia and diabetes. Continuing to elucidate the mechanisms and pathogenesis behind the progression of NAFLD is critical to being able to design therapeutic interventions that can directly target the liver.
References
Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol 2010;5:145–171
Milic S, Stimac D. Nonalcoholic fatty liver disease/steatohepatitis: epidemiology, pathogenesis, clinical presentation and treatment. Dig Dis 2012;30:158–162
Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 2010;52:1836–1846
Bugianesi E, Leone N, Vanni E, Marchesini G, Brunello F, Carucci P, et al. Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology 2002;123:134–140
Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int 2006;26:1175–1186
Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest 2011;121:2111–2117
Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab 2008;7:496–507
Baffy G. Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J Hepatol 2009;51:212–223
Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol 2007;47:571–579
Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010;59:347–357
Nijhuis J, Rensen SS, Slaats Y, van Dielen FM, Buurman WA, Greve JW. Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity (Silver Spring) 2009;17:2014–2018
Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol 2009;175:1473–1482
Alkhouri N, Morris-Stiff G, Campbell C, Lopez R, Tamimi TA, Yerian L, et al. Neutrophil to lymphocyte ratio: a new marker for predicting steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease. Liver Int 2012;32:297–302
Henning JR, Graffeo CS, Rehman A, Fallon NC, Zambirinis CP, Ochi A, et al. Dendritic cells limit fibro-inflammatory injury in NASH. Hepatology 2013;58(2):589–602
Bahjat KS, Prell RA, Allen HE, Liu W, Lemmens EE, Leong ML, et al. Activation of immature hepatic NK cells as immunotherapy for liver metastatic disease. J Immunol 2007;179:7376–7384
Csak T, Dolganiuc A, Kodys K, Nath B, Petrasek J, Bala S, et al. Mitochondrial antiviral signaling protein defect links impaired antiviral response and liver injury in steatohepatitis in mice. Hepatology 2011;53:1917–1931
O’Shea D, Cawood TJ, O’Farrelly C, Lynch L. Natural killer cells in obesity: impaired function and increased susceptibility to the effects of cigarette smoke. PLoS One 2010;5:e8660
Lamas O, Martinez JA, Marti A. Energy restriction restores the impaired immune response in overweight (cafeteria) rats. J Nutr Biochem 2004;15:418–425
Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 2006;130:435–452
Ronchi F, Falcone M. Immune regulation by invariant NKT cells in autoimmunity. Front Biosci 2008;13:4827–4837
Godfrey DI, Hammond KJ, Poulton LD, Smyth MJ, Baxter AG. NKT cells: facts, functions and fallacies. Immunol Today 2000;21:573–583
Kremer M, Thomas E, Milton RJ, Perry AW, van Rooijen N, Wheeler MD, et al. Kupffer cell and interleukin-12-dependent loss of natural killer T cells in hepatosteatosis. Hepatology 2010;51:130–141
Li Z, Soloski MJ, Diehl AM. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology 2005;42:880–885
Xu CF, Yu CH, Li YM, Xu L, Du J, Shen Z. Association of the frequency of peripheral natural killer T cells with nonalcoholic fatty liver disease. World J Gastroenterol 2007;13:4504–4508
Ma X, Hua J, Li Z. Probiotics improve high fat diet-induced hepatic steatosis and insulin resistance by increasing hepatic NKT cells. J Hepatol 2008;49:821–830
Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010;51:1998–2007
Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H, et al. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut 2012;61:1323–1329
Unitt E, Rushbrook SM, Marshall A, Davies S, Gibbs P, Morris LS, et al. Compromised lymphocytes infiltrate hepatocellular carcinoma: the role of T-regulatory cells. Hepatology 2005;41:722–730
Stoop JN, van der Molen RG, Baan CC, van der Laan LJ, Kuipers EJ, Kusters JG, et al. Regulatory T cells contribute to the impaired immune response in patients with chronic hepatitis B virus infection. Hepatology 2005;41:771–778
Cabrera R, Tu Z, Xu Y, Firpi RJ, Rosen HR, Liu C, et al. An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes in hepatitis C virus infection. Hepatology 2004;40:1062–1071
Soderberg C, Marmur J, Eckes K, Glaumann H, Sallberg M, Frelin L, et al. Microvesicular fat, inter cellular adhesion molecule-1 and regulatory T-lymphocytes are of importance for the inflammatory process in livers with non-alcoholic steatohepatitis. APMIS 2011;119:412–420
Speletas M, Argentou N, Germanidis G, Vasiliadis T, Mantzoukis K, Patsiaoura K, et al. Foxp3 expression in liver correlates with the degree but not the cause of inflammation. Mediators Inflamm 2011;2011:827565
Ma X, Hua J, Mohamood AR, Hamad AR, Ravi R, Li Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 2007;46:1519–1529
Zhao L, de Qiu K, Ma X. Th17 cells: the emerging reciprocal partner of regulatory T cells in the liver. J Dig Dis 2010;11:126–133
Ye C, Li WY, Zheng MH, Chen YP. T-helper 17 cell: a distinctive cell in liver diseases. Hepatol Res 2011;41:22–29
Hammerich L, Heymann F, Tacke F. Role of IL-17 and Th17 cells in liver diseases. Clin Dev Immunol 2011;2011:345803
Tang Y, Bian Z, Zhao L, Liu Y, Liang S, Wang Q, et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol 2011;166:281–290
Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol 2002;20:197–216
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010;11:373–384
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006;124:783–801
Alisi A, Carsetti R, Nobili V. Pathogen- or damage-associated molecular patterns during nonalcoholic fatty liver disease development. Hepatology 2011;54:1500–1502
Szabo G, Velayudham A, Romics L Jr, Mandrekar P. Modulation of non-alcoholic steatohepatitis by pattern recognition receptors in mice: the role of Toll-like receptors 2 and 4. Alcohol Clin Exp Res 2005;29:140S–145S
Rivera CA, Gaskin L, Allman M, Pang J, Brady K, Adegboyega P, et al. Toll-like receptor-2 deficiency enhances non-alcoholic steatohepatitis. BMC Gastroenterol 2010;10:52
Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 2013;57:577–589
Ehses JA, Meier DT, Wueest S, Rytka J, Boller S, Wielinga PY, et al. Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetologia 2010;53:1795–1806
Harii N, Lewis CJ, Vasko V, McCall K, Benavides-Peralta U, Sun X, et al. Thyrocytes express a functional Toll-like receptor 3: overexpression can be induced by viral infection and reversed by phenylmethimazole and is associated with Hashimoto’s autoimmune thyroiditis. Mol Endocrinol 2005;19:1231–1250
Wen L, Peng J, Li Z, Wong FS. The effect of innate immunity on autoimmune diabetes and the expression of Toll-like receptors on pancreatic islets. J Immunol 2004;172:3173–3180
Wu LH, Huang CC, Adhikarakunnathu S, San Mateo LR, Duffy KE, Rafferty P, et al. Loss of Toll-like receptor 3 function improves glucose tolerance and reduces liver steatosis in obese mice. Metabolism 2012;61:1633–1645
Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009;49:1877–1887
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007;56:1761–1772
Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009;50:1094–1104
Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011;54:133–144
Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J, et al. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology 2011;54:1620–1630
Guo J, Friedman SL. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair 2010;3:21
Schroder K, Tschopp J. The inflammasomes. Cell 2010;140:821–832
Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol 2009;27:229–265
Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol 2012;57:642–654
Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol 2011;55:1086–1094
de Roos B, Rungapamestry V, Ross K, Rucklidge G, Reid M, Duncan G, et al. Attenuation of inflammation and cellular stress-related pathways maintains insulin sensitivity in obese type I interleukin-1 receptor knockout mice on a high-fat diet. Proteomics 2009;9:3244–3256
Isoda K, Sawada S, Ayaori M, Matsuki T, Horai R, Kagata Y, et al. Deficiency of interleukin-1 receptor antagonist deteriorates fatty liver and cholesterol metabolism in hypercholesterolemic mice. J Biol Chem 2005;280:7002–7009
Elinav E, Strowig T, Henao-Mejia J, Flavell RA. Regulation of the antimicrobial response by NLR proteins. Immunity 2011;34:665–679
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 2011;17:179–188
Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci USA 2011;108:15324–15329
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012;482:179–185
Younossi ZM, McCullough AJ. Metabolic syndrome, non-alcoholic fatty liver disease and hepatitis C virus: impact on disease progression and treatment response. Liver Int 2009;29(Suppl 2):3–12
Kahraman A, Miller M, Gieseler RK, Gerken G, Scolaro MJ, Canbay A. Non-alcoholic fatty liver disease in HIV-positive patients predisposes for acute-on-chronic liver failure: two cases. Eur J Gastroenterol Hepatol 2006;18:101–105
Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005;122:669–682
Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2012. (in press)
Makiura N, Ojima M, Kou Y, Furuta N, Okahashi N, Shizukuishi S, et al. Relationship of Porphyromonas gingivalis with glycemic level in patients with type 2 diabetes following periodontal treatment. Oral Microbiol Immunol 2008;23:348–351
Xie N, Cui Y, Yin YN, Zhao X, Yang JW, Wang ZG, et al. Effects of two Lactobacillus strains on lipid metabolism and intestinal microflora in rats fed a high-cholesterol diet. BMC Complement Altern Med 2011;11:53
Velayudham A, Dolganiuc A, Ellis M, Petrasek J, Kodys K, Mandrekar P, et al. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology 2009;49:989–997
Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol 2010;28:321–342
Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology 2012;143:1158–1172
Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta 2010;1799:101–113
Yanai H, Ban T, Taniguchi T. High-mobility group box family of proteins: ligand and sensor for innate immunity. Trends Immunol 2012;33:633–640
Bianchi ME. HMGB1 loves company. J Leukoc Biol 2009;86:573–576
Vuppalanchi R, Chalasani N. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: selected practical issues in their evaluation and management. Hepatology 2009;49:306–317
Rizki G, Arnaboldi L, Gabrielli B, Yan J, Lee GS, Ng RK, et al. Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. J Lipid Res 2006;47:2280–2290
Oosterveer MH, van Dijk TH, Tietge UJ, Boer T, Havinga R, Stellaard F, et al. High fat feeding induces hepatic fatty acid elongation in mice. PLoS One 2009;4:e6066
Cohen P, Miyazaki M, Socci ND, Hagge-Greenberg A, Liedtke W, Soukas AA, et al. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science 2002;297:240–243
Abergel A, Sapin V, Dif N, Chassard C, Darcha C, Marcand-Sauvant J, et al. Growth arrest and decrease of alpha-SMA and type I collagen expression by palmitic acid in the rat hepatic stellate cell line PAV-1. Dig Dis Sci 2006;51:986–995
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010;328:228–231
Frances R, Zapater P, Gonzalez-Navajas JM, Munoz C, Cano R, Moreu R, et al. Bacterial DNA in patients with cirrhosis and noninfected ascites mimics the soluble immune response established in patients with spontaneous bacterial peritonitis. Hepatology 2008;47:978–985
Guarner C, Gonzalez-Navajas JM, Sanchez E, Soriando G, Frances R, Chiva M, et al. The detection of bacterial DNA in blood of rats with CCl4-induced cirrhosis with ascites represents episodes of bacterial translocation. Hepatology 2006;44:633–639
Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest 2009;119:305–314
Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med 2012;52:59–69
Feve B, Bastard JP. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 2009;5:305–311
Netea MG, Joosten LA, Lewis E, Jensen DR, Voshol PJ, Kullberg BJ, et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med 2006;12:650–656
Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 2007;56:16–23
Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 2011;12:408–415
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011;12:222–230
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012;36:401–414
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003;112:1796–1808
Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology 2012;142(711–725):e6
Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 2007;117:2621–2637
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993;259:87–91
Moschen AR, Molnar C, Geiger S, Graziadei I, Ebenbichler CF, Weiss H. Anti-inflammatory effects of excessive weight loss: potent suppression of adipose interleukin 6 and tumour necrosis factor alpha expression. Gut 2010;59:1259–1264
Senn JJ, Klover PJ, Nowak IA, Zimmers TA, Koniaris LG, Furlanetto RW, et al. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J Biol Chem 2003;278:13740–13746
Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Umemura A, et al. Blockade of IL-6 signaling exacerbates liver injury and suppresses antiapoptotic gene expression in methionine choline-deficient diet-fed db/db mice. Lab Invest 2011;91:609–618
Matthews VB, Allen TL, Risis S, Chan MH, Henstridge DC, Watson N, et al. Interleukin-6-deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia 2010;53:2431–2441
Yamaguchi K, Itoh Y, Yokomizo C, Nishimura T, Niimi T, Fujii H, et al. Blockade of interleukin-6 signaling enhances hepatic steatosis but improves liver injury in methionine choline-deficient diet-fed mice. Lab Invest 2010;90:1169–1178
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.