
Abstract
Aims/hypothesis
Inflammation contributes to both insulin resistance and pancreatic beta cell failure in human type 2 diabetes. Toll-like receptors (TLRs) are highly conserved pattern recognition receptors that coordinate the innate inflammatory response to numerous substances, including NEFAs. Here we investigated a potential contribution of TLR2 to the metabolic dysregulation induced by high-fat diet (HFD) feeding in mice.
Methods
Male and female littermate Tlr2 +/+ and Tlr2 −/− mice were analysed with respect to glucose tolerance, insulin sensitivity, insulin secretion and energy metabolism on chow and HFD. Adipose, liver, muscle and islet pathology and inflammation were examined using molecular approaches. Macrophages and dendritic immune cells, in addition to pancreatic islets were investigated in vitro with respect to NEFA-induced cytokine production.
Results
While not showing any differences in glucose homeostasis on chow diet, both male and female Tlr2 −/− mice were protected from the adverse effects of HFD compared with Tlr2 +/+ littermate controls. Female Tlr2 −/− mice showed pronounced improvements in glucose tolerance, insulin sensitivity, and insulin secretion following 20 weeks of HFD feeding. These effects were associated with an increased capacity of Tlr2 −/− mice to preferentially burn fat, combined with reduced tissue inflammation. Bone-marrow-derived dendritic cells and pancreatic islets from Tlr2 −/− mice did not increase IL-1β expression in response to a NEFA mixture, whereas Tlr2 +/+ control tissues did.
Conclusion/interpretation
These data suggest that TLR2 is a molecular link between increased dietary lipid intake and the regulation of glucose homeostasis, via regulation of energy substrate utilisation and tissue inflammation.
Similar content being viewed by others
Introduction
Type 2 diabetes develops as a result of a combination of both insulin resistance and pancreatic beta cell failure, resulting in hyperglycaemia. Chronic activation of the innate immune system is associated with type 2 diabetes, and evidence now suggests that both insulin resistance and beta cell failure are regulated by this inflammatory state in humans [1–3]. Indeed, clinical data have shown improved insulin resistance after treatment of type 2 diabetes patients with salicylate (via inhibition of nuclear factor κB [NF-κB]) [4, 5], and improved beta cell insulin secretion in patients after treatment with the IL-1 receptor antagonist (IL-1Ra) [6].
Local tissue inflammation is a characteristic of the human pathology of insulin resistance in obesity and beta cell failure in type 2 diabetes [1, 2, 7]. Animal models of type 2 diabetes also show increased expression of inflammatory markers specific for cytokines, chemokines and immune cells in both insulin-responsive tissues (liver, muscle, adipose tissue) and pancreatic islets [3, 8–11]. This local tissue inflammation has now been causally linked to insulin resistance and beta cell function in a number of genetic knockout models and therapeutic intervention studies [3, 11]. Genetic ablation studies in the high-fat diet (HFD) model have recently shown that pro-inflammatory CD11c+ cells are responsible for this inflammatory response in insulin sensitive tissues, impacting on insulin sensitivity [12]. However, the upstream/cell surface molecular mechanisms responsible for triggering increased tissue inflammation in obesity and type 2 diabetes are still not known.
Toll-like receptors (TLRs) are expressed in multiple tissues and are integral to mounting an immediate defence against the presence of pathogens [13, 14]. Upon binding to their cognate ligands, TLRs recruit intracellular signalling molecules (e.g. MYD88), leading to the activation of NF-κB and the secretion of proinflammatory cytokines and chemokines. Intriguingly, both TLR2 (which heterodimers with TLR1, or TLR6) and TLR4 recognise lipid-based structures; classically bacterial lipopeptides and lipopolysaccharide, respectively. Recent studies have shown that NEFAs, including palmitate and oleate, can activate both TLR2 and TLR4 signalling to induce proinflammatory cytokine production in various tissues, leading to an impairment of tissue-specific effects [15–19]. In addition, both Tlr2 −/− and Tlr4 −/− mice show improvements in the metabolic syndrome associated with HFD-induced obesity [17, 20–22]. Despite this, the mechanism underlying resistance to HFD-induced metabolic dysregulation in Tlr2 −/− mice is unclear.
Here we show that TLR2 signalling contributes to the adverse effects of HFD feeding in mice, impacting on insulin resistance, beta cell insulin secretion, energy substrate utilisation and tissue inflammation. These data suggest that TLR2 is a molecular link between increased dietary lipid intake and the regulation of glucose homeostasis.
Methods
Please see the Electronic supplementary material (ESM) for further detailed methods.
Materials and animals
Tlr2 −/− mice backcrossed on C57BL/6J for four to five generations were from the Jackson Laboratory (Strain B6.129-Tlr2 tm1Kir/J) (Bar Harbor, ME, USA) and bred in house with C57BL/6J mice to generate all Tlr2 +/+ and Tlr2 −/− sex- and age-matched littermate mice used for in vitro and in vivo studies.
Animals were housed under specific-pathogen-free conditions at the Institute of Labortierkunde, Vetsuisse faculty of the University of Zurich (Zurich, Switzerland). Experiments were performed according to Swiss veterinary law and institutional guidelines. The committee for animal welfare at the Katholieke Universiteit Leuven approved all tissue isolation protocols from mice used for Tlr2 tissue expression profiling.
Tissue expression of Tlr2
Tissue expression analysis using Affymetrix gene arrays was performed as previously described [23].
Metabolic studies
Animals were fed a high-fat diet (HFD; Diet D12331) manufactured by Research Diets (New Brunswick, NJ, USA) as previously described [10, 23]. For glucose-tolerance testing, mice were injected i.p. with 2 mg/g body weight glucose (IPGTT) and insulin was measured by ELISA (Mercodia, Uppsala, Sweden). Insulin-tolerance testing (ITT) was performed by injecting female mice i.p. with 0.85 U/kg human insulin (Novo Nordisk, Denmark), and male mice i.p. with 1.0 U/kg insulin, as previously described [23].
Isolated adipocyte experiments
Adipocyte isolation and glucose incorporation were performed as described previously [24]. Aliquots of all adipocyte fractions were used to determine mean cell diameters, as previously described [25]. At least 100 adipocytes per fraction of four independent experiments were analysed.
Indirect calorimetry and physical activity
Energy expenditure and respiratory quotient (RQ) were measured for 48 h (two dark and light phases) by using two open-circuit calorimetry systems (Integra system, AccuScan Instruments, Columbus OH, USA). Physical activity was measured by telemetry every 5 min using Dataquest A.R.T. 3.1 software.
Cytokines and chemokines
Plasma samples were assayed using mouse Luminex kits (Millipore, Billerica, MA, USA) as previously described [11].
Glucose clamp studies
After 5 h fast, hyperinsulinaemic–euglycaemic clamps were performed in freely moving mice as previously described [24].
Total liver lipid extraction
Total liver lipids were determined as recently described [24].
RNA extraction and real-time PCR
Total tissue RNA was prepared as described [11] from adipose tissue, liver, muscle, isolated islets, bone-marrow-derived macrophages (BMDMs), and bone-marrow-derived dendritic cells (BMDCs) according to the manufacturer’s instructions (Qiagen, Hombrechtikon, Switzerland). Commercially available mouse primers (Applied Biosystems, CA, USA) were used. For ex vivo detection of islet inflammatory genes after HFD feeding, islets were initially allowed to recover for 4 h in suspension before extraction of total RNA.
NEFA preparation
NEFAs were prepared using endotoxin-free BSA as recently described [26].
Immunohistochemistry
Mouse liver and pancreatic cryosections were incubated with an anti-F4/80 primary Ab (clone CI:A3-1, BMA Biomedicals, Switzerland), or isotype control rat IgG2b (AbD Serotec) and counterstained with haematoxylin and eosin. F4/80 was visualised using goat anti-rat Ab (112-005-167) (Jackson ImmunoResearch, Newmarket, UK) and a donkey anti-goat Ab conjugated to HRP (Jackson 705-035-147). Images were captured on an Axioplan 2 imaging system (Zeiss, Feldbach, Switzerland) and F4/80 positive area/islet positive area was quantified using Image J software (NIH). For liver sections, three random fields of view were analysed in two sections per animal separated by 200 μm (an average of 3,600 ± 100 cells per animal). For the pancreas, all islets in four sections separated by 200 μm were analysed per animal (an average of 47 ± 7 islets per animal).
Pancreatic islet isolation and BMDM/BMDC preparation
Mouse islets were isolated by collagenase digestion, followed by Histopaque gradient centrifugation and cultured as previously described [10]. For in vitro islet experiments, islets were plated on extracellular matrix (Novamed, Jerusalem, Israel). Islets were allowed to adhere and spread on the extracellular matrix dishes for 48 h before initiation of experiments.
BMDMs and BMDCs were prepared as previously described [27, 28]. Briefly, bone marrow was isolated from femurs and tibias of 8- to 10-week-old female mice and cultured in macrophage medium (50% DMEM) supplemented with 20% horse serum and 30% L929 conditioned medium (a source of macrophage colony-stimulating factor). After 7 days cells were plated into 24-well plates (5 × 106 cells/well) for experiments and extraction of RNA. To generate BMDCs, bone marrow prepared from femurs and tibias was cultured in non-tissue-culture-treated Petri dishes with RPMI medium supplemented with 2-mercaptoethanol (50 μM), 10% FCS and 200 U/ml granulocyte macrophage colony-stimulating factor, as previously described [28]. After 7 days, non-adherent cells were plated into 24-well plates (5 × 106 cells/well) for experiments and extraction of RNA. The specificity of the two differentiation protocols to generate BMDMs (CD45+F4/80+CD11b+Cd11c−) and BMDCs (CD45+CD11b+CD11c+) was verified by FACS analysis. BMDM and BMDC were resuspended in FACS buffer (2% FCS, 10 mmol/l EDTA in PBS) and incubated with anti-mouse CD16/32 (Fc-block, BD Biosciences, Heidelberg, Germany) for 5 min, then stained with anti-mouse CD45-biotin/SAV-APC/Cy7 (BD Biosciences), F4/80-FITC (BD Biosciences), CD11c-PE (Invitrogen) and CD11b-APC (BD Biosciences) for 20 min. The cells were analysed with a Partec FloMax flow cytometer (Munster, Germany). 7-AAD (7-amino-actinomycin D, Invitrogen) was used to exclude nonviable cells in flow cytometric analysis. Cells were treated for 6 h with BSA or a 0.5 mmol/l palmitate:oleate (2:1 molar ratio) preparation before RNA extraction.
Statistics
Data are expressed as means ± S.E. with the number of individual experiments presented in the figure legends. All data were analysed using the nonlinear regression analysis programme PRISM (GraphPad, CA, USA), and significance was tested using Student’s t test and analysis of variance (ANOVA) with Dunnett’s or Bonferroni’s post-hoc test for multiple comparison analysis. Significance was set at p < 0.05.
Results
Tlr2 tissue expression and regulation by HFD feeding
We initially profiled mouse tissue expression of Tlr2 mRNA (Fig. 1a). These data indicated that Tlr2 has a broad tissue expression profile, which includes not only organs classically described as being heavily populated by immune cells (spleen), but also insulin-sensitive tissues and the pancreatic islet. Based on this information, we analysed mRNA expression of Tlr1, -2, -4 and -6 in response to 20 weeks of HFD feeding in female B6 mice in perigonadal adipose tissue, skeletal quadriceps muscle, liver tissue, pancreatic islets and circulating leucocytes (Fig. 1b). Indeed, HFD feeding resulted in increased mRNA expression of some of Tlr1, -2, -4 and -6 in all these tissues.

HFD regulation of tissue Tlr1, -2, -4 and -6 mRNA expression. a Relative tissue expression profile of mouse Tlr2 mRNA based on 430 2.0 mouse Affymetrix gene arrays (n = 3 for all tissues except islet, bone marrow and pituitary, where n = 4). b Female C57BL/6J littermate mice were fed an HFD for 20 weeks starting at 10–12 weeks of age. Perigonadal adipose tissue, quadriceps skeletal muscle, liver tissue, isolated pancreatic islets and circulating leucocytes were isolated and analysed for Tlr expression by quantitative PCR (chow n = 7, HFD n = 8). White bars, Tlr1; light grey bars, Tlr2; black bars, Tlr6; dark grey bars, Tlr4. *p < 0.05, as tested by Student’s t test compared with chow-fed age-matched control (1.0 ± SEM)
Tlr2 −/− mice are resistant to the adverse effects of HFD feeding
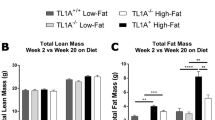
Based on published data [15, 18, 19] and the upregulation of TLR expression by HFD, we reasoned that mice lacking Tlr2 (which are unable to signal via TLR1/2, or TLR2/6 heterodimers) might show differences in susceptibility to HFD-induced glucose intolerance. Tlr2 −/− mice were confirmed to be unresponsive to the synthetic TLR2 ligand, Pam2CSK4, in vivo (ESM Fig. 1). Further, neither female nor male Tlr2 −/− mice showed any difference in glucose tolerance or insulin sensitivity at 10–12 weeks of age when compared with littermate controls (Tlr2 +/+) (ESM Fig. 2a–d). Thereafter, female and male Tlr2 +/+ and Tlr2 −/− mice were put on HFD for 20 weeks, until 32 weeks of age. HFD-induced weight gain was not different between genotypes in both female (Fig. 2a) and male mice (ESM Fig. 3a). After 12 weeks of HFD, female Tlr2 −/− mice already showed improved glucose tolerance, insulin sensitivity and beta cell insulin secretion in response to a glucose challenge (ESM Fig. 4). At this age male Tlr2 −/− mice were also more insulin sensitive compared with Tlr2 +/+ littermates, while showing no differences in glucose tolerance or insulin secretion (ESM Fig. 3b–d). After 20 weeks of HFD, female Tlr2 −/− mice (aged 32 weeks) showed markedly improved glucose tolerance, insulin sensitivity and beta cell insulin secretion in response to a glucose challenge (Fig. 2b–e). Supporting improved peripheral insulin sensitivity, fasting circulating insulin levels were reduced both at 12 and 20 weeks of HFD feeding in female Tlr2 −/− mice compared with Tlr2 +/+ controls (Fig. 2d and ESM Fig. 4c). Age-matched 32-week-old chow-fed female Tlr2 +/+ and Tlr2 −/− mice showed no differences in glucose homeostasis (ESM Fig. 2e–f). Based on these data, we focused on female Tlr2 −/− mice for the remainder of the study.

Tlr2 −/− mice are resistant to the adverse effects of HFD feeding. a Female Tlr2 +/+ and Tlr2 −/− littermate mice were fed an HFD starting at 10–12 weeks of age, and body weight was monitored (n = 7 for chow groups, n = 31 for HFD Tlr2 +/+ and n = 32 for HFD Tlr2 −/−). b An IPGTT (2 g/kg glucose) was performed after 20 weeks of HFD feeding (32-week-old mice; n = 21 for HFD Tlr2 +/+ and n = 22 for HFD Tlr2 −/−). c An ITT (0.85 U/kg insulin) was performed after 20 weeks of HFD feeding (32-week-old mice; n = 8 for HFD Tlr2 +/+ and n = 7 for HFD Tlr2 −/−). d, e Insulin secretion during an IPGTT performed after 20 weeks of HFD feeding (32-week-old mice; n = 8 for HFD Tlr2 +/+ and n = 7 for HFD Tlr2 −/−). Black triangles, chow Tlr2 +/+; white triangles, chow Tlr2 −/−; black squares and unbroken line, HFD Tlr2 +/+; white squares and broken line, HFD Tlr2 −/− .*p < 0.05 and **p < 0.01, as tested by Student’s t test compared with Tlr2 +/+ control
We went on to investigate insulin target tissue inflammation and insulin sensitivity in addition to examining islet inflammation and islet insulin secretion. HFD-fed female Tlr2 −/− mice had reduced perigonadal fat pad weight compared with wild-type animals, in addition to having smaller adipocytes that displayed improved insulin-stimulated glucose uptake (Fig. 3a–d). This was consistent with increased GLUT4 protein levels in whole adipose tissue of Tlr2 −/− mice (ESM Fig. 5). Interestingly, HFD-induced adipose tissue cytokine mRNA expression was similar between genotypes, with only the chemokine, Mcp-1, and the macrophage marker, Cd68, showing reductions in mRNA in Tlr2 −/− mice on HFD (Fig. 3e–g). Further, mRNA expression of Cd36, the fatty acid translocase known to associate with TLR2 [29], was unchanged in adipose tissue (Fig. 3h).

Adipose tissue in female Tlr2 +/+ and Tlr2 −/− mice after 20 weeks of HFD feeding. a Perigonadal fat pad weight as % body weight in chow- and HFD-fed female Tlr2 +/+ and Tlr2 −/− mice (n = 13 chow Tlr2 +/+, n = 15 chow Tlr2 −/−, n = 21 HFD Tlr2 +/+, n = 23 HFD Tlr2 −/−).*p < 0.05 relative to Tlr2 +/+ chow control and †p < 0.05 relative to Tlr2 +/+ HFD, as tested by ANOVA with Newman–Keuls post hoc test. b Insulin (INS)-stimulated glucose incorporation in isolated perigonadal adipocytes (n = 4 per group). *p < 0.05, as tested by Student’s t test. c Perigonadal adipocyte size distribution (n = 4 per group). d Representative image of perigonadal adipose tissue. e, f, g Relative cytokine, chemokine and macrophage/dendritic cell marker mRNA expression in perigonadal adipose tissue compared with chow-fed Tlr2 +/+ animals (n = 8 for HFD Tlr2 +/+ and n = 7 for HFD Tlr2 −/−). h Cd36 mRNA expression compared with chow-fed Tlr2 +/+ animals (n = 8 for HFD Tlr2 +/+ and n = 7 for HFD Tlr2 −/−). Black bars, Tlr2 +/+; white bars, Tlr2 −/−. *p < 0.05 as tested by Student’s t test
In agreement with reduced adiposity, circulating leptin, MCP-1 and TNF-α levels were significantly reduced in female Tlr2 −/− animals after 20 weeks of HFD feeding (ESM Table 1). However, we did not detect differences in circulating resistin, IL-6, cholesterol, NEFAs or ketones. In support of improved insulin sensitivity, circulating triacylglycerol levels were significantly reduced in Tlr2 −/− mice on HFD (ESM Table 1).
Given the reduced adiposity of female Tlr2 −/− mice on HFD, we investigated energy homeostasis in these animals during HFD feeding. Despite reduced adiposity, Tlr2 −/− mice tended to consume more calories (10% more) (Fig. 4a). At the same time, we observed a parallel 10% increase in total energy expenditure, an effect that was more pronounced during dark phases (Fig. 4b,c). Increased total energy expenditure was not due to increased physical activity (Fig. 4d,e). Interestingly, Tlr2 −/− mice preferentially used lipids as an energy source as indicated by a significantly reduced RQ, especially during the light phases (Fig. 4f,g). This was consistent with increased skeletal muscle Acox1 and Mcad mRNA expression (enzymes involved in beta-oxidation) in Tlr2 −/− mice, while other genes involved in fatty acid uptake (Cd36) and inflammation were unchanged (Fig. 4h–k). However, at this time point we did not detect differences in skeletal muscle triacylglycerols or glycogen content between genotypes (ESM Fig. 6). In summary, female Tlr2 −/− mice show reduced adiposity on HFD compared with Tlr2 +/+ littermates. This was consistent with a reduced RQ, and increased mRNA expression of some genes involved in beta-oxidation in skeletal muscle. Overall energy intake and expenditure were balanced, possibly explaining why body weight did not differ between groups. Finally, adipose tissue and skeletal muscle inflammation due to HFD feeding was not strongly impacted by lack of TLR2 signalling.

Indirect calorimetry and muscle in female Tlr2 +/+ and Tlr2 −/− mice after 20 weeks of HFD feeding. a Food intake monitored over four consecutive days as normalised to body weight († p = 0.24). Energy expenditure calculated continuously over 48 h (b) and total summed energy expenditure during dark and light phases (c). Time: 0 h = 18:00 h, and represents lights off; 12 h dark and light phases are separated by vertical lines. Physical activity monitored by telemetry continuously over 48 h (d) and averaged during dark and light phases (e). Respiratory quotient (RQ) calculated continuously over 48 h (f) and averaged during dark and light phases (g). n = 6 per group for all measurements. *p < 0.05 between genotypes as tested by Student’s t test. h Relative mRNA expression of genes involved in fatty acid metabolism in quadriceps muscle in female Tlr2 +/+ and Tlr2 −/− mice after 20 weeks of HFD feeding (n = 8 for HFD Tlr2 +/+ and n = 7 for HFD Tlr2 −/−). *p < 0.05 between genotypes as tested by Student’s t test, † p = 0.067. Relative cytokine (i), chemokine (j) and macrophage/dendritic (k) cell marker mRNA expression (n = 8 for HFD Tlr2 +/+ and n = 7 for HFD Tlr2 −/−). Black bars, or black squares with unbroken line, Tlr2 +/+; white bars, or white squares with broken line, Tlr2 −/−. BW, body weight; ND, not detectable
Tlr2 −/− mice are protected from liver insulin resistance, hepatosteatosis and liver inflammation on HFD
We performed hyperinsulinaemic–euglycaemic clamps to assess peripheral insulin sensitivity in female Tlr2 −/− and Tlr2 +/+ mice after HFD feeding for 20 weeks. In agreement with enhanced whole body insulin sensitivity observed during the ITT (Fig. 2c), glucose infusion rates were increased in Tlr2 −/− mice concomitant with insulin-stimulated glucose turnover (Fig. 5a–c and ESM Fig. 7). Further, liver insulin sensitivity was preserved in Tlr2 −/− mice, as demonstrated by a significant reduction in hepatic glucose production during the hyperinsulinaemic clamp (Fig. 5d). Lipid content was reduced in livers of Tlr2 −/− mice, suggesting protection against the development of hepatosteatosis under HFD (Fig. 5f). Consistent with this, mRNA of the fatty acid translocase, Cd36, was strongly reduced in Tlr2 −/− mice, whereas genes involved in beta-oxidation (Acox1, Cpt1a), oxidative metabolism (Pgc1α [also known as Ppargc1a]) or lipogenesis (Fas) were unchanged relative to controls (Fig. 5g). Indeed, Tlr2 −/− mice were protected from HFD-induced liver Il-1β (also known as Il1b), MCP-1 and Cd68 mRNA expression compared with Tlr2 +/+ controls (Fig. 5h–j). However, other HFD-induced cytokines (IL-6 [also known as Il6], Tnf-α [also known as Tnf], IL-10 [also known as Il10]) and chemokines (MIP-1α [also known as Clcl3], Kc [also known as Cxcl1]) were unchanged between genotypes (Fig. 5h–j). Consistent with mRNA data, numbers of liver F4/80+ cells (a macrophage marker) were unchanged between genotypes on HFD (13.7 ± 1.1% in Tlr2 +/+ [n = 8] vs 13.5 ± 1.3% F4/80+ cells/total cells in Tlr2 −/− [n = 7]). Thus, female Tlr2 −/− mice are protected from liver insulin resistance, hepatosteatosis and some markers of inflammation on HFD.

Liver in female Tlr2 +/+ and Tlr2 −/− mice after 20 weeks of HFD feeding. Hyperinsulinaemic–euglycaemic clamps were performed after 20 weeks of HFD feeding in female Tlr2 +/+ and Tlr2 −/− littermates. Blood glucose during clamping (a) and glucose infusion rate (b) (n = 4). c Insulin-stimulated glucose turnover in female Tlr2 +/+ and Tlr2 −/− littermates (n = 4). d Suppression of hepatic glucose production (HGP) during clamp (n = 4). Representative liver stained with haematoxylin and eosin (e) and total liver lipids (f) (n = 7 for HFD Tlr2 +/+ and n = 8 for HFD Tlr2 −/−). g Relative mRNA expression of genes involved in fatty acid transport (Cd36), beta-oxidation (Acox1, Cpt1a, Pgc1α) and lipogenesis (Fas). Relative cytokine (f), chemokine (i) and macrophage/dendritic (j) cell marker mRNA expression in liver isolated from female HFD Tlr2 +/+ and Tlr2 −/− littermates compared with chow Tlr2 +/+ controls (n = 8 for HFD Tlr2 +/+ and n = 7 for HFD Tlr2 −/−). Black bars, Tlr2 +/+; white bars, Tlr2 −/−. *p < 0.05 and **p < 0.01 as tested by Student’s t test compared with Tlr2 +/+ control
Tlr2 −/− mice are protected from impaired beta cell insulin secretion and islet inflammation on HFD
We isolated pancreatic islets after 20 weeks of HFD feeding from female littermate Tlr2 +/+ and Tlr2 −/− mice. Islets from Tlr2 −/− mice responded to an increased glucose concentration by increasing insulin secretion while Tlr2 +/+ islets showed no response to glucose (Fig. 6a). There was no difference in islet insulin content between genotypes (not shown). There was also no difference in glucose-stimulated insulin secretion between age-matched 32-week-old chow-fed Tlr2 +/+ and Tlr2 −/− mice (not shown). This supports our in vivo findings (Fig. 2d) that female Tlr2 −/− mice on HFD show improved beta cell insulin secretion compared with littermate Tlr2 +/+ controls.

Pancreatic islet in female Tlr2 +/+ and Tlr2 −/− mice after 20 weeks of HFD feeding. a Pancreatic islets were isolated after 20 weeks of HFD feeding in Tlr2 +/+ and Tlr2 −/− littermates and static glucose-stimulated insulin secretion was performed using 2.8 mmol/l glucose and 16.7 mmol/l glucose (1 h each, 15 islets/well) and normalised for total islet insulin content. Content was unchanged between genotypes (n = 8 for HFD Tlr2 +/+ and n = 7 for HFD Tlr2 −/−). *p < 0.05 as tested by Student’s t test. b Pancreatic islets isolated from chow- and HFD-fed Tlr2 +/+ mice were analysed for Il-1β, Cd68, F4/80 and Cd11c mRNA expression by quantitative PCR. There is a strong positive correlation between islet Il-1β mRNA and islet Cd68, F4/80 or Cd11c mRNA under HFD conditions (for Cd68 r 2 = 0.91, p = 0.0003, black squares; for F4/80 r 2 = 0.92, p = 0.0006, black circles; for Cd11c r 2 = 0.60, p = 0.02, white triangles; n = 8). Relative cytokine (c), chemokine (d) and macrophage (e) marker mRNA expression in pancreatic islets isolated from female HFD-fed Tlr2 +/+ and Tlr2 −/− littermates compared with chow Tlr2 +/+ controls (n = 8 for HFD Tlr2 +/+ and n = 7 for HFD Tlr2 −/−). f Relative islet Cd36 mRNA expression in HFD-fed Tlr2 +/+ and Tlr2 −/− mice (n = 8 for HFD Tlr2 +/+ and n = 7 for HFD Tlr2 −/−). Black bars, Tlr2 +/+; white bars, Tlr2 −/−. *p < 0.05 as tested by Student’s t test compared with Tlr2 +/+ control
Next, we analysed islet inflammation due to HFD feeding in female Tlr2 +/+ and Tlr2 −/− mice. Increases in islet Il-1β mRNA owing to HFD positively correlated with increased macrophage/dendritic cell marker expression: Cd68, F4/80 (also known as Emr1) and Cd11c (also known as Itgax) (Fig. 6b) (for Cd68 r 2 = 0.91, p = 0.0003; for F4/80 r 2 = 0.92, p = 0.0006; for Cd11c r 2 = 0.60, p = 0.02; n = 8). By contrast, islets from Tlr2 −/− mice on HFD were protected from increased Il-1β, Cd68 and F4/80 mRNA (Fig. 6c–e). Furthermore, Il-6, Tnf-α, Mcp-1 and Kc mRNA was also significantly reduced in Tlr2 −/− vs Tlr2 +/+ islets on HFD, with no effect on islet Cd36 expression (Fig. 6c–f). No reductions in these inflammatory markers were observed in islets isolated from sex- and age-matched chow-fed Tlr2 −/− compared with Tlr2 +/+ mice (not shown). In contrast to F4/80 mRNA data, and similar to the liver, the amount of islet F4/80+ cells were unchanged in HFD Tlr2 −/− mice (percentage area of F4/80+ cells of total islet area: 1.9 ± 0.4% in Tlr2 +/+ [n = 4] vs 1.8 ± 0.4% in Tlr2 −/− [n = 4]), possibly suggesting an altered activation status of liver and islet macrophages/dendritic cells in Tlr2 −/− vs Tlr2 +/+ mice on HFD.
Finally, analysis of total peripheral leucocytes did not show differences in cytokine mRNA (Il-1β, Tnf-α) or macrophage marker expression (Cd68, F4/80) between Tlr2 genotypes on HFD (data not shown), supporting the tissue specificity of the inflammatory response.
Impaired inflammatory response of BMDCs and islets to NEFAs in Tlr2 −/− mice
Numbers of F4/80+ cells in the liver and islets were unchanged, despite reductions in Il-1β mRNA in these tissues in Tlr2 −/− mice. Thus, we hypothesised that tissue immune cells (macrophages and/or dendritic cells) from Tlr2 −/− mice and/or parenchymal tissue itself may display a reduced ability to mount an inflammatory response to certain ligands present during HFD feeding. BMDMs and BMDCs from Tlr2 +/+ and Tlr2 −/− mice were cultured for 6 h in the presence of endotoxin-free BSA control or a mix of 0.5 mM palmitate:oleate (2:1). Indeed, BMDCs from Tlr2 −/− mice displayed an attenuated Il-1β mRNA response to the NEFAs, whereas BMDMs did not (Fig. 7a,b). Both BMDMs and BMDCs from Tlr2 −/− mice were unable to respond to the Tlr2 ligand Pam2CSK4 (ESM Fig. 8). As a representative tissue, we also investigated the response of pancreatic islets from Tlr2 +/+ and Tlr2 −/− mice to NEFAs. Palmitate (0.1 mmol/l) induced Il-1β mRNA 2.5-fold in Tlr2 +/+ islets, whereas this effect was absent in islets from mice lacking Tlr2 (Tlr2 −/−) (Fig. 7c). This effect was specific for the NEFA palmitate, as oleate (18:1) alone was unable to increase islet Il-1β mRNA expression (not shown). The IL-1β-induced Il-1β auto-amplification system [30] was unchanged in Tlr2 −/− islets vs Tlr2 +/+ controls, indicating no developmental defect in Tlr2 −/− islets with respect to the induction of Il-1β mRNA expression (Fig. 7d).

Impaired inflammatory response to NEFAs in BMDCs and islets of Tlr2 −/− mice. BMDMs (a) and BMDCs (b) were prepared from Tlr2 +/+ and Tlr2 −/− mice as described and treated for 6 h with BSA control or palmitate:oleate (16:0:18:1, 2:1 molar ratio, 0.5 mmol/l). RNA was extracted and IL-1β mRNA is shown relative to 18S (n = 3 for Tlr2 +/+, n = 4 for Tlr2 −/−). c Regulation of islet IL-1β mRNA by 48 h treatment with 0.1 mmol/l palmitate in Tlr2 +/+ and Tlr2 −/− islets (n = 3–7). d Regulation of islet IL-1β mRNA by 48 h treatment with 0.2 ng/ml IL-1β in Tlr2 +/+ and Tlr2 −/− islets (n = 3–7). Black bars, Tlr2 +/+; white bars, Tlr2 −/−.*p < 0.05 as tested by Student’s t test compared with BSA genotype control
Discussion
The impact of inflammation on insulin resistance and beta cell dysfunction has been confirmed clinically in patients with type 2 diabetes [3, 6]. Nevertheless, the triggering mechanism responsible for induction of the inflammatory response in obesity and type 2 diabetes is not known. Recent studies have implicated TLR4 and its associated co-receptor, CD14, in HFD- and NEFA-stimulated tissue inflammation and insulin resistance [17, 21, 22, 31]. Here, we show that TLR2 signalling is also detrimental for proper glucose homeostasis under HFD conditions. Overall, TLR2 deficiency protected from HFD-induced insulin resistance and beta cell dysfunction via regulation of energy substrate utilisation and tissue inflammation.
During the preparation of this manuscript, another study on the role of genetic deletion of TLR2 during HFD feeding was published [20]. Overall, their data on improved whole body glucose homeostasis and liver lipid content are consistent with our data. Our data extends on these data, however, indicating that lack of TLR2 positively regulates energy-substrate utilisation, hepatic and adipose tissue insulin sensitivity, beta cell insulin secretion and liver and islet tissue inflammation. Furthermore, our in vitro data suggest a role for TLR2 in tissue immune cells (BMDCs) or parenchymal tissue itself (islets) in regulating the inflammatory response to elevated fatty acids during HFD feeding.
Our male and female Tlr2 −/− mice were more prone to obesity on HFD than the male Tlr2 −/− mice used in the study of Himes and Smith [20]. This may be explained by differences in diets used, or the use of littermate controls in our study, which was not explicitly stated in the other study. Indeed, we also performed our experiments on non-littermate-controlled Tlr2 +/+ and Tlr2 −/− mice initially, with data matching those of Himes and Smith in male mice (not shown). Repeating the study on littermate mice resulted in the sexually dimorphic phenotype shown here in response to HFD, and a milder resistance to obesity phenotype than published [20]. Interestingly, our data are similar to a published report on Tlr4 −/− mice [17], which also showed sexual dimorphism in their resistance to the adverse effects of HFD feeding, with males showing only a partial resistance compared with females. We cannot explain these differences, but it will be interesting to investigate the effects of sex on tissue inflammation with respect to obesity and diabetes.
A reduced RQ, together with a reduction in adiposity and liver lipid content in Tlr2 −/− animals, suggests that fatty acids are being shunted away from the liver to be increasingly oxidised in other tissues (such as skeletal muscle) in HFD Tlr2 −/− mice. This would be consistent with a diminished capacity of the liver to take up lipids due to suppressed Cd36 mRNA expression. TLR2 has been shown to associate with CD36 at the plasma membrane following receptor ligand activation, suggesting that the absence of TLR2 activation in the liver may protect from fatty acid uptake [29].
Reduced adiposity and reduced hepatosteatosis were also consistent with overall improvements in whole body and hepatic insulin sensitivity as determined by the hyperinsulinaemic–euglycaemic clamp. Thus, our data show for the first time that TLR2 regulates hepatic insulin sensitivity during HFD, extending findings that reduced TLR2 expression by antisense oligonucleotides improved insulin signalling in muscle and white adipose tissue of HFD-fed mice [32].
Beyond improved insulin sensitivity, we also observed improvements in beta cell insulin secretion in response to glucose load, both in vivo and in vitro. Thus, the overall improved glucose tolerance of HFD Tlr2 −/− mice was due to improved insulin sensitivity and beta cell insulin secretion, which correlated well with a partially attenuated tissue inflammatory response in the liver and islet.
We went on to investigate why tissue inflammation might be suppressed in Tlr2 −/− mice on HFD. Reductions in tissue inflammation may be due to reduced immune cell content of tissues, a reduced activation status of tissue immune cells or reduced inflammation of the parenchymal tissue. Despite reductions in Cd68, and/or F4/80 mRNA in adipose tissue, liver and in islets, we did not observe differences between genotypes in the number of F4/80+ cells by immunohistochemistry in the liver or islet. Thus, we hypothesised that the activation status of proinflammatory immune cells in Tlr2 −/− mice or the parenchymal tissue itself had a reduced capacity to mount an inflammatory response under HFD conditions. Pro-inflammatory CD11c+ cells have been causally linked to tissue inflammation and the induction of insulin resistance in response to HFD in the B6 mouse [12]. Our data showing increased Cd11c mRNA expression during HFD feeding in adipose tissue, liver tissue and islets are consistent with the notion that CD11c+ cells are recruited to these tissues during obesity [19, 33]. Indeed, Tlr2 −/− BMDCs (CD45+CD11b+Cd11c+) showed a refractory response to NEFA-induced Il-1β mRNA in vitro, whereas BMDMs (CD45+F4/80+CD11b+Cd11c−) did not. The pancreatic islet was also tested as a representative tissue with respect to NEFA induction of Il-1β mRNA. Consistent with the effects seen in BMDCs and our in vivo effects, Tlr2 −/− islets were also refractory to palmitate-induced IL-1β mRNA. We previously found no effect of oleate on mouse islet Il-1β mRNA (not shown) [26]. These data suggest that Tlr2 −/− CD11c+ cells are resistant to the effects of NEFAs present in HFD, possibly resulting in the reduced tissue inflammation seen in the liver and islets of HFD Tlr2 −/− mice. However, given the broad tissue expression of Tlr2 mRNA, we cannot conclude whether the observed reductions in tissue inflammation in vivo are due to an immune cell, or parenchymal cell, origin.
Recent data also suggest a role for TLR2 in human type 2 diabetes. One study has shown increased production of TLR2 protein on circulating monocytes following feeding of a high-fat high-carbohydrate meal in healthy lean human participants [34]. Evidence for increased TLR2 protein levels on monocytes and increased circulating TLR2 ligands in recently diagnosed type 2 diabetes patients has also been reported [35]. Monocytes from these type 2 diabetes individuals showed increased proinflammatory cytokine secretion following TLR2 stimulation [35]. Further, adipose tissue from participants with obesity and type 2 diabetes compared with controls also showed increased protein levels of TLR2 [36]. Whether TLR2 activation also contributes to insulin resistance and beta cell dysfunction in humans with type 2 diabetes awaits further investigation.
In summary, these data show that deficiency in TLR2-mediated signalling has a positive impact on glucose homeostasis, insulin sensitivity, insulin secretion and energy-substrate utilisation during HFD feeding. The overall improved metabolic phenotype of Tlr2 −/− mice on HFD is likely to be due to both reduced fat accumulation and reduced tissue inflammation, impacting on tissue-specific functions to improve both insulin sensitivity and secretion.
Abbreviations
- BMDC:
-
Bone-marrow-derived dendritic cell
- BMDM:
-
Bone-marrow-derived macrophage
- GSIS:
-
Glucose-stimulated insulin secretion
- HFD:
-
High-fat diet
- IL-1Ra:
-
IL-1 receptor antagonist
- IPGTT:
-
I.p. glucose tolerance test
- ITT:
-
Insulin tolerance test
- NF-κB:
-
Nuclear factor κB
- TLR:
-
Toll-like receptor
References
Donath MY, Schumann DM, Faulenbach M et al (2008) Islet inflammation in type 2 diabetes: from metabolic stress to therapy. Diabetes Care 31(Suppl 2):S161–S164
Wellen KE, Hotamisligil GS (2005) Inflammation, stress, and diabetes. J Clin Invest 115:1111–1119
Shoelson SE, Goldfine AB (2009) Getting away from glucose: fanning the flames of obesity-induced inflammation. Nat Med 15:373–374
Hundal RS, Petersen KF, Mayerson AB et al (2002) Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest 109:1321–1326
Yuan M, Konstantopoulos N, Lee J et al (2001) Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 293:1673–1677
Larsen CM, Faulenbach M, Vaag A et al (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356:1517–1526
Ehses JA, Ellingsgaard H, Boni-Schnetzler M, Donath MY (2009) Pancreatic islet inflammation in type 2 diabetes: from alpha and beta cell compensation to dysfunction. Arch Physiol Biochem 115:240–247
Chiang SH, Bazuine M, Lumeng CN et al (2009) The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 138:961–975
Hong EG, Ko HJ, Cho YR et al (2009) Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 58:2525–2535
Ehses JA, Perren A, Eppler E et al (2007) Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56:2356–2370
Ehses JA, Lacraz G, Giroix MH et al (2009) IL-1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proc Natl Acad Sci U S A 106:13998–14003
Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG (2008) Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab 8:301–309
Medzhitov R (2001) Toll-like receptors and innate immunity. Nat Rev Immunol 1:135–145
Takeda K, Akira S (2004) TLR signaling pathways. Semin Immunol 16:3–9
Lee JY, Zhao L, Youn HS et al (2004) Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem 279:16971–16979
Lee JY, Sohn KH, Rhee SH, Hwang D (2001) Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem 276:16683–16689
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116:3015–3025
Senn JJ (2006) Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J Biol Chem 281:26865–26875
Nguyen MT, Favelyukis S, Nguyen AK et al (2007) A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem 282:35279–35292
Himes RW, Smith CW (2010) Tlr2 is critical for diet-induced metabolic syndrome in a murine model. Faseb J 24:731–739
Poggi M, Bastelica D, Gual P et al (2007) C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 50:1267–1276
Tsukumo DM, Carvalho-Filho MA, Carvalheira JB et al (2007) Loss-of-function mutation in toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 56:1986–1998
Ellingsgaard H, Ehses JA, Hammar EB et al (2008) Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc Natl Acad Sci U S A 105:13163–13168
Wueest S, Rapold RA, Schumann DM et al (2010) Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J Clin Invest 120:191–202
Wueest S, Rapold RA, Rytka JM, Schoenle EJ, Konrad D (2009) Basal lipolysis, not the degree of insulin resistance, differentiates large from small isolated adipocytes in high-fat fed mice. Diabetologia 52:541–546
Boni-Schnetzler M, Boller S, Debray S et al (2009) Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 150:5218–5229
Vats D, Mukundan L, Odegaard JI et al (2006) Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 4:13–24
Suter T, Biollaz G, Gatto D et al (2003) The brain as an immune privileged site: dendritic cells of the central nervous system inhibit T cell activation. Eur J Immunol 33:2998–3006
Triantafilou M, Gamper FG, Haston RM et al (2006) Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem 281:31002–31011
Boni-Schnetzler M, Thorne J, Parnaud G et al (2008) Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J Clin Endocrinol Metab 93:4065–4074
Cani PD, Amar J, Iglesias MA et al (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56:1761–1772
Caricilli AM, Nascimento PH, Pauli JR et al (2008) Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J Endocrinol 199:399–406
Lumeng CN, Bodzin JL, Saltiel AR (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117:175–184
Ghanim H, Abuaysheh S, Sia CL et al (2009) Increase in plasma endotoxin concentrations and the expression of toll like receptors and suppressor of cytokine signaling-3 in mononuclear cells following a high fat high carbohydrate meal: implications for insulin resistance. Diabetes Care 32:2281–2287
Dasu MR, Devaraj S, Park S, Jialal I (2010) Increased toll-like receptor activation and tlr ligands in recently diagnosed type 2 diabetes subjects. Diabetes Care. doi:10.2337/dc09-1799
Creely SJ, McTernan PG, Kusminski CM et al (2007) Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab 292:E740–E747
Acknowledgements
We thank R.M. Zinkernagel and A.A. Navarini (University Hospital of Zurich) for the Tlr2 −/− mice. We thank M. Borsig, A. Fitsche and G. Kristiansen for technical assistance. This work was supported by grants from the Swiss National Science Foundation (M. Y. Donath, D. Konrad), the European Foundation for the Study of Diabetes (EASD/MSD to M. Y. Donath), the Juvenile Diabetes Research Foundation (M. Y. Donath), the Katholieke Universiteit Leuven (GOA 2004/11 and GOA 2008/16, to F.S.), the Gebert Rüf Stiftung (GRS-027/06 to O. Tschopp), the Swiss SystemsX.ch Initiative to the project ‘LiverX’ of the Competence Center for Systems Physiology and Metabolic Diseases (O. Tschopp and S. M. Schultze) and the University Research Priority Program ‘Integrative Human Physiology’ at the University of Zürich (J.A. Ehses and M. Y. Donath).
Duality of interest
M. Y. Donath is a consultant for Amgen, XOMA, Novartis, Merck, Solianis, and Nycomed. M. Y. Donath is listed as the inventor on a patent (WO6709) filed in 2003 for the use of an IL-1 receptor antagonist for the treatment of or prophylaxis against type 2 diabetes. The patent is owned by the University of Zurich, and M. Y. Donath has no financial interest in the patent. All other co-authors have no duality of interest to report.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. A. Ehses and D. T. Meier contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Pam2CSK4 does not induce SAA in Tlr2 −/− mice. 5 μg Pam2CSK4 or saline was injected i.p. into Tlr2 +/+ (n = 7–8) and Tlr2 −/− mice (n = 3–7) and plasma serum amyloid A was analysed by ELISA after 24 h. *p < 0.05 compared with saline control and #p < 0.05 compared with Tlr2 +/+ Pam2CSK4 as tested by ANOVA with Newman–Keuls post hoc test. Black bars, Tlr2 +/+; white bars Tlr2 −/−. SAA, serum amyloid A (PDF 32 kb)
ESM Fig. 2
Glucose homeostasis in chow-fed Tlr2 +/+ and Tlr2 −/− mice. a, b IPGTT (left, 2 g/kg glucose) and ITT (right, 0.85 U/kg insulin) in female mice at 12 weeks of age (n = 12 Tlr2 +/+, n = 10 Tlr2 −/−). c, d IPGTT (left, 2 g/kg glucose) and ITT (right, 1.0 U/kg insulin) in male mice at 12 weeks of age (n = 6 Tlr2 +/+, n = 4 Tlr2 −/−). e, f IPGTT (left, 2 g/kg glucose) and ITT (right, 0.85 U/kg insulin) in female mice at 32 weeks of age (IPGTT: n = 4 Tlr2 +/+, n = 4 Tlr2 −/−; ITT: n = 10 Tlr2 +/+, n = 7 Tlr2 −/−). Black triangles, unbroken line: Tlr2 +/+; white triangles, broken line: Tlr2 −/− (PDF 110 kb)
ESM Fig. 3
Glucose homeostasis in male Tlr2 +/+ and Tlr2 −/− mice fed an HFD. a Male Tlr2 +/+ and Tlr2 −/− littermate controls were fed an HFD starting at 10–12 weeks of age and body weight was monitored. b An IPGTT (2 g/kg glucose) was performed after 12 weeks of HFD feeding. c An ITT (1.0 U/kg insulin) was performed after 12 weeks of HFD feeding. d Insulin secretion during an IPGTT performed after 12 weeks of HFD feeding (n = 10 Tlr2 +/+, n = 12 Tlr2 −/− for all data). Black squares, unbroken line, Tlr2 +/+; white squares, broken line, Tlr2 −/−. *p < 0.05 as tested by Student’s t test compared with Tlr2 +/+ control (PDF 68 kb)
ESM Fig. 4
Glucose homeostasis in female Tlr2 +/+ and Tlr2 −/− mice fed HFD for 12 weeks. a An IPGTT (2 g/kg glucose) was performed after 12 weeks of HFD feeding (n = 13 Tlr2 +/+, n = 15 Tlr2 −/−). b An ITT (0.85 U/kg insulin) was performed after 12 weeks of HFD feeding (n = 12 Tlr2 +/+, n = 11 Tlr2 −/−). c Insulin secretion during the IPGTT (n = 8 Tlr2 +/+, n = 7 Tlr2 −/−). Black squares, unbroken line, Tlr2 +/+; white squares, broken line, Tlr2 −/−. *p < 0.05 and **p < 0.01 as tested by Student’s t test compared with Tlr2 +/+ control (PDF 100 kb)
ESM Fig. 5
Adipose tissue GLUT4 expression in female Tlr2 +/+ and Tlr2 −/− mice fed HFD for 20 weeks. Protein was extracted from whole perigonadal adipose tissue and analysed using anti-GLUT4 and anti-actin antibodies as shown. Four representative samples are shown from each genotype, and quantification was performed on n = 9 Tlr2 +/+ and n = 9 Tlr2 −/− . Black bars, Tlr2 +/+; white bars, Tlr2 −/−. *p < 0.05 as tested by Student’s t test compared with Tlr2 +/+ control (PDF 42 kb)
ESM Fig. 6
Skeletal muscle triacylglycerols and glycogen content in female Tlr2 +/+ and Tlr2 −/− mice fed HFD for 20 weeks. a Skeletal muscle triacylglycerols were quantified as described (n = 9 Tlr2 +/+, n = 10 Tlr2 −/−). b Skeletal muscle glycogen content was quantified as described (n = 9 Tlr2 +/+, n = 10 Tlr2 −/−). Black bars, Tlr2 +/+; white bars, Tlr2 −/− (PDF 43 kb)
ESM Fig. 7
Blood glucose and glucose infusion rate during hyperinsulinaemic–euglycaemic clamp. Hyperinsulinaemic–euglycaemic clamps were performed after 20 weeks of HFD feeding in female Tlr2 +/+ and Tlr2 −/− littermates. a Blood glucose levels were clamped between 5 and 6 mmol/l in both Tlr2 +/+ and Tlr2 −/− mice. b To maintain euglycaemia, glucose infusion rate was adjusted over time (n = 4). Black squares, unbroken line, Tlr2 +/+; white squares, broken line, Tlr2 −/− (PDF 81 kb)
ESM Fig. 8
Pam2CSK4-induced Il-1β mRNA in BMDMs and BMDCs derived from Tlr2 +/+ and Tlr2 −/− mice. a BMDMs and b BMDCs were prepared from Tlr2 +/+ and Tlr2 −/− mice as described and treated for 6 h with 100 ng/ml Pam2CSK4. RNA was extracted and Il-1β mRNA is shown relative to 18S (n = 3 for Tlr2 +/+, n = 4 for Tlr2 −/−). Black bars, Tlr2 +/+; white bars, Tlr2 −/−. *p < 0.05 compared with control and #p < 0.05 compared with Tlr2 +/+ Pam2CSK4 as tested by ANOVA with Newman–Keuls post hoc test (PDF 91 kb)
ESM Table 1
Plasma analysis of age-matched chow and 20-week-old HFD-fed female Tlr2+/+ and Tlr2−/− mice (PDF 77 kb)
ESM 1
(PDF 66 kb)
Rights and permissions
About this article
Cite this article
Ehses, J.A., Meier, D.T., Wueest, S. et al. Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetologia 53, 1795–1806 (2010). https://doi.org/10.1007/s00125-010-1747-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1747-3