
Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by the cardinal features of tremor, bradykinesia, rigidity, and postural instability, in addition to other non-motor symptoms. Pathologically, PD is attributed to the loss of dopaminergic neurons in the substantia nigra pars compacta, with the hallmark of the presence of intracellular protein aggregates of α-synuclein in the form of Lewy bodies. The pathogenesis of PD is still yet to be fully elucidated due to the multifactorial nature of the disease. However, a myriad of studies has indicated several intracellular events in triggering apoptotic neuronal cell death in PD. These include oxidative stress, mitochondria dysfunction, endoplasmic reticulum stress, alteration in dopamine catabolism, inactivation of tyrosine hydroxylase, and decreased levels of neurotrophic factors. Laboratory studies using the herbicide paraquat in different in vitro and in vivo models have demonstrated the induction of many PD pathological features. The selective neurotoxicity induced by paraquat has brought a new dawn in our perspectives about the pathophysiology of PD. Epidemiological data have suggested an increased risk of developing PD in the human population exposed to paraquat for a long term. This model has opened new frontiers in the quest for new therapeutic targets for PD. The purpose of this review is to synthesize the relationship between the exposure of paraquat and the pathogenesis of PD in in vitro and in vivo models.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
PD is the second most common and progressive neurodegenerative disease after Alzheimer’s disease, affecting approximately six million people over the age of 60, worldwide [1]. The cardinal features of PD include tremor, bradykinesia, rigor, and postural instability [2]. These motor symptoms are due to the selective degeneration of the dopaminergic neuronal cells located in the substantia nigra pars compacta (SNpc), in addition to increased dopamine deficit in the striatal axonal projection area. However, PD is also often associated with other non-motor symptoms that commonly manifest in the gastrointestinal tract, such as gastric reflux, constipation, and swallowing difficulties, in addition to cognitive impairment and neuropsychiatric symptoms, such as depression, anxiety, sleep behaviors, and olfactory dysfunction [3]. These non-motor signs and symptoms are effects of deficits in other neurotransmitters implicated by different brain regions, such as the olfactory bulb, basal ganglia, and frontal cortex, and can occur before the appearance of motor symptoms [3]. Pathologically, the presence of neuronal inclusions known as Lewy bodies in many dopaminergic cells of the SNpc has been seen in many post-mortem findings in patients with PD.
Due to the pathogenesis of PD being multifactorial and the fact that the cause of PD remains unclear, the currently available treatments include pharmacological therapy such as using medications to increase dopamine levels in the brain, deep brain stimulation, and physiotherapy [4]. Although these treatments can reduce the motor symptoms and improve the quality of life of PD patients, there is an urgent need for new therapeutic strategies to prevent, slow, or halt the progression of the disease. Current diagnostic modalities of PD are circumscribed by the fact that there are no specific tests to diagnose PD other than identifying the motor symptoms that the PD patients developed [5]. However, it is estimated that motor symptoms begin to appear when > 30% of dopaminergic neurons have been degenerated [6]. Thus, early diagnosis of PD was always thought to have crucial implications for disease-modifying strategies. For the development of such strategies, the use of appropriate in vitro and in vivo models becomes inevitably valuable to obtain a greater insight into its cause and pathogenesis of PD, in addition to reproducing all clinical and pathological characteristics of PD.
Current in vitro and in vivo PD models can be broadly categorized into genetic and neurotoxin models. Genetic models are used by manipulating genes that have been causally linked to the development of familial PD. However, the neuropathological and behavioral changes evocative to human PD cannot be fully recapitulated in this sophisticated model [7]. On the other hand, neurotoxin models are the most widely used classical PD model, due to their low cost, easy handling, and rapid development in the progression of PD [7]. Various neurotoxin-based models exhibiting degeneration of dopaminergic cells in the SNpc and inducing PD-like phenotypes have been reported by using 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), in addition to other herbicides, such as maneb, rotenone, and paraquat. The use of pesticides and herbicides to model PD has become increasingly important in recent years with the goal of developing neuroprotective agents to halt the progression of PD. This is because exposure to pesticides and herbicides by living in rural areas, farming, or well-water consumption has been implicated with increased risk and incidence for the development of PD [8, 9].
Paraquat is an important member of the bipyridylium family of broad-spectrum, a nonselective fast-acting herbicide that disrupts the intracellular electron transfer system in plants, resulting in the disruption of the plant organelles and ultimately leading to cell death [10]. It is widely used worldwide in many agricultural and non-agricultural settings to control broad-leaved weeds and grasses in many crops, such as cotton, soybeans, sugar cane, and corn. Paraquat has been reported to cause acute poisoning and death due to its toxicity. The common exposure routes of paraquat that would lead to poisoning, either accidentally or intentionally, are ingestion, skin exposure, and inhalation. Paraquat has been used in experimental studies focusing on its pathological effects on the brain, heart, lungs, kidneys, liver, and muscle due to the systemic toxicity and fatality after acute exposure. The interest in using paraquat as a neurotoxin to model PD started since its discovery due to its similarity in terms of its molecular structure and biochemistry with 1-methyl-4-phenylpyridinium (MPP+), the active metabolite of MPTP, a neurotoxin that can induce PD-like features in animal models and humans [11]. For many years, studies have demonstrated that individuals exposed to paraquat had a higher risk of developing PD [12,13,14]. In this article, we will collate evidence of paraquat exposure in relation to PD and discuss paraquat-induced alterations at both cellular and molecular levels. We will first conceptualize paraquat-induced alterations around different pathogenic mechanisms of PD, which can potentially lead to the activation of the apoptotic cell death machinery.
Paraquat-Induced α-Synuclein Pathology
Neuronal inclusions known as Lewy bodies are present in many dopaminergic cells of the SNpc in many post-mortem findings in patients with PD [15]. The major component of a Lewy body consists of an aggregated intracytoplasmic protein known as α-synuclein [16]. α-Synuclein is an intrinsically disordered and highly dynamic protein that can exist in either soluble monomers or an α-helical multimeric conformation [17]. However, α-synuclein may have the ability to convert from the monomeric form to different oligomeric and aggregated configurations, including spherical and fibrils that are built by recruiting additional α-synuclein monomeric subunits [18]. α-Synuclein is expressed abundantly throughout the brain with a higher concentration in the SNpc [19]. The exact function of α-synuclein is still inconclusive. However, the protein might be involved in synaptic plasticity and neurotransmitter release since it is mainly located in the presynaptic terminal of neurons [20]. At the cellular level, α-synuclein is expressed primarily in the presynaptic terminal of neurons, mitochondria, endoplasmic reticulum (ER), Golgi apparatus, and in the endo-lysosomal system [21]. The exact physiological function of α-synuclein at each subcellular compartment is still poorly understood. However, α-synuclein has been demonstrated to interact strongly with synphilin-1, an adaptor molecule that anchors α-synuclein to cytoplasmic proteins involved in vesicular transport and cytoskeletal function [22].
Cumulative studies suggested the accumulation and aggregation of α-synuclein affect the functional integrity of neurons and can contribute to neurotoxicity [23]. This can be seen in a study by Powers et al. [19], where overexpression of α-synuclein in N27 dopaminergic cells potentiated the paraquat-induced toxicity and metabolic dysfunction compared to normal cells. The role of α-synuclein in contributing to cell vulnerability arises from the misfolding and accumulation of the protein into a membrane-bound pore-like structure resulting in membrane leakage and altered intracellular ionic balance [24]. In addition, α-synuclein interacts with complex I of the electron transport chain (ETC), resulting in higher production of reactive oxygen species (ROS), which in turn alters the expression of mitochondrial genome-encoded genes and induces mitochondrial fragmentation [25].
In recent years, a great deal of evidence has suggested the interaction of α-synuclein with environmental toxicants such as paraquat, resulting in the increased α-synuclein propensity to oligomerize and accumulate. In vitro studies using paraquat were reported to induce a conformational change in α-synuclein and significantly accelerated the α-synuclein fibrillation rate in a dose-dependent manner [26, 27]. In addition, Chorfa et al. [28] reported that paraquat increased the intracellular concentration of α-synuclein in SH-SY5Y cells when compared to other pesticides, such as maneb and rotenone. However, only α-synuclein monomers, as opposed to high molecular mass oligomers or fibrils, were detected [28]. Nevertheless, the protein expression of α-synuclein was upregulated (~ 1.5-fold increase) in the mouse frontal cortex and ventral mesencephalon after 48 h of paraquat (10 mg/kg, i.p., once weekly for 3 weeks) post-treatment; however, the expression returned to baseline levels within a week [27]. A 2.1-fold increase in the α-synuclein protein expression level in the striata was also observed in mice treated with paraquat (10 mg/kg, i.p., twice weekly for 4 weeks) [29]. Fernagut et al. [30] demonstrated the administration of paraquat (10 mg/kg, i.p., once weekly for 3 weeks) caused a 1.9-fold increase in the number of α-synuclein aggregates in the SNpc of mice overexpressing human α-synuclein. However, the α-synuclein aggregates were not detected in saline- and paraquat-treated wild-type mice [30]. In that study, the histological sections of the SNpc had been pretreated with proteinase K to selectively visualize insoluble α-synuclein aggregates [30]. Nonetheless, the findings suggest that α-synuclein over-expression can act synergistically with paraquat to elevate the aggregation of α-synuclein.
Although the underlying initiating mechanism of α-synuclein oligomerization has not been completely deciphered, studies have indicated that the formation of α-synuclein radicals might be a key mechanism. Paraquat has been demonstrated to form α-synuclein radicals in the mid-brain of mice through two distinct mechanisms; (1) the activation of NADPH oxidase and induced nitric oxide synthase (iNOS) in microglia to produce peroxynitrite (ONOO−) and (2) leakage of cytochrome C out from the mitochondria into the cytosol to activate the peroxidase activity [31, 32].
There has been a lot of debate over whether the aggregation of α-synuclein is a key feature that contributes to the dysregulation of various cellular processes and cellular toxicity. Manning-Bog et al. [33] demonstrated that mice overexpressing human wild-type or mutated A53T α-synuclein can resist dopaminergic cell degeneration against paraquat and is attributed to the increased expression of heat-shock-protein 70 (Hsp70). This is also consistent with another study where nigral degeneration was not found in mice expressing human wild-type or A53T α-synuclein administered with paraquat (10 mg/kg, i.p., twice weekly for 3 weeks) when compared to their non-transgenic littermates [34]. Moreover, another study demonstrated that MN9D dopaminergic cells overexpressing human wild-type α-synuclein treated with paraquat alone did not exhibit cytotoxicity or compromised membrane integrity [35]. Thus, more studies are required to establish the role of α-synuclein in paraquat-induced neurodegeneration.
Paraquat-Induced Oxidative Stress
Increased Lipid Peroxidation
Oxidative stress plays a crucial role in the progressive deterioration of dopaminergic neurons in PD. Efforts have been made to study the oxidative stress markers present in PD patients compared to healthy controls. Recent advancement in diagnostic testing has provided a new approach to analyze biomarkers that are involved in oxidative stress in the blood and cerebrospinal fluid (CSF). Oxidative stress can trigger lipid peroxidation, which can damage cellular membranes, lipoproteins, and other molecules that contain lipids. The brain is particularly susceptible to lipid peroxidation due to its high unsaturated fatty acid levels. A meta-analysis reported by Wei et al. [36] concluded that malondialdehyde (MDA), an end product of lipid peroxidation, was increased in the blood of PD patients. The presence of other end products of lipid peroxidation, such as 4-hydroxynonenal (HNE) and Nε-(carboxymethyl)lysine, were also found in the Lewy bodies of post-mortem PD brain tissues [37]. In addition, it has been shown that MDA levels were doubled in SK-N-SH cells treated with paraquat (14 μM, 24 h) [38]. MDA levels were also increased in the brain of paraquat-treated Drosophila flies [39, 40]. Since MDA is a well-known biomarker of lipid peroxidation [41], the results above suggest that paraquat may induce oxidative stress leading to increased lipid peroxidation in the brain.
Increased Intracellular ROS Levels
Oxidative stress defines a disequilibrium between the generation and accumulation of ROS in the cells and tissues, and the ability of the biological system to detoxify them through the production of antioxidants. Enhanced ROS production has been implicated in the development of many pathologies, including neurodegenerative diseases such as PD [42]. Accumulating evidence has shown that toxins such as paraquat have been linked to increased oxidative stress. For instance, Alural et al. [43] demonstrated a 1.5-fold increase in ROS levels in SH-SY5Y cells treated with paraquat (500 μM, 24 h). In accordance, Ravi et al. [38] showed a 2-fold increase in ROS generation in SK-N-SH cells treated with paraquat (14 μM, 24 h). Niso-Santano et al. [44] also reported a dramatic increase in the superoxide anion (O2•−) radicals when SH-SY5Y cells were treated with paraquat (100 µM, 4 h). An in vivo study using Drosophila flies treated with paraquat (20 mM, p.o., 24 h) indicated a 3.7-fold increase of O2•− radical in the brain [39]. ROS such as hydrogen peroxide was also doubled in the brain of Drosophila flies when treated with paraquat (20 mM, p.o., 48 h) [40]. The data show that paraquat causes neurodegeneration by increasing intracellular ROS levels.
Impaired Antioxidant Defense
Glutathione (GSH) is a natural antioxidant found in the body, which plays a significant role in protecting the cells against ROS and reactive nitrogen species (RNS) [45]. A decrease in GSH level in the SNpc of the post-mortem brain of PD patients has been documented [46,47,48,49,50]. GSH level in the blood was also lower in patients with PD [36]. Nevertheless, the deficiency in GSH has been reported to impair the cellular antioxidant defense mechanism. Re-exposure to paraquat (5 mg/kg, i.p., twice weekly for 12 weeks) during adulthood caused a 45% decrease of GSH content in the nigrostriatal tissue of rats that had been previously exposed to paraquat, postnatally [51]. Depletion of GSH in the brain of PD patients may be due to the decrease in the synthesis of GSH. It would be foreseen that the activity of glutamate-cysteine ligase (GCL), the rate-limiting enzyme in GSH synthesis [52], decreases in the brain of PD patients if the alteration in the synthesis of GSH was the cause of GSH depletion. Indeed, the activity of GCL was found to be lower throughout the brain as a result of the aging process [53]. Liang et al. [54] reported a 60% reduction in GSH level in the striatum of GCL knockout mice. Lam, Ko [55] reported a 30% reduction in the GCL activity in differentiated PC12 cells treated with paraquat (150 μM, 24 h). GSH depletion may be contributed by increased efflux of glutathione disulfide (GSSG) mainly out of glial cells [56]. Intracellular glutathione levels are maintained at a redox equilibrium between GSSG and GSH [57]. However, oxidative stress will occur if the equilibrium is disrupted and altered toward GSSG [58]. This was seen in a study by Djukic et al. [59], where the authors reported a significantly higher GSSG and GSSG/GSH ratio in the bilateral cortex of adult Wistar rats treated with paraquat (2.5 μg/10 μL, 24 h), intrastriatally.
Other antioxidant enzymes, such as catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (GPx), play a prominent role in antioxidant defense [60]. Kish et al. [61] concluded a slight but significant decrease in GPx activity in several post-mortem brain regions, including SNpc of PD patients. The results were consistent with other animal models resembling PD, such as in Tang et al. [62], where the authors signified a reduction in GPx activity in the midbrain of Sprague–Dawley rats treated with paraquat (10 mg/kg, i.p., once weekly for 4 weeks). Similar to the peroxidase activity, the activity of CAT was also decreased in the SNpc of PD patients [63]. CAT level in the blood was also reduced in patients with PD [36]. Shukla et al. [39] reported that paraquat (20 mM, p.o., 24 h) induced a significant decrease in SOD activity by ~ 50% in the brain of the Drosophila flies. These findings indicate that paraquat causes oxidative damage by lowering the antioxidant defense.
In contrast, Srivastav et al. [40] showed a 2.2-fold upregulation in SOD activity in the head of the Drosophila flies when the flies were treated with paraquat (20 mM, p.o., 48 h). CAT activity in the brain of paraquat-treated flies was also higher (~ 1.6-fold) [40]. The increase in SOD and CAT activities may be a protective response to elevated levels of free radicals in paraquat-treated flies [39, 40]. In accordance, several studies showed that SOD activity was also higher in the SNpc and erythrocytes of PD patients [64,65,66].
Downregulated Nrf2-Keap1-ARE Signaling Pathway
Nuclear factor erythroid 2-related factor 2 (Nrf2) is an essential transcription factor that regulates a wide range of antioxidant defense pathways, leading to the production of various antioxidant enzymes [67]. Under normal conditions, the abundance of intracellular Nrf2 present in the cytoplasm is consistently low due to the rapid degradation of Nrf2 via the ubiquitin-proteasomal pathway [68]. The degradation of Nrf2 occurs when Nrf2 is bound to Kelch-like ECH-associated protein 1 (Keap1) located in the cytoplasm [68]. In response to oxidative stress, Keap1-mediated degradation of Nrf2 is inhibited, resulting in the translocation and accumulation of Nrf2 in the nucleus [69]. Nrf2 then binds to antioxidant response element (ARE), activating many antioxidant and cytoprotective genes, such as GSH, SOD, CAT, glutathione-S-transferase (GST), NAD(P)H dehydrogenase (quinone) 1 (NQO1), and heme oxygenase-1 (HO-1) [68]. Petrillo et al. [70] demonstrated a significant increase in Nrf2 mRNA and protein expression in leukocytes of PD patients compared to healthy individuals. In autopsy brain tissue obtained from PD patients, nuclear translocation of Nrf2 was found to be more abundant in the dopaminergic neurons in the SNpc, but this response may not be sufficient to protect neurons from cell death [71]. An in vivo study using Drosophila flies showed that treatment with paraquat (20 mM, p.o., 48 h) resulted in the upregulation of Nrf2 mRNA expression by 1.6-fold [40]. Also, HO-1 cDNA expression by immunoblotting was found to be upregulated (~ 4-fold higher) in SH4741 dopaminergic neuronal cell line treated with paraquat (800 μM, 20 h) [72]. Paraquat (500 μM, 24 h) caused more cell death in Nrf2 knockdown SH-SY5Y cells than control cells [43], suggesting that Nrf2 plays an essential role in neuroprotection. Alural et al. [43] reported a reduction in the expression of NQO1 and HO-1 mRNAs in Nrf2 knockdown SH-SY5Y cells. These studies indicated that paraquat-induced oxidative stress could trigger the Nrf2-Keap1-ARE pathway to initiate the downstream antioxidant responsive elements, including NQO1 and HO-1 mRNA expression, to prevent oxidative injury.
Paraquat-Induced Nitrosative Stress
More evidence has suggested that RNS is involved in mediating nitrosative stress. RNS is generated by the rapid reaction between O2•− radicals and nitric oxide, which results in the production of ONOO− [73]. The instability of ONOO− stabilizes itself by donating the -NO2 functional group to Tyr residues of proteins that form the neuronal cytoskeleton, resulting in the formation of 3-nitrotyrosine [74]. This results in structural alteration of proteins, which ultimately leads to the death of dopaminergic neurons [75]. 3-Nitrotyrosine has been well known as a potential biomarker of oxidative and nitrosative stress associated with numerous pathological conditions and disorders of the central nervous system, including PD [74]. Fernández et al. [76] reported elevated levels of free 3-nitrotyrosine and nitroalbumin in the serum and CSF of patients with early PD. In addition, the authors reported the presence of nitro-α-synuclein in serum but not in CSF. Paraquat (10 mg/kg, s.c., twice weekly for 3 weeks) induced a higher magnitude of increase in 3-nitrotyrosine level in GCL knockout mice compared to their wild-type counterpart [54]. In addition, paraquat (20 mM, p.o., 24 h) has also been shown to cause a 2.6-fold increase in the concentration of ONOO- in the brain of Drosophila flies [39]. Thus, oxidative and nitrosative stress plays a crucial role in paraquat-induced neurodegeneration.
Paraquat-Induced Impairment of Dopamine Catabolism
Several lines of evidence have suggested that biochemical defect in dopamine catabolism is implicated in patients with PD. The concentration of dopamine in the SNpc is strictly regulated. Increased dopamine levels, dopamine oxidation, and its reactive catabolites have been suggested as the major oxidative stressor resulting in neuronal death in PD [77]. Dopamine catabolism starts a reaction known as oxidative deamination catalyzed by monoamine oxidase, producing hydrogen peroxide, ammonia, and 3,4-dihydroxyphenylacetaldehyde (DOPAL) [78]. DOPAL is further metabolized to 3,4-dihydroxyphenylacetic acid (DOPAC) or 3,4-dihydroxyphenylethanol (DOPET) by aldehyde dehydrogenase [78]. A post-mortem examination on the brain of sporadic PD patients revealed a decrease in dopamine, DOPAL, and DOPAC levels in the putamen, caudate, and cortex [79]. Other studies also reported elevated DOPAL:DOPAC ratio, in addition to decreased aldehyde dehydrogenase activity in the putamen [80]. Furthermore, a reduced level of DOPAC was also seen in the CSF of PD patients [81]. Although DOPAL is a physiological intermediate in dopamine catabolism, it can also act as a potent neurotoxin. DOPAL injection into the SNpc resulted in a more prominent loss of dopaminergic neurons when compared to dopamine, DOPAC, or DOPET [82].
Apoptosis of the dopaminergic neurons is associated with reducing dopamine and increasing DOPAC levels [83]. Drosophila flies treated with paraquat (20 mM, p.o., 24 h) showed a decrease in dopamine level by 63% and a 2.9-fold increase in DOPAC level in the brain [39]. In addition, a concentration-dependent decrease in the number of dopaminergic neurons was seen at 24 h and 48 h. Tyrosine hydroxylase (TH) is a rate-limiting enzyme in the synthesis of dopamine and TH expression is decreased when the dopaminergic neuronal loss occurred [77]. Exposure to paraquat, for example, can generate ONOO−, which leads to the nitration of the tyrosine residue in TH, resulting in the loss of its enzymatic activity and subsequently permanent loss of TH-positive neurons [84, 85]. Dwyer et al. [86] showed that the loss of TH-positive neurons persisted throughout the 6 months after the last paraquat injection (10 mg/kg, i.p., every other day for 2 weeks) on C57Bl6 mice. The data above suggest that paraquat-induced dopaminergic cell death may be linked to the alteration of dopamine catabolism.
Paraquat-Induced Reduction of Brain-Derived Neurotrophic Factor
Among the proteins putatively involved in the pathogenesis of PD, neurotrophic factors play an important role in the differentiation, maturation, maintenance, and survival of mammalian neurons [87]. These factors have also been shown to have neurogenic and neuroprotective effects under adverse conditions, such as cerebral ischemia [88], neurotoxicity [89], glutamatergic stimulation [90], and neurodegenerative diseases [90]. One of the well-studied neurotrophic factors is brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, identified in most brain regions, including the ventral midbrain, ventral tegmental area, and SNpc [91]. The biosynthesis of BDNF involves the precursor protein pre-pro-BDNF in the ER to be processed to pro-BDNF upon the cleavage of the signal peptide [92]. Pro-BDNF is transported to the Golgi apparatus to be packaged into secretory vesicles and is released from the neurons, either constitutively or in an activity-dependent manner. It may also be converted into mature BDNF by members of the subtilisin/kexin family of endoproteases such as furin, or by the action of plasmin through tissue plasminogen activator (tPA) catalysis [93]. Mature BDNF has been known to augment neuronal synaptogenesis and dendritogenesis and to improve synaptic plasticity upon binding with TrkB receptors, resulting in neuronal development and survival [94]. On the contrary, pro-BDNF acts through the p75 neurotrophin receptor (p75NTR), and the activation of the receptor can oppose those effects elicited by the mature BDNF/TrkB signaling, such as cell death, retraction of the neuronal growth cone, and pruning of axonal processes [95]. For many years, evidence has suggested the importance of BDNF as one of the critical factors in establishing the proper number and function of dopaminergic neurons in the SNpc [96].
Downregulation of BDNF mRNA and protein specifically in the SNpc of patients with PD might participate in the death of the nigral dopaminergic neurons [97,98,99]. Alural et al. [43] demonstrated that BDNF mRNA expression and secreted protein levels were decreased by > 60% in SH-SY5Y cells treated with paraquat (500 μM, 24 h). The study also showed a decrease in the neurite number and length by 50% and 15%, respectively, which indicates the importance of BDNF in promoting neuron survival and growth [43]. Moreover, paraquat has been reported to downregulate BDNF protein expression in the hippocampus [100, 101] and mRNA expression in the striatum of C57BL/6 male mice after repeated administration for a total duration of 3 weeks [102]. Regardless, the action of paraquat on pro-BDNF and mature BDNF was not specified from the studies above. Moyano et al. [103], however, reported a concentration-dependent decrease in the protein concentration of both pro-BDNF and mature BDNF, in addition to TrkB and tPA in primary hippocampal cells treated with paraquat. Moreover, upregulation of the protein expression of p75NTR can be observed in the study [103].
Nonetheless, there are conflicting reports on the circulating level of BDNF in patients with PD [98, 99]. The discrepancy could be due to the vast clinical heterogeneity in PD, particularly the variability in disease severity, subtypes, and duration. Moreover, many factors, including gender, exposure to various medications, and individual cognitive performances, have been documented to affect circulating BDNF levels in PD patients [100,101,102]. In accordance, hippocampal BDNF protein expression was increased by > 65% in female C57BL/6 mice treated with paraquat (10 mg/kg, i.p., 3 times a week for 3 consecutive weeks) [104]. However, there was no significant difference in the protein expression of CREB, the primary mediator of BNDF transcriptional regulation [104]. Thus, it is tempting to speculate that sexual dimorphism may play a role in paraquat-associated BDNF expression occurring in the hippocampus and potentially in other areas of the mouse brain. The female sex hormone estrogen has been well documented to upregulate transcription of BDNF by interacting with the estrogen response element in the BDNF gene [105]. The mRNA expression of BNDF was upregulated at 48 h and 72 h after exposure of U118 astroglia to paraquat (250 μM) [106, 107]. However, paraquat also upregulates the expression of other pro-inflammatory astrocytic factors such as interleukin (IL)-1β and IL-6 resulting in cell cycle arrest, indicating the increase in the expression of neurotrophic factors could be a compensatory mechanism to neuronal insult [106]. Moreover, upregulation of BDNF as a compensatory mechanism against neuronal damage has also been reported in other neurodegenerative diseases. In Alzheimer’s disease, for instance, the compensatory increase of BDNF occurs at the early stage and is then followed by a drop as the disease progresses to the advanced stage [108].
Paraquat-Induced ER Stress
The ER is the largest membrane-closed cellular organelle in all eukaryotes, which plays a crucial role in the synthesis of protein and lipid, as well as functioning as a free calcium reservoir [109]. Initial protein maturation steps occur in the ER where the synthesized proteins in the secretory pathways are precisely folded. However, under certain circumstances, physiological stresses such as glucose starvation, hypoxia, oxidative stress, and disruption of calcium homeostasis can disrupt the protein folding at the ER, leading to the accumulation of unfolded and misfolded proteins, a cellular condition termed as ER stress [110]. The accumulation of misfolded protein in the ER that cannot be effectively removed by the protein degradation mechanism such as ubiquitin–proteasome system can ultimately result in neuronal death [111]. Paraquat has been demonstrated to impair the activity of the proteasomal system, which is a late event in the progression of cell death [112, 113].
Upon ER stress, cells can activate a cascade of mechanisms to cope with protein folding alterations, which is termed the unfolded protein response (UPR). Activation of the UPR transduce information about the status of protein folding in the ER lumen to other organelles, such as the nucleus and cytosol, to mitigate any further accumulation of unfolded protein load [114]. The consequences of activating the UPR are the enervation in the rate of protein synthesis, upregulation of genes encoding chaperones and other proteins involved in the protein degradation or prevention of protein aggregation, participation of protein folding and stabilization, and lastly, translocation of proteins to other cellular compartments [115]. The UPR is mainly controlled by three major transmembrane stress sensors, namely protein kinase RNA-like endoplasmic reticulum kinase (PERK) or eIF2α kinase, activating transcriptional factor 6 (ATF6), and inositol-requiring transmembrane kinase/endoribonuclease 1 (IRE1), which orientate with their luminal domain in the ER lumen and their signal transduction domain toward their cytoplasm [116].
Under physiological conditions, an ER-resident chaperone known as glucose-regulated protein of 78 kDa (GRP78) binds to the luminal domains of PERK, which keeps the kinase in an inactive state [117]. However, when unfolded and misfolded proteins accumulate in the ER, GRP78 dissociates from PERK and allows the dimerization and autophosphorylation of PERK, thus activating it [117]. The activation of PERK phosphorylates the α subunit of the eIF2α, consequently resulting in the induction of signal transduction events that activate downstream UPR target genes, such as GRP78 and activating transcriptional factor 4 (ATF4) [110]. On the contrary, the ATF6 sensor protein is triggered due to the accumulation of misfolded proteins and UPR activation. This results in the translocation of the protein to the Golgi apparatus, where it undergoes proteolysis producing a free cytosolic domain that triggers transcriptional regulation of ER chaperone proteins, such as GRP78 and growth arrest- and DNA damage-inducible gene 153 (GADD153)/C/EBP-homologous protein (CHOP) [118, 119]. Nonetheless, IRE1 also monitors ER homeostasis and activates its intrinsic RNase activity upon ER stress through conformational change, homodimerization, and autophosphorylation [120]. The activation of the RNAse activity of IRE1 results in the generation of an active spliced isoform of the transcription factor, X-box-binding protein 1 (XBP-1) [121]. Subsequently, XBP-1 translocates to the nucleus and modulates gene expression of molecular chaperones and proteins, attributing to ER-associated degradation by binding to the promoters of its target gene [122].
In human PD post-mortem brain tissue, the expression of UPR activation markers, such as p-PERK and p-eIF2α, were upregulated in the dopaminergic neurons of the SNpc [123, 124]. In another study, Baek et al. [125] showed an upregulation of the GRP78 and p-PERK protein levels in the cingulate gyrus of PD patients. More recently, Baek et al. [126] concluded a significant upregulation of GRP78 mRNA level in all brain regions in PD patients compared to control subjects. Nonetheless, the eIF2α mRNA level was not significantly different in any of the brain regions compared to the control group [126]. However, in a study by Esteves, Cardoso [127], the protein expression levels of GRP78 and ATF4 were found to be downregulated in the SNpc of PD-post-mortem brain samples. Interestingly, a study reported colocalization of p-IRE1 with α-synuclein in the SNpc of PD patients, indicating that the accumulation of α-synuclein contributes to the activation of IRE1/XBP-1 of the UPR [128]. In line with these studies, a time-dependent increase in the protein expression of GRP78, glucose-regulated protein of 78 kDa (GRP94), and p-eIF2α in the dopaminergic N27 cells treated with paraquat (500 μM, 12–48 h) was also observed [129]. Moreover, protein expression of p-PERK, p-eIF2α, ATF6, p-IRE1, XBP-1, and immunoglobulin heavy chain binding protein (BiP) was found to be upregulated in adrenal pheochromocytoma PC12 cells treated with paraquat (1 mM, 24 h) [130], suggesting that paraquat plays an important role in causing ER stress.
Physiological processes requiring a high demand for protein synthesis can activate the UPR without triggering the apoptotic pathway [131]. However, conditions that lead to prolonged ER stress can often cause cellular dysfunction and cell death. Chronically sustained activation of eIF2α upregulates the pro-apoptotic transcription factor GADD153/CHOP [132]. Upregulation of both the mRNA and protein expression of GRP78 and CHOP was observed in the SNpc region of PD post-mortem brains compared to the age-matched control group [133]. However, another study by Baek et al. [126] reported that CHOP mRNA levels in all brain regions were not significantly different between PD patients and control subjects. Paraquat has been demonstrated to increase the mRNA expression of CHOP in a concentration-dependent manner in SH-SY5Y cells [134]. Overexpression of CHOP has been reported to result in cell cycle arrest and ER stress-induced apoptosis [135]. This was demonstrated in two independent studies by Chinta et al. [129] and Huang et al. [130], where an upregulation of protein expression of CHOP and other apoptotic markers, such as caspase-3, caspase-7, and cleaved poly(ADP-ribose) polymerase (PARP) in vitro, indicating the activation of ER stress-induced apoptosis.
Paraquat-Induced Mitochondrial Dysfunction
Many lines of evidence suggested that impairment of the mitochondrial function has been linked to the pathogenesis of PD [136]. An increased level of deleted mitochondrial DNA was observed in the SNpc of PD patients, suggesting respiratory chain deficiency and mitochondrial dysfunction [137]. A reduction in the metabolic activity and protein level of NADH dehydrogenase or mitochondrial complex I in the SNpc and frontal cortex of post-mortem examination in PD patients has been reported [138, 139]. In addition, a recent study has shown the deficiency in the mitochondrial complex I throughout the brain of PD patients [140]. Furthermore, brain mitochondria from PD patients showed functional impairment and mis-assembly of complex I [141]. Mitochondrial complex I is the first enzyme present in the mitochondrial ETC, which functions to translocate protons from the mitochondrial matrix to the intermembrane space, generating an electrochemical gradient to produce ATP. The disruption of mitochondrial complex I activity leads to the inefficient generation of ATP and an increased level of ROS in the neurons. Therefore, complex I is considered to be an important site of ROS generation. Nonetheless, the causes and consequences of mitochondrial complex I deficiency in SNpc neuronal cells remain to be explored in the future. Regardless, Choi et al. [142] demonstrated that dopaminergic neurons from NADH: ubiquinone oxidoreductase subunit S4 (NDUFS4) knockout mice that are complex I-deficient appeared normal and healthy with no decrease in survival when compared to neurons from wild-type mice.
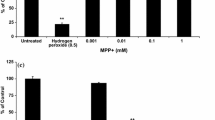
The direct linkage of mitochondrial dysfunction with PD came from neurotoxins such as paraquat. A study conducted by Fukushima et al. [143] demonstrated a decrease in mitochondrial complex I activity and an increase in lipid peroxidation in the brain of bovine treated with 500 μM paraquat. Mitochondrial complex I transfers two electrons from NADPH to ubiquinone, resulting in the oxidation of NADPH to NADP+. Upon entry into cells, paraquat dication (PQ2+) undergoes redox cycling where it disrupts the oxidation of NADPH by accepting electrons to form paraquat mono-cation radical (PQ•+) through NADPH-cytochrome P450 reductase, ultimately inhibiting mitochondrial complex I activity [144, 145]. In a study by Srivastav et al. [40], ATP levels were decreased in the Drosophila flies when treated with paraquat (20 mM, p.o., 48 h). Choi et al. [142] have suggested that the inhibition of mitochondrial complex I is not a pivotal factor to paraquat-induced dopaminergic neuronal death. In that study, the dopaminergic neurons from NDUFS4 knockout mice did not show increased sensitivity to paraquat (50 μM, 24 h) or MPP+ [142]. However, another PD-mimicking neurotoxin, rotenone, showed increased TH-positive neuronal loss in NDUFS4 knockout mice compared to wild-type mice [142]. Nonetheless, the potential for paraquat to induce mitochondrial dysfunction on dopaminergic neurons warranted further investigation.
The mitochondrial membrane potential (ΔΨm) generated by the proton pumps of the ETC, i.e., complex I, complex III, and complex IV, plays a crucial role in storing energy in the form of ATP during oxidative phosphorylation. Under basal conditions, cells maintain a stable intracellular ΔΨm, which is essential for maintaining normal cellular homeostasis [146]. Alteration to the ΔΨm can have deleterious effects on cells. Paraquat has been shown to impair the ΔΨm, as evidenced in a study by Kang et al. [147], where a decrease in the ΔΨm in PC12 cells by 75% was seen when the cells were treated with paraquat (300 μM, 24 h). The loss of ΔΨm has been widely due to the opening of the mitochondrial permeability transition pore, a transmembrane protein residing in the inner mitochondrial membrane [148]. Once the ΔΨm has collapsed, the cells are committed to the apoptotic pathway due to energy depletion [149].
Cytochrome c, a protein located in the inner mitochondrial membrane, plays a vital role in shuttling electrons from complex III and complex IV in the ETC [150]. Cytochrome c is undoubtedly an essential player in the apoptotic pathway of cells. Although it is a ‘quiet worker’ in the ETC, the induction of apoptotic stimuli causes cytochrome c to be released from the mitochondria to the cytosol to initiate the recruitment of caspase-9 and maturation of other caspases that eventually mediates the biochemical and morphological features of apoptosis [150]. Studies have shown that paraquat can induce cytochrome c release from the mitochondria to the cytosol of neuroblastoma cell lines in a concentration-dependent fashion [44, 151]. In addition, Yang et al. [152] co-transfected cells with a vector designed for fluorescent labeling of mitochondria and another vector for the fluorescent labeling of pro-apoptotic proteins. SH-SY5Y cells treated with paraquat were then fixed and subjected to confocal microscopy. Besides cytochrome c, Yang et al. [152] also reported that other mitochondrial pro-apoptotic proteins, such as DIABLO and HTRA2, were also released upon paraquat exposure (300 μM, 24 h). DIABLO and cytochrome c are released from the intermembrane space of the mitochondria into the cytosol [153]. While cytochrome c directly activates Apaf-1 and caspase-9, DIABLO interacts and removes multiple inhibitor of apoptosis proteins (IAPs) that inhibit both initiator and effector caspases [153]. Thus, removing IAPs by DIABLO frees up the caspases and consequently activates the apoptotic mechanism.
Paraquat-Induced Apoptosis
Dopaminergic neuronal cell death in the SNpc is a defining feature of PD [154]. Apoptosis or programmed cell death is thought to be the primary mechanism contributing to dopaminergic neuronal cell death [155]. Neuronal apoptosis is an evolutionarily well-conserved process where it plays a vital role in many physiological processes, particularly in the development and maturation of the nervous system [156]. Several studies have suggested that paraquat can induce apoptosis in the dopaminergic neuronal cells in vitro and in vivo, ultimately resulting in cell death. Flow cytometric assessment by Ju et al. [157] using Annexin V-FITC dye reported a 3.8-fold increase in the total percentages of early and apoptotic SH-SY5Y cells when treated with paraquat (300 μM, 24 h). Another study by Alural et al. [43] showed that paraquat (500 μM, 24 h) caused a 1.8-fold increase in the percentage of SH-SY5Y apoptotic cells in the sub G1 phase of the cell cycle.
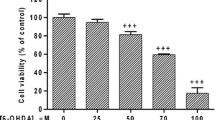
Apoptosis is characterized by a series of specific morphological events in which the onset begins with the cell and nuclear shrinkage in addition to condensation of the chromatin in the nucleus [158]. Extensive plasma membrane blebbing also occurs during this stage. Later on, the nucleus of the cells progressively condenses and fragments [158]. Apoptotic characteristics, such as nuclear condensation, chromatin fragmentation, and apoptotic chromatic changes, were reported in the SNpc dopaminergic neurons of PD patients [159]. Chen et al. [160] demonstrated that paraquat (400 μM, 24 h) significantly increased the number of N27 dopaminergic neuronal cells with fragmented apoptotic nuclei and multiple chromatin condensation. Nonetheless, one of the hallmarks of apoptosis is internucleosomal DNA fragmentation, which has been demonstrated together with the typical morphological events described above [158]. DNA fragmentation occurs when endogenous DNases excise the internucleosomal region into double-stranded DNA fragments of 180–200 bps [161]. This was confirmed by Chun et al. [72] in which the nuclear DNA of dopaminergic neuronal cells was isolated and separated on an agarose gel and found out that paraquat (800 μM, 12 h) resulted in a significant number of cells undergoing DNA fragmentation as indicated by the presence of multiple bands on the gel. Another method of detecting DNA fragmentation is known as the TUNEL assay. A study showed that exposure of PC12 cells to paraquat (300 μM, 24 h) caused a significant 3.7-fold increase in the number of TUNEL-positive cells, indicating that paraquat induces apoptosis in PC12 cells [147]. Similarly, a lower dose of paraquat (14 µM, 24 h) also caused a 5.5-fold increase in the number of TUNEL-positive SK-N-SH neuroblastoma cells [38].
The initiation of apoptosis is a tightly regulated and controlled process since it is irreversible once activated. To date, research has indicated that there are two main pathways to initiate apoptosis; extrinsic (death receptor pathway) and intrinsic (mitochondrial pathway) [162]. While there is some consensus that the extrinsic pathway contributes to the mechanism of neuronal loss in PD, its role remains unclear; thus, it will not be explored in this review. Regardless, the intrinsic pathway is initiated inside the cells by many endogenous and exogenous stimuli, including ischemia, oxidative stress, and DNA damage. A key player to the intrinsic pathway is the mitochondria, where the outer membrane of the mitochondria membrane becomes permeable and releases apoptogenic proteins such as cytochrome c, which normally exists in the mitochondrial intermembrane space, to the cytosol [163]. This process, which is also known as mitochondrial outer membrane permeabilization (MOMP), is mediated and controlled by the balance between pro-apoptotic (i.e., Bak and Bax) and anti-apoptotic Bcl-2 family proteins (i.e., Bcl-2) [163]. Elevated Bax level has been reported in the SNpc of PD patients [149]. The level of Bcl-2 mRNA expression was reduced in SK-N-SH and SH-SY5Y neuroblastoma cell lines upon exposure to paraquat [151, 164]. Exposure of paraquat (500 μM, 24 h) in SH-SY5Y cells resulted in a 2.6-fold increase in the pro-apoptotic Bax mRNA expression and a 1.7-fold increase in Bax protein expression [43]. Fei et al. [151] demonstrated a higher level of the pro-apoptotic protein Bak in SK-N-SH cells and SNpc of C57BL/6 mice, by 220% and 30%, respectively, when treated with paraquat.
Caspases are widely expressed in most cells as inactive zymogens and activate other procaspases, allowing the protease cascade initiation [165]. The activation of caspases results in the amplification of the apoptotic signaling pathway, leading to rapid programmed cell death. Caspases-8 and -9 are initiator caspases that initiate the entire apoptotic cascade. In contrast, caspase-3, -6, and -7 are executioner caspases that carry out mass proteolysis by degrading cellular components [166]. Furthermore, increased activity and protein expression of caspase-3 was reported in the SNpc of PD patients [167, 168]. In addition, active caspase-8 and caspase-9 were also detected in the SNpc from autopsied PD patients but were not detected in normal controls [169]. Ju et al. [157] reported a 1.9-fold increase in caspase-9 protein expression in SH-SY5Y cells treated with paraquat (300 μM, 24 h). In vitro studies using N27 rat dopaminergic neuronal cells have demonstrated that caspase-3 and -7 protein expression in addition to caspase activity were upregulated in dopaminergic N27 cells upon exposure to paraquat (500 μM, 48 h) [129]. Srivastav et al. [40] reported a 4-fold increase in the mRNA expression and a 3-fold increase in the protein expression of caspase-3 in the head section of Drosophila flies after treatment with paraquat (20 mM, p.o., 48 h).
One of the several vital proteins responsible for cellular functioning and survival is PARP. PARP is a highly conserved and multifunctional nuclear protein that plays a vital role in repairing single- and double-stranded bases in DNA [170]. Activation of PARP-1 catalyzes the transfer of negatively charged ADP-ribose moieties from cellular NAD+ to many proteins on specific amino acid residues, generating nicotinamide and ADP-ribose as by-products [170]. Successive addition of ADP-ribose unit to form a long and branched-chain of poly(ADP-ribose), also known as the poly(ADP-ribosylation), forms a scaffold. It then recruits other proteins critical in the DNA repair mechanism. Cleavage of PARP-1 by caspases is considered a prominent hallmark of apoptosis and is responsible for the inactivation of the poly(ADP-ribosylation) process [171]. Cleavage of PARP-1 by caspase-3 has been implicated in several neurological diseases such as PD [171]. A large body of evidence has shown that PARP-1 is cleaved by caspase-3, -7, and -9. Nevertheless, it has also been described that paraquat induces PARP activation in vitro. Chinta et al. [129] reported a 4.8-fold increase in the protein expression of cleaved PARP after paraquat treatment (500 μM, 24 h); however, the protein expression was observed to decrease at 48 h.
Conclusions
The mechanisms of PQ-induced toxicity are summarized in Fig. 1. The synthesized review has strongly demonstrated the relationship between paraquat and PD as a strong inducer of oxidative stress, which contributes ROS formation. PD is a multifactorial disease involving many biochemical pathways, such as oxidative injury, mitochondrial dysfunction, ER stress, alteration in dopamine catabolism, inactivation of TH, and decrease in the neurotrophic factor BDNF, ultimately resulting in apoptosis of the dopaminergic neurons in the SNpc. Thus, the use of in vitro and in vivo models using paraquat, which directly or indirectly contributes to the pathogenesis of the disease under the exacerbated condition of oxidative stress, may provide us with a larger picture to develop new therapeutic targets in the near future. The production of ROS and RNS such as O2•− and ONOO− via redox cycling of paraquat is widely proposed to be the key mechanism of oxidative stress resulting in the apoptosis of dopaminergic neurons. Like innumerable reviews, we identified consistency among studies that investigate the cellular processes and in vivo organisms, in addition to the human population in the framework of PD pathogenesis. The pathophysiological processes involving laboratory animals and the affected signaling cassettes we reviewed in this paper identifying the development of PD characteristics following exposure to paraquat provides imperative evidence to expand this research and solve this problem.

The major molecular targets of paraquat that lead to cellular damage and apoptosis. (1) Paraquat crosses the blood–brain barrier via LAAT. (2) It then enters neuronal cells via transporters such as dopamine transporter DAT, OCT2, and OCT3. (3) Upon entry to the cells, paraquat undergoes a process of redox cycling, a process of alternate reduction and reoxidation. Paraquat is reduced by enzymes present in the mitochondria to form a monocation free radical, PQ•+. PQ•+ is then rapidly reoxidized in the presence of oxygen to generate O2•− and regenerates its parent compound PQ2+. If there is sufficient NADPH as an electron donor and O2 as an electron acceptor, paraquat will repeatedly undergo the reduction–oxidation cycle, generating O2•−. This results in the initiation of a reaction cascade leading to ROS generation, such as H2O2 and OH−, in addition to RNS such as ONOO−. (4) The generation of ROS and RNS are counterbalanced by the activation of the Nrf2-Keap1-ARE signaling pathway to activate endogenous antioxidant enzyme genes, such as HO-1 and NQO1. (5) Paraquat increase α-synuclein modifications, misfolding, and fibrillation rate resulting in aggregation to form Lewy bodies which are toxic to the cell. (6) Paraquat can also induce ER stress by activating the UPR signal proteins, such as PERK, ATF6, and IRE1, consequently leading to the upregulation of the pro-apoptotic transcription factor, CHOP. (7) Ultimately, the activation of various pathological cellular processes, such as oxidative and nitrosative stress, ER stress, and mitochondrial dysfunction, results in the apoptotic pathway activation, leading to the cell death of dopaminergic neurons. ARE antioxidant response element, ATF6 activating transcription factor 6, BiP binding immunoglobulin protein, CHOP C/EBP-homologous protein, DAT dopamine transporter, ER endoplasmic reticulum, GPx glutathione peroxidase, GRP78 glucose regulatory protein 78, GRP94 glucose regulatory protein 94, GSH glutathione, GSSG glutathione disulfide, H2O water, H2O2 hydrogen peroxide, HO-1 heme oxygenase-1, HtrA2 HtrA serine peptidase 2, IAPs inhibitor of apoptosis proteins, IRE1 inositol-requiring transmembrane kinase/endoribonuclease 1, Keap1 Kelch-like ECH-associated protein 1, LAAT l-neutral amino acid, NO nitric oxide, NQO1 NAD(P)H dehydrogenase (quinone) 1, Nrf2 nuclear factor erythroid 2-related factor 2, O2 oxygen, O2•− superoxide anion, OCT2 organic cation transporter 2, OCT3 organic cation transporter 3, ONOO− peroxynitrite, PARP-1 poly (ADP-ribose) polymerase-1, PERK protein kinase RNA-like endoplasmic reticulum kinase, PQ paraquat, PQ•+ paraquat monocation free radical, PQ2+ paraquat dication, ROS reactive oxygen species, Smac/DIABLO second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein, SOD superoxide dismutase, Ub ubiquitin, UPR unfolded protein response
Data Availability
Not applicable.
References
Dauer W, Przedborski S (2003) Parkinson’s disease: mechanisms and models. Neuron 39(6):889–909. https://doi.org/10.1016/s0896-6273(03)00568-3
Gazewood JD, Richards DR, Clebak K (2013) Parkinson disease: an update. Am Fam Physician 87(4):267–273
Schapira AHV, Chaudhuri KR, Jenner P (2017) Non-motor features of Parkinson disease. Nat Rev Neurosci 18(7):435–450. https://doi.org/10.1038/nrn.2017.62
Kaur R, Mehan S, Singh S (2019) Understanding multifactorial architecture of Parkinson’s disease: pathophysiology to management. Neurol Sci 40(1):13–23. https://doi.org/10.1007/s10072-018-3585-x
Armstrong MJ, Okun MS (2020) Diagnosis and treatment of Parkinson disease: a review. JAMA 323(6):548–560. https://doi.org/10.1001/jama.2019.22360
Cheng H-C, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67(6):715–725. https://doi.org/10.1002/ana.21995
Blesa J, Przedborski S (2014) Parkinson’s disease: animal models and dopaminergic cell vulnerability. Front Neuroanat 8:155. https://doi.org/10.3389/fnana.2014.00155
Breckenridge CB, Berry C, Chang ET, Sielken RL Jr, Mandel JS (2016) Association between Parkinson’s disease and cigarette smoking, rural living, well-water consumption, farming and pesticide use: systematic review and meta-analysis. PLoS ONE 11(4):e0151841. https://doi.org/10.1371/journal.pone.0151841
Vaccari C, El Dib R, Gomaa H, Lopes LC, de Camargo JL (2019) Paraquat and Parkinson’s disease: a systematic review and meta-analysis of observational studies. J Toxicol Environ Health B Crit Rev 22(5–6):172–202. https://doi.org/10.1080/10937404.2019.1659197
Tsai W-T (2013) A review on environmental exposure and health risks of herbicide paraquat. Toxicol Environ Chem 95(2):197–206. https://doi.org/10.1080/02772248.2012.761999
Zhang X-f, Thompson M, Xu Y-h (2016) Multifactorial theory applied to the neurotoxicity of paraquat and paraquat-induced mechanisms of developing Parkinson’s disease. Lab Invest 96(5):496–507. https://doi.org/10.1038/labinvest.2015.161
Tanner CM, Ross GW, Jewell SA, Hauser RA, Jankovic J, Factor SA, Bressman S, Deligtisch A, Marras C, Lyons KE, Bhudhikanok GS, Roucoux DF, Meng C, Abbott RD, Langston JW (2009) Occupation and risk of parkinsonism: a multicenter case-control study. Arch Neurol 66(9):1106–1113. https://doi.org/10.1001/archneurol.2009.195
Costello S, Cockburn M, Bronstein J, Zhang X, Ritz B (2009) Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am J Epidemiol 169(8):919–926. https://doi.org/10.1093/aje/kwp006
Kamel F, Tanner C, Umbach D, Hoppin J, Alavanja M, Blair A, Comyns K, Goldman S, Korell M, Langston J, Ross G, Sandler D (2007) Pesticide exposure and self-reported Parkinson’s disease in the agricultural health study. Am J Epidemiol 165(4):364–374. https://doi.org/10.1093/aje/kwk024
Wakabayashi K, Tanji K, Mori F, Takahashi H (2007) The Lewy body in Parkinson’s disease: molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology 27(5):494–506. https://doi.org/10.1111/j.1440-1789.2007.00803.x
Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M (1997) α-Synuclein in Lewy bodies. Nature 388(6645):839–840. https://doi.org/10.1038/42166
Zhang G, Xia Y, Wan F, Ma K, Guo X, Kou L, Yin S, Han C, Liu L, Huang J, Xiong N, Wang T (2018) New Perspectives on roles of alpha-synuclein in Parkinson’s disease. Front Aging Neurosci 10:370. https://doi.org/10.3389/fnagi.2018.00370
Hijaz BA, Volpicelli-Daley LA (2020) Initiation and propagation of α-synuclein aggregation in the nervous system. Mol Neurodegener 15(1):19. https://doi.org/10.1186/s13024-020-00368-6
Powers R, Lei S, Anandhan A, Marshall DD, Worley B, Cerny RL, Dodds ED, Huang Y, Panayiotidis MI, Pappa A, Franco R (2017) Metabolic investigations of the molecular mechanisms associated with Parkinson’s disease. Metabolites 7 (2). https://doi.org/10.3390/metabo7020022
Gómez-Benito M, Granado N, García-Sanz P, Michel A, Dumoulin M, Moratalla R (2020) Modeling Parkinson’s disease with the alpha-synuclein protein. Front Pharmacol 11:356–356. https://doi.org/10.3389/fphar.2020.00356
Bernal-Conde LD, Ramos-Acevedo R, Reyes-Hernández MA, Balbuena-Olvera AJ, Morales-Moreno ID, Argüero-Sánchez R, Schüle B, Guerra-Crespo M (2019) Alpha-synuclein physiology and pathology: a perspective on cellular structures and organelles. Front Neurosci 13:1399. https://doi.org/10.3389/fnins.2019.01399
Engelender S, Kaminsky Z, Guo X, Sharp AH, Amaravi RK, Kleiderlein JJ, Margolis RL, Troncoso JC, Lanahan AA, Worley PF, Dawson VL, Dawson TM, Ross CA (1999) Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat Genet 22(1):110–114. https://doi.org/10.1038/8820
Fusco G, Chen SW, Williamson PTF, Cascella R, Perni M, Jarvis JA, Cecchi C, Vendruscolo M, Chiti F, Cremades N, Ying L, Dobson CM, De Simone A (2017) Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 358(6369):1440–1443. https://doi.org/10.1126/science.aan6160
Feng LR, Federoff HJ, Vicini S, Maguire-Zeiss KA (2010) α-Synuclein mediates alterations in membrane conductance: a potential role for α-synuclein oligomers in cell vulnerability. Eur J Neurosci 32(1):10–17. https://doi.org/10.1111/j.1460-9568.2010.07266.x
Bernal-Conde LD, Ramos-Acevedo R, Reyes-Hernández MA, Balbuena-Olvera AJ, Morales-Moreno ID, Argüero-Sánchez R, Schüle B, Guerra-Crespo M (2020) Alpha-synuclein physiology and pathology: a perspective on cellular structures and organelles. Front Neurosci 13:1399–1399. https://doi.org/10.3389/fnins.2019.01399
Uversky VN, Li J, Fink AL (2001) Pesticides directly accelerate the rate of α-synuclein fibril formation: a possible factor in Parkinson’s disease. FEBS Lett 500(3):105–108. https://doi.org/10.1016/S0014-5793(01)02597-2
Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA (2002) The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem 277(3):1641–1644. https://doi.org/10.1074/jbc.C100560200
Chorfa A, Bétemps D, Morignat E, Lazizzera C, Hogeveen K, Andrieu T, Baron T (2013) Specific pesticide-dependent increases in alpha-synuclein levels in human neuroblastoma (SH-SY5Y) and melanoma (SK-MEL-2) cell lines. Toxicol Sci 133(2):289–297. https://doi.org/10.1093/toxsci/kft076
Su C, Niu P (2015) Low doses of single or combined agrichemicals induces α-synuclein aggregation in nigrostriatal system of mice through inhibition of proteasomal and autophagic pathways. Int J Clin Exp Med 8(11):20508–20515
Fernagut PO, Hutson CB, Fleming SM, Tetreaut NA, Salcedo J, Masliah E, Chesselet MF (2007) Behavioral and histopathological consequences of paraquat intoxication in mice: effects of alpha-synuclein over-expression. Synapse 61(12):991–1001. https://doi.org/10.1002/syn.20456
Kumar A, Ganini D, Mason RP (2016) Role of cytochrome c in α-synuclein radical formation: implications of α-synuclein in neuronal death in Maneb- and paraquat-induced model of Parkinson’s disease. Mol Neurodegener 11(1):70. https://doi.org/10.1186/s13024-016-0135-y
Kumar A, Leinisch F, Kadiiska MB, Corbett J, Mason RP (2016) Formation and implications of alpha-synuclein radical in Maneb- and paraquat-induced models of Parkinson’s disease. Mol Neurobiol 53(5):2983–2994. https://doi.org/10.1007/s12035-015-9179-1
Manning-Bog AB, McCormack AL, Purisai MG, Bolin LM, Di Monte DA (2003) Alpha-synuclein overexpression protects against paraquat-induced neurodegeneration. J Neurosci 23(8):3095–3099. https://doi.org/10.1523/JNEUROSCI.23-08-03095.2003
Norris EH, Uryu K, Leight S, Giasson BI, Trojanowski JQ, Lee VM (2007) Pesticide exposure exacerbates alpha-synucleinopathy in an A53T transgenic mouse model. Am J Pathol 170(2):658–666. https://doi.org/10.2353/ajpath.2007.060359
Feng LR, Maguire-Zeiss KA (2011) Dopamine and paraquat enhance α-synuclein-induced alterations in membrane conductance. Neurotox Res 20(4):387. https://doi.org/10.1007/s12640-011-9255-x
Wei Z, Li X, Li X, Liu Q, Cheng Y (2018) Oxidative stress in Parkinson’s disease: a systematic review and meta-analysis. Front Mol Neurosci 11:236. https://doi.org/10.3389/fnmol.2018.00236
Castellani RJ, Perry G, Siedlak SL, Nunomura A, Shimohama S, Zhang J, Montine T, Sayre LM, Smith MA (2002) Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci Lett 319(1):25–28. https://doi.org/10.1016/s0304-3940(01)02514-9
Ravi SK, Narasingappa RB, Joshi CG, Girish TK, Vincent B (2018) Neuroprotective effects of Cassia tora against paraquat-induced neurodegeneration: relevance for Parkinson’s disease. Nat Prod Res 32(12):1476–1480. https://doi.org/10.1080/14786419.2017.1353504
Shukla AK, Pragya P, Chaouhan HS, Patel DK, Abdin MZ, Kar Chowdhuri D (2014) Mutation in Drosophila methuselah resists paraquat induced Parkinson-like phenotypes. Neurobiol Aging 35(10):2419.e2411-2419.e2416. https://doi.org/10.1016/j.neurobiolaging.2014.04.008
Srivastav S, Fatima M, Mondal AC (2018) Bacopa monnieri alleviates paraquat induced toxicity in Drosophila by inhibiting jnk mediated apoptosis through improved mitochondrial function and redox stabilization. Neurochem Int 121:98–107. https://doi.org/10.1016/j.neuint.2018.10.001
Tsikas D (2017) Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: analytical and biological challenges. Anal Biochem 524:13–30. https://doi.org/10.1016/j.ab.2016.10.021
Puspita L, Chung SY, Shim J-w (2017) Oxidative stress and cellular pathologies in Parkinson’s disease. Mol Brain 10(1):53. https://doi.org/10.1186/s13041-017-0340-9
Alural B, Ozerdem A, Allmer J, Genc K, Genc S (2015) Lithium protects against paraquat neurotoxicity by NRF2 activation and miR-34a inhibition in SH-SY5Y cells. Front Cell Neurosci 9:209. https://doi.org/10.3389/fncel.2015.00209
Niso-Santano M, Gonzalez-Polo RA, Bravo-San Pedro JM, Gomez-Sanchez R, Lastres-Becker I, Ortiz-Ortiz MA, Soler G, Moran JM, Cuadrado A, Fuentes JM (2010) Activation of apoptosis signal-regulating kinase 1 is a key factor in paraquat-induced cell death: modulation by the Nrf2/Trx axis. Free Radic Biol Med 48(10):1370–1381. https://doi.org/10.1016/j.freeradbiomed.2010.02.024
Gaucher C, Boudier A, Bonetti J, Clarot I, Leroy P, Parent M (2018) Glutathione: antioxidant properties dedicated to nanotechnologies. Antioxidants 7(5):62. https://doi.org/10.3390/antiox7050062
Perry TL, Godin DV, Hansen S (1982) Parkinson’s disease: a disorder due to nigral glutathione deficiency? Neurosci Lett 33(3):305–310. https://doi.org/10.1016/0304-3940(82)90390-1
Sofic E, Lange KW, Jellinger K, Riederer P (1992) Reduced and oxidized glutathione in the substantia nigra of patients with Parkinson’s disease. Neurosci Lett 142(2):128–130. https://doi.org/10.1016/0304-3940(92)90355-B
Pearce RK, Owen A, Daniel S, Jenner P, Marsden CD (1997) Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J Neural Transm 104(6–7):661–677. https://doi.org/10.1007/bf01291884
Fitzmaurice PS, Ang L, Guttman M, Rajput AH, Furukawa Y, Kish SJ (2003) Nigral glutathione deficiency is not specific for idiopathic Parkinson’s disease. Mov Disord 18(9):969–976. https://doi.org/10.1002/mds.10486
Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD (1994) Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol 36(3):348–355. https://doi.org/10.1002/ana.410360305
Mittra N, Chauhan AK, Singh G, Patel DK, Singh C (2020) Postnatal zinc or paraquat administration increases paraquat or zinc-induced loss of dopaminergic neurons: insight into augmented neurodegeneration. Mol Cell Biochem 467(1–2):27–43. https://doi.org/10.1007/s11010-020-03694-x
Franklin CC, Backos DS, Mohar I, White CC, Forman HJ, Kavanagh TJ (2009) Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol Aspects Med 30(1–2):86–98. https://doi.org/10.1016/j.mam.2008.08.009
Zhu Y, Carvey PM, Ling Z (2006) Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res 1090(1):35–44. https://doi.org/10.1016/j.brainres.2006.03.063
Liang L-P, Kavanagh TJ, Patel M (2013) Glutathione deficiency in Gclm null mice results in complex I inhibition and dopamine depletion following paraquat administration. Toxicol Sci 134(2):366–373. https://doi.org/10.1093/toxsci/kft112
Lam PY, Ko KM (2011) (-)Schisandrin B ameliorates paraquat-induced oxidative stress by suppressing glutathione depletion and enhancing glutathione recovery in differentiated PC12 cells. BioFactors 37(1):51–57. https://doi.org/10.1002/biof.136
Sian J, Dexter DT, Lees AJ, Daniel S, Jenner P, Marsden CD (1994) Glutathione-related enzymes in brain in Parkinson’s disease. Ann Neurol 36(3):356–361. https://doi.org/10.1002/ana.410360306
Jones DP (2002) Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol 348:93–112. https://doi.org/10.1016/S0076-6879(02)48630-2
Martin HL, Teismann P (2009) Glutathione—a review on its role and significance in Parkinson’s disease. FASEB J 23(10):3263–3272. https://doi.org/10.1096/fj.08-125443
Djukic MM, Jovanovic MD, Ninkovic M, Stevanovic I, Ilic K, Curcic M, Vekic J (2012) Protective role of glutathione reductase in paraquat induced neurotoxicity. Chem Biol Interact 199(2):74–86. https://doi.org/10.1016/j.cbi.2012.05.008
Ighodaro OM, Akinloye OA (2018) First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alexandria J Med 54(4):287–293. https://doi.org/10.1016/j.ajme.2017.09.001
Kish SJ, Morito C, Hornykiewicz O (1985) Glutathione peroxidase activity in Parkinson’s disease brain. Neurosci Lett 58(3):343–346. https://doi.org/10.1016/0304-3940(85)90078-3
Tang SP, Kuttulebbai Nainamohamed Salam S, Jaafar H, Gan SH, Muzaimi M, Sulaiman SA (2017) Tualang honey protects the rat midbrain and lung against repeated paraquat exposure. Oxid Med Cell Longev 2017:4605782–4605782. https://doi.org/10.1155/2017/4605782
Ambani LM, Van Woert MH, Murphy S (1975) Brain peroxidase and catalase in Parkinson disease. Arch Neurol 32(2):114–118. https://doi.org/10.1001/archneur.1975.00490440064010
Marttila RJ, Lorentz H, Rinne UK (1988) Oxygen toxicity protecting enzymes in Parkinson’s disease. Increase of superoxide dismutase-like activity in the substantia nigra and basal nucleus. J Neurol Sci 86(2–3):321–331. https://doi.org/10.1016/0022-510x(88)90108-6
Saggu H, Cooksey J, Dexter D, Wells FR, Lees A, Jenner P, Marsden CD (1989) A selective increase in particulate superoxide dismutase activity in parkinsonian substantia nigra. J Neurochem 53(3):692–697. https://doi.org/10.1111/j.1471-4159.1989.tb11759.x
Younes-Mhenni S, Frih-Ayed M, Kerkeni A, Bost M, Chazot G (2007) Peripheral blood markers of oxidative stress in Parkinson’s disease. Eur Neurol 58(2):78–83. https://doi.org/10.1159/000103641
Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426. https://doi.org/10.1146/annurev-pharmtox-011112-140320
Brandes MS, Gray NE (2020) NRF2 as a therapeutic target in neurodegenerative diseases. ASN Neuro 12:1759091419899782–1759091419899782. https://doi.org/10.1177/1759091419899782
Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M (2006) Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol 26(1):221–229. https://doi.org/10.1128/mcb.26.1.221-229.2006
Petrillo S, Schirinzi T, Di Lazzaro G, D’Amico J, Colona VL, Bertini E, Pierantozzi M, Mari L, Mercuri NB, Piemonte F, Pisani A (2020) Systemic activation of Nrf2 pathway in Parkinson’s disease. Mov Disord 35(1):180–184. https://doi.org/10.1002/mds.27878
Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL (2007) Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol 66(1):75–85. https://doi.org/10.1097/nen.0b013e31802d6da9
Chun HS, Gibson GE, DeGiorgio LA, Zhang H, Kidd VJ, Son JH (2001) Dopaminergic cell death induced by MPP(+), oxidant and specific neurotoxicants shares the common molecular mechanism. J Neurochem 76(4):1010–1021. https://doi.org/10.1046/j.1471-4159.2001.00096.x
Liu F, Dong H, Tian Y (2019) Real-time monitoring of peroxynitrite (ONOO−) in the rat brain by developing a ratiometric electrochemical biosensor. Analyst 144(6):2150–2157. https://doi.org/10.1039/C9AN00079H
Bandookwala M, Sengupta P (2020) 3-Nitrotyrosine: a versatile oxidative stress biomarker for major neurodegenerative diseases. Int J Neurosci 130(10):1047–1062. https://doi.org/10.1080/00207454.2020.1713776
Blanchard-Fillion B, Prou D, Polydoro M, Spielberg D, Tsika E, Wang Z, Hazen SL, Koval M, Przedborski S, Ischiropoulos H (2006) Metabolism of 3-nitrotyrosine induces apoptotic death in dopaminergic cells. J Neurosci 26(23):6124–6130. https://doi.org/10.1523/jneurosci.1038-06.2006
Fernández E, García-Moreno JM, Martín de Pablos A, Chacón J (2013) May the evaluation of nitrosative stress through selective increase of 3-nitrotyrosine proteins other than nitroalbumin and dominant tyrosine-125/136 nitrosylation of serum α-synuclein serve for diagnosis of sporadic Parkinson’s disease? Antioxid Redox Signal 19(9):912–918. https://doi.org/10.1089/ars.2013.5250
Bisaglia M, Filograna R, Beltramini M, Bubacco L (2014) Are dopamine derivatives implicated in the pathogenesis of Parkinson’s disease? Ageing Res Rev 13:107–114. https://doi.org/10.1016/j.arr.2013.12.009
Masato A, Plotegher N, Boassa D, Bubacco L (2019) Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol Neurodegener 14(1):35. https://doi.org/10.1186/s13024-019-0332-6
Goldstein DS, Sullivan P, Holmes C, Kopin IJ, Basile MJ, Mash DC (2011) Catechols in post-mortem brain of patients with Parkinson disease. Eur J Neurol 18(5):703–710. https://doi.org/10.1111/j.1468-1331.2010.03246.x
Goldstein DS, Sullivan P, Holmes C, Miller GW, Alter S, Strong R, Mash DC, Kopin IJ, Sharabi Y (2013) Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J Neurochem 126(5):591–603. https://doi.org/10.1111/jnc.12345
Goldstein DS, Holmes C, Sullivan P, Jinsmaa Y, Kopin IJ, Sharabi Y (2016) Elevated cerebrospinal fluid ratios of cysteinyl-dopamine/3,4-dihydroxyphenylacetic acid in parkinsonian synucleinopathies. Parkinsonism Relat Disord 31:79–86. https://doi.org/10.1016/j.parkreldis.2016.07.009
Burke WJ, Li SW, Williams EA, Nonneman R, Zahm DS (2003) 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: implications for Parkinson’s disease pathogenesis. Brain Res 989(2):205–213. https://doi.org/10.1016/s0006-8993(03)03354-7
Peng J, Mao XO, Stevenson FF, Hsu M, Andersen JK (2004) The herbicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway. J Biol Chem 279(31):32626–32632. https://doi.org/10.1074/jbc.M404596200
Dinis-Oliveira RJ, Remião F, Carmo H, Duarte JA, Navarro AS, Bastos ML, Carvalho F (2006) Paraquat exposure as an etiological factor of Parkinson’s disease. Neurotoxicology 27(6):1110–1122. https://doi.org/10.1016/j.neuro.2006.05.012
Park S, Geddes TJ, Javitch JA, Kuhn DM (2003) Dopamine prevents nitration of tyrosine hydroxylase by peroxynitrite and nitrogen dioxide: is nitrotyrosine formation an early step in dopamine neuronal damage? J Biol Chem 278(31):28736–28742. https://doi.org/10.1074/jbc.M304362200
Dwyer Z, Rudyk C, Farmer K, Beauchamp S, Shail P, Derksen A, Fortin T, Ventura K, Torres C, Ayoub K, Hayley S (2021) Characterizing the protracted neurobiological and neuroanatomical effects of paraquat in a murine model of Parkinson’s disease. Neurobiol Aging 100:11–21. https://doi.org/10.1016/j.neurobiolaging.2020.11.013
Vilar M, Mira H (2016) Regulation of neurogenesis by neurotrophins during adulthood: expected and unexpected roles. Front Neurosci 10:26. https://doi.org/10.3389/fnins.2016.00026
Harris NM, Ritzel R, Mancini NS, Jiang Y, Yi X, Manickam DS, Banks WA, Kabanov AV, McCullough LD, Verma R (2016) Nano-particle delivery of brain derived neurotrophic factor after focal cerebral ischemia reduces tissue injury and enhances behavioral recovery. Pharmacol Biochem Behav 150–151:48–56. https://doi.org/10.1016/j.pbb.2016.09.003
Apfel SC (1996) Neurotrophic factors in the treatment of neurotoxicity: an overview. Neurotoxicology 17(3–4):839–844
Mattson MP (2008) Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci 1144:97–112. https://doi.org/10.1196/annals.1418.005
Seroogy KB, Lundgren KH, Tran TM, Guthrie KM, Isackson PJ, Gall CM (1994) Dopaminergic neurons in rat ventral midbrain express brain-derived neurotrophic factor and neurotrophin-3 mRNAs. J Comp Neurol 342(3):321–334. https://doi.org/10.1002/cne.903420302
Greenberg ME, Xu B, Lu B, Hempstead BL (2009) New insights in the biology of BDNF synthesis and release: implications in CNS function. J Neurosci 29(41):12764–12767. https://doi.org/10.1523/jneurosci.3566-09.2009
Mowla SJ, Pareek S, Farhadi HF, Petrecca K, Fawcett JP, Seidah NG, Morris SJ, Sossin WS, Murphy RA (1999) Differential sorting of nerve growth factor and brain-derived neurotrophic factor in hippocampal neurons. J Neurosci 19(6):2069–2080. https://doi.org/10.1523/jneurosci.19-06-02069.1999
Kowiański P, Lietzau G, Czuba E, Waśkow M, Steliga A, Moryś J (2018) BDNF: a key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol Neurobiol 38(3):579–593. https://doi.org/10.1007/s10571-017-0510-4
Hempstead BL (2015) Brain-derived neurotrophic factor: three ligands, many actions. Trans Am Clin Climatol Assoc 126:9–19
Baquet ZC, Bickford PC, Jones KR (2005) Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci 25(26):6251–6259. https://doi.org/10.1523/jneurosci.4601-04.2005
Parain K, Murer MG, Yan Q, Faucheux B, Agid Y, Hirsch E, Raisman-Vozari R (1999) Reduced expression of brain-derived neurotrophic factor protein in Parkinson’s disease substantia nigra. NeuroReport 10(3):557–561. https://doi.org/10.1097/00001756-199902250-00021
Howells DW, Porritt MJ, Wong JY, Batchelor PE, Kalnins R, Hughes AJ, Donnan GA (2000) Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp Neurol 166(1):127–135. https://doi.org/10.1006/exnr.2000.7483
Nagatsu T, Sawada M (2007) Biochemistry of postmortem brains in Parkinson’s disease: historical overview and future prospects. J Neural Transm Suppl 72:113–120. https://doi.org/10.1007/978-3-211-73574-9_14
Mangano EN, Peters S, Litteljohn D, So R, Bethune C, Bobyn J, Clarke M, Hayley S (2011) Granulocyte macrophage-colony stimulating factor protects against substantia nigra dopaminergic cell loss in an environmental toxin model of Parkinson’s disease. Neurobiol Dis 43(1):99–112. https://doi.org/10.1016/j.nbd.2011.02.011
Rudyk C, Dwyer Z, McNeill J, Salmaso N, Farmer K, Prowse N, Hayley S (2019) Chronic unpredictable stress influenced the behavioral but not the neurodegenerative impact of paraquat. Neurobiol Stress 11:100179. https://doi.org/10.1016/j.ynstr.2019.100179
Mangano EN, Litteljohn D, So R, Nelson E, Peters S, Bethune C, Bobyn J, Hayley S (2012) Interferon-γ plays a role in paraquat-induced neurodegeneration involving oxidative and proinflammatory pathways. Neurobiol Aging 33(7):1411–1426. https://doi.org/10.1016/j.neurobiolaging.2011.02.016
Moyano P, Sanjuan J, García JM, Anadon MJ, Naval MV, Sola E, García J, Frejo MT, Pino Jd (2020) Dysregulation of prostaglandine E2 and BDNF signaling mediated by estrogenic dysfunction induces primary hippocampal neuronal cell death after single and repeated paraquat treatment. Food Chem Toxicol 144:111611. https://doi.org/10.1016/j.fct.2020.111611
Litteljohn D, Nelson E, Bethune C, Hayley S (2011) The effects of paraquat on regional brain neurotransmitter activity, hippocampal BDNF and behavioural function in female mice. Neurosci Lett 502(3):186–191. https://doi.org/10.1016/j.neulet.2011.07.041
Brann DW, Lu Y, Wang J, Zhang Q, Thakkar R, Sareddy GR, Pratap UP, Tekmal RR, Vadlamudi RK (2021) Brain-derived estrogen and neural function. Neuroscience & Biobehavioral Reviews. https://doi.org/10.1016/j.neubiorev.2021.11.014
Zhan X, Li F, Chu Q, Pang H (2018) Effects of PQ’s cytotoxicity on secretory vesicles in astroglia: expression alternation of secretogranin II and its potential interaction with intracellular factors. Biochem Biophys Res Commun 497(2):675–682. https://doi.org/10.1016/j.bbrc.2018.02.130
Zhan X, Li F, Chu Q, Pang H (2018) Secretogranin III may be an indicator of paraquat-induced astrocyte activation and affects the recruitment of BDNF during this process. Int J Mol Med 42(6):3622–3630. https://doi.org/10.3892/ijmm.2018.3909
Merlo S, Spampinato SF, Sortino MA (2019) Early compensatory responses against neuronal injury: a new therapeutic window of opportunity for Alzheimer’s Disease? CNS Neurosci Ther 25(1):5–13. https://doi.org/10.1111/cns.13050
Schwarz DS, Blower MD (2016) The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci 73(1):79–94. https://doi.org/10.1007/s00018-015-2052-6
Omura T, Kaneko M, Okuma Y, Matsubara K, Nomura Y (2013) Endoplasmic reticulum stress and Parkinson’s disease: the role of HRD1 in averting apoptosis in neurodegenerative disease. Oxid Med Cell Longev 2013:239854. https://doi.org/10.1155/2013/239854
Xu C, Bailly-Maitre B, Reed JC (2005) Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115(10):2656–2664. https://doi.org/10.1172/JCI26373
Yang W, Tiffany-Castiglioni E (2007) The bipyridyl herbicide paraquat induces proteasome dysfunction in human neuroblastoma SH-SY5Y cells. J Toxicol Environ Health A 70(21):1849–1857. https://doi.org/10.1080/15287390701459262
Navarro-Yepes J, Anandhan A, Bradley E, Bohovych I, Yarabe B, de Jong A, Ovaa H, Zhou Y, Khalimonchuk O, Quintanilla-Vega B, Franco R (2016) Inhibition of protein ubiquitination by paraquat and 1-methyl-4-phenylpyridinium impairs ubiquitin-dependent protein degradation pathways. Mol Neurobiol 53(8):5229–5251. https://doi.org/10.1007/s12035-015-9414-9
Hetz C, Martinon F, Rodriguez D, Glimcher LH (2011) The unfolded protein response: integrating stress signals through the stress sensor IRE1α. Physiol Rev 91(4):1219–1243. https://doi.org/10.1152/physrev.00001.2011
Hetz C, Papa FR (2018) The unfolded protein response and cell fate control. Mol Cell 69(2):169–181. https://doi.org/10.1016/j.molcel.2017.06.017
Mou Z, Yuan Y-h, Zhang Z, Song L-k, Chen N-H (2020) Endoplasmic reticulum stress, an important factor in the development of Parkinson’s disease. Toxicol Lett 324:20–29. https://doi.org/10.1016/j.toxlet.2020.01.019
Rao RV, Bredesen DE (2004) Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol 16(6):653–662. https://doi.org/10.1016/j.ceb.2004.09.012
Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, Brown MS, Goldstein JL (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 6(6):1355–1364. https://doi.org/10.1016/S1097-2765(00)00133-7
Chen X, Shen J, Prywes R (2002) The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J Biol Chem 277(15):13045–13052. https://doi.org/10.1074/jbc.M110636200
Ali MMU, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, McAndrews C, Rowlands MG, Morgan GJ, Aherne W, Collins I, Davies FE, Pearl LH (2011) Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J 30(5):894–905. https://doi.org/10.1038/emboj.2011.18
Chen Y, Brandizzi F (2013) IRE1: ER stress sensor and cell fate executor. Trends Cell Biol 23(11):547–555. https://doi.org/10.1016/j.tcb.2013.06.005
Ni H, Rui Q, Li D, Gao R, Chen G (2018) The role of IRE1 signaling in the central nervous system diseases. Curr Neuropharmacol 16(9):1340–1347. https://doi.org/10.2174/1570159X16666180416094646
Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W (2007) Activation of the unfolded protein response in Parkinson’s disease. Biochem Biophys Res Commun 354(3):707–711. https://doi.org/10.1016/j.bbrc.2007.01.043
Hoozemans JJ, Van Haastert ES, Nijholt DA, Rozemuller AJ, Scheper W (2012) Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener Dis 10(1–4):212–215. https://doi.org/10.1159/000334536
Baek JH, Whitfield D, Howlett D, Francis P, Bereczki E, Ballard C, Hortobágyi T, Attems J, Aarsland D (2016) Unfolded protein response is activated in Lewy body dementias. Neuropathol Appl Neurobiol 42(4):352–365. https://doi.org/10.1111/nan.12260
Baek JH, Mamula D, Tingstam B, Pereira M, He Y, Svenningsson P (2019) GRP78 level is altered in the brain, but not in plasma or cerebrospinal fluid in Parkinson’s disease patients. Front Neurosci 13:697. https://doi.org/10.3389/fnins.2019.00697
Esteves AR, Cardoso SM (2020) Differential protein expression in diverse brain areas of Parkinson’s and Alzheimer’s disease patients. Sci Rep 10(1):13149. https://doi.org/10.1038/s41598-020-70174-z
Heman-Ackah SM, Manzano R, Hoozemans JJ, Scheper W, Flynn R, Haerty W, Cowley SA, Bassett AR, Wood MJ (2017) Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum Mol Genet 26(22):4441–4450. https://doi.org/10.1093/hmg/ddx331
Chinta SJ, Rane A, Poksay KS, Bredesen DE, Andersen JK, Rao RV (2008) Coupling endoplasmic reticulum stress to the cell death program in dopaminergic cells: effect of paraquat. Neuromolecular Med 10(4):333–342. https://doi.org/10.1007/s12017-008-8047-9
Huang C-L, Lee Y-C, Yang Y-C, Kuo T-Y, Huang N-K (2012) Minocycline prevents paraquat-induced cell death through attenuating endoplasmic reticulum stress and mitochondrial dysfunction. Toxicol Lett 209(3):203–210. https://doi.org/10.1016/j.toxlet.2011.12.021
Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13(2):89–102. https://doi.org/10.1038/nrm3270
Oyadomari S, Mori M (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11(4):381–389. https://doi.org/10.1038/sj.cdd.4401373
Selvaraj S, Sun Y, Watt JA, Wang S, Lei S, Birnbaumer L, Singh BB (2012) Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J Clin Invest 122(4):1354–1367. https://doi.org/10.1172/JCI61332
Li F, Ge B, Damirin A (2017) Overexpression of p58ipk protects neuroblastoma against paraquat-induced toxicity. Int J Clin Exp Pathol 10(8):8233–8242
Hu H, Tian M, Ding C, Yu S (2019) The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front Immunol 9 (3083). https://doi.org/10.3389/fimmu.2018.03083
Chen C, Turnbull DM, Reeve AK (2019) Mitochondrial dysfunction in Parkinson's disease-cause or consequence? Biology 8 (2). https://doi.org/10.3390/biology8020038
Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38(5):515–517. https://doi.org/10.1038/ng1769
Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem 54(3):823–827. https://doi.org/10.1111/j.1471-4159.1990.tb02325.x
Parker WD Jr, Parks JK, Swerdlow RH (2008) Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res 1189:215–218. https://doi.org/10.1016/j.brainres.2007.10.061
Flønes IH, Fernandez-Vizarra E, Lykouri M, Brakedal B, Skeie GO, Miletic H, Lilleng PK, Alves G, Tysnes O-B, Haugarvoll K, Dölle C, Zeviani M, Tzoulis C (2018) Neuronal complex I deficiency occurs throughout the Parkinson’s disease brain, but is not associated with neurodegeneration or mitochondrial DNA damage. Acta Neuropathol 135(3):409–425. https://doi.org/10.1007/s00401-017-1794-7
Keeney PM, Xie J, Capaldi RA, Bennett JP Jr (2006) Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci 26(19):5256–5264. https://doi.org/10.1523/jneurosci.0984-06.2006
Choi WS, Kruse SE, Palmiter RD, Xia Z (2008) Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc Natl Acad Sci U S A 105(39):15136–15141. https://doi.org/10.1073/pnas.0807581105
Fukushima T, Tawara T, Isobe A, Hojo N, Shiwaku K, Yamane Y (1995) Radical formation site of cerebral complex I and Parkinson’s disease. J Neurosci Res 42(3):385–390. https://doi.org/10.1002/jnr.490420313
Fukushima T, Yamada K, Isobe A, Shiwaku K, Yamane Y (1993) Mechanism of cytotoxicity of paraquat: I.NADH oxidation and paraquat radical formation via complex I. Exp Toxicol Pathol 45(5):345–349. https://doi.org/10.1016/S0940-2993(11)80424-0
Fussell KC, Udasin RG, Gray JP, Mishin V, Smith PJ, Heck DE, Laskin JD (2011) Redox cycling and increased oxygen utilization contribute to diquat-induced oxidative stress and cytotoxicity in Chinese hamster ovary cells overexpressing NADPH-cytochrome P450 reductase. Free Radic Biol Med 50(7):874–882. https://doi.org/10.1016/j.freeradbiomed.2010.12.035
Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G (1995) Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med 182(2):367–377. https://doi.org/10.1084/jem.182.2.367
Kang X, Chen J, Xu Z, Li H, Wang B (2007) Protective effects of Ginkgo biloba extract on paraquat-induced apoptosis of PC12 cells. Toxicol In Vitro 21(6):1003–1009. https://doi.org/10.1016/j.tiv.2007.02.004
Tatton WG, Chalmers-Redman RME, Rideout HJ, Tatton NA (1999) Mitochondrial permeability in neuronal death: possible relevance to the pathogenesis of Parkinson’s disease. Parkinsonism Relat Disord 5(4):221–229. https://doi.org/10.1016/S1353-8020(99)00041-3
Tatton NA (2000) Increased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson’s disease. Exp Neurol 166(1):29–43. https://doi.org/10.1006/exnr.2000.7489
Cai J, Yang J, Jones DP (1998) Mitochondrial control of apoptosis: the role of cytochrome c. Biochim Biophys Acta 1366(1–2):139–149. https://doi.org/10.1016/s0005-2728(98)00109-1
Fei Q, McCormack AL, Di Monte DA, Ethell DW (2008) Paraquat neurotoxicity is mediated by a Bak-dependent mechanism. J Biol Chem 283(6):3357–3364. https://doi.org/10.1074/jbc.M708451200
Yang JM, Huang HM, Cheng JJ, Huang CL, Lee YC, Chiou CT, Huang HT, Huang NK, Yang YC (2018) LGK974, a PORCUPINE inhibitor, mitigates cytotoxicity in an in vitro model of Parkinson’s disease by interfering with the WNT/beta-CATENIN pathway. Toxicology 410:65–72. https://doi.org/10.1016/j.tox.2018.09.003
Srinivasula SM, Datta P, Fan XJ, Fernandes-Alnemri T, Huang Z, Alnemri ES (2000) Molecular determinants of the caspase-promoting activity of Smac/DIABLO and its role in the death receptor pathway. J Biol Chem 275(46):36152–36157. https://doi.org/10.1074/jbc.C000533200
Kostrzewa RM (2000) Review on apoptosis vs. necrosis of substantia nigrapars compacta in parkinson’s disease. Neurotox Res 2(2):239–250. https://doi.org/10.1007/BF03033797
Barzilai A, Melamed E (2003) Molecular mechanisms of selective dopaminergic neuronal death in Parkinson’s disease. Trends Mol Med 9(3):126–132. https://doi.org/10.1016/s1471-4914(03)00020-0
Jacobson MD, Weil M, Raff MC (1997) Programmed cell death in animal development. Cell 88(3):347–354. https://doi.org/10.1016/S0092-8674(00)81873-5
Ju DT, Sivalingam K, Kuo WW, Ho TJ, Chang RL, Chung LC, Day CH, Viswanadha VP, Liao PH, Huang CY (2019) Effect of vasicinone against paraquat-induced MAPK/p53-mediated apoptosis via the IGF-1R/PI3K/AKT Pathway in a Parkinson's disease-associated SH-SY5Y cell model. Nutrients 11 (7). https://doi.org/10.3390/nu11071655
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4):495–516. https://doi.org/10.1080/01926230701320337
Tompkins MM, Basgall EJ, Zamrini E, Hill WD (1997) Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigral neurons. Am J Pathol 150(1):119–131
Chen P, Li A, Zhang M, He M, Chen Z, Wu X, Zhao C, Wang S, Liang L (2008) Protective effects of a new metalloporphyrin on paraquat-induced oxidative stress and apoptosis in N27 cells. Acta Biochim Biophys Sin 40(2):125–132. https://doi.org/10.1111/j.1745-7270.2008.00386.x
Matassov D, Kagan T, Leblanc J, Sikorska M, Zakeri Z (2004) Measurement of apoptosis by DNA fragmentation. Methods Mol Biol 282:1–17. https://doi.org/10.1385/1-59259-812-9:001
Erekat NS (2018) Apoptosis and its role in Parkinson’s disease. In: Stoker TB, Greenland JC (eds) Parkinson’s disease: pathogenesis and clinical aspects. Codon Publications, Brisbane (AU). https://doi.org/10.15586/codonpublications.parkinsonsdisease.2018.ch4
Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305(5684):626–629. https://doi.org/10.1126/science.1099320
Moran JM, Gonzalez-Polo RA, Ortiz-Ortiz MA, Niso-Santano M, Soler G, Fuentes JM (2008) Identification of genes associated with paraquat-induced toxicity in SH-SY5Y cells by PCR array focused on apoptotic pathways. J Toxicol Environ Health A 71(22):1457–1467. https://doi.org/10.1080/15287390802329364
Donepudi M, Grütter MG (2002) Structure and zymogen activation of caspases. Biophys Chem 101–102:145–153. https://doi.org/10.1016/s0301-4622(02)00151-5
Chowdhury I, Tharakan B, Bhat GK (2008) Caspases—an update. Comp Biochem Physiol B Biochem Mol Biol 151(1):10–27. https://doi.org/10.1016/j.cbpb.2008.05.010
Hartmann A, Hunot S, Michel PP, Muriel MP, Vyas S, Faucheux BA, Mouatt-Prigent A, Turmel H, Srinivasan A, Ruberg M, Evan GI, Agid Y, Hirsch EC (2000) Caspase-3: A vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson’s disease. Proc Natl Acad Sci U S A 97(6):2875–2880. https://doi.org/10.1073/pnas.040556597
Mogi M, Togari A, Kondo T, Mizuno Y, Komure O, Kuno S, Ichinose H, Nagatsu T (2000) Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J Neural Transm 107(3):335–341. https://doi.org/10.1007/s007020050028
Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F, Yang L, Beal MF, Andersen JK (2001) Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease. J Neurosci 21(24):9519–9528. https://doi.org/10.1523/jneurosci.21-24-09519.2001
Ray Chaudhuri A, Nussenzweig A (2017) The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 18(10):610–621. https://doi.org/10.1038/nrm.2017.53
Chaitanya GV, Steven AJ, Babu PP (2010) PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal 8:31–31. https://doi.org/10.1186/1478-811X-8-31
Acknowledgements
Not applicable.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Contributions
W.Z.C.S. performed the literature search and wrote the first draft of the manuscript. R.N. revised the manuscript and gave critical comments. K.S.T. contributed to the conception and extensive revision of the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
See, W.Z.C., Naidu, R. & Tang, K.S. Cellular and Molecular Events Leading to Paraquat-Induced Apoptosis: Mechanistic Insights into Parkinson’s Disease Pathophysiology. Mol Neurobiol 59, 3353–3369 (2022). https://doi.org/10.1007/s12035-022-02799-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-02799-2